
The BET Protein Inhibitor JQ1 Decreases Hypoxia and Improves the Therapeutic Benefit of Anti-PD-1 in a High-Risk Neuroblastoma Mouse Model
 , and
, and
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Culture
2.2. Protein Extraction for Western Blotting
2.3. RNA Extraction, Reverse Transcription, and RT-qPCR
2.4. In Vivo Experiments and Transgenic TH-MYCN Mouse Model
2.5. Immunohistochemistry on Tumors
2.6. Magnetic Resonance Imaging (MRI) and Images Acquisition
2.7. Data Analysis and Statistics
2.8. Fluorescence-Activated Cell Sorting (FACS) In Vivo
2.9. Survival Curves
3. Results
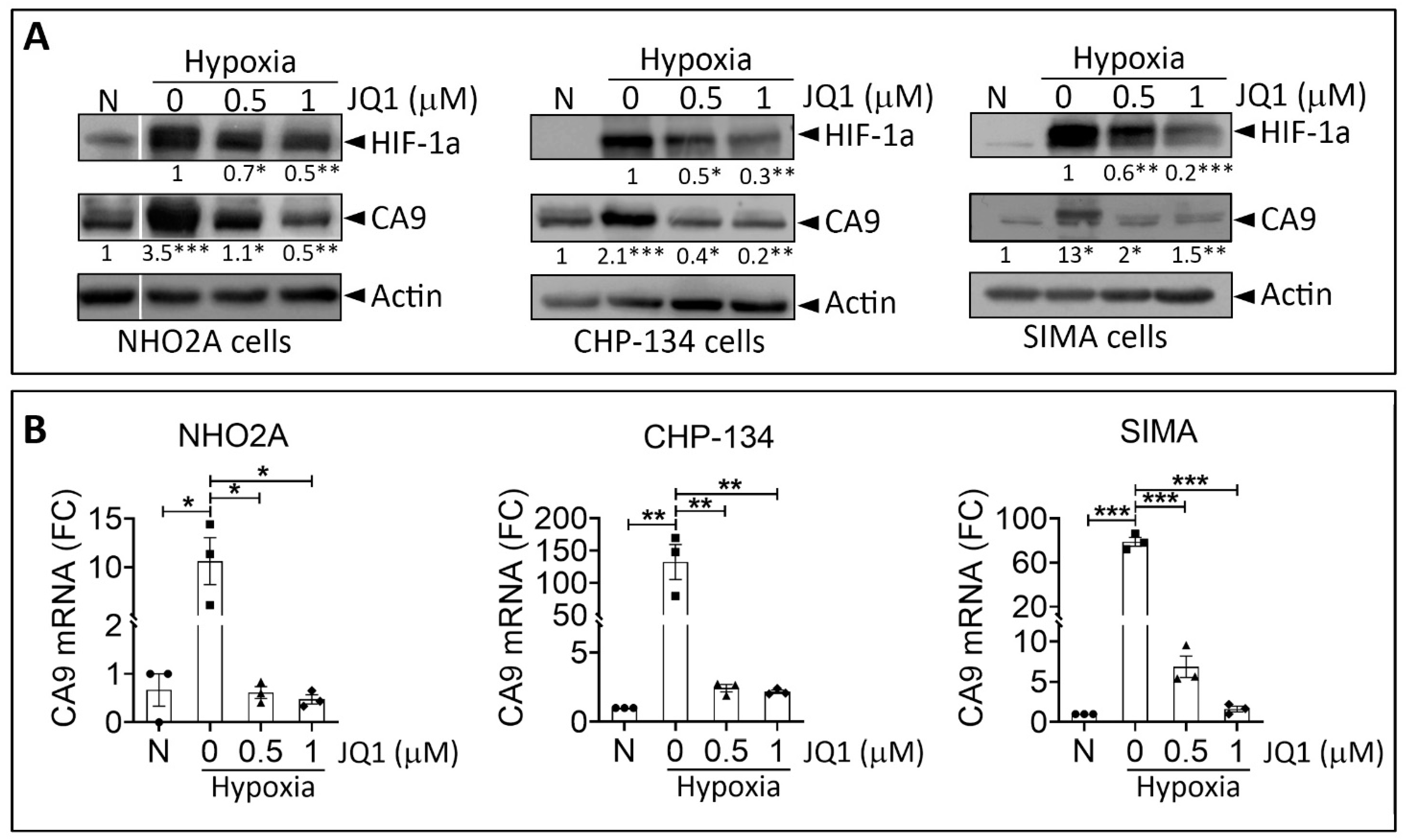
3.1. JQ1 Impairs Hypoxic Responses in Neuroblastoma Cells In Vitro
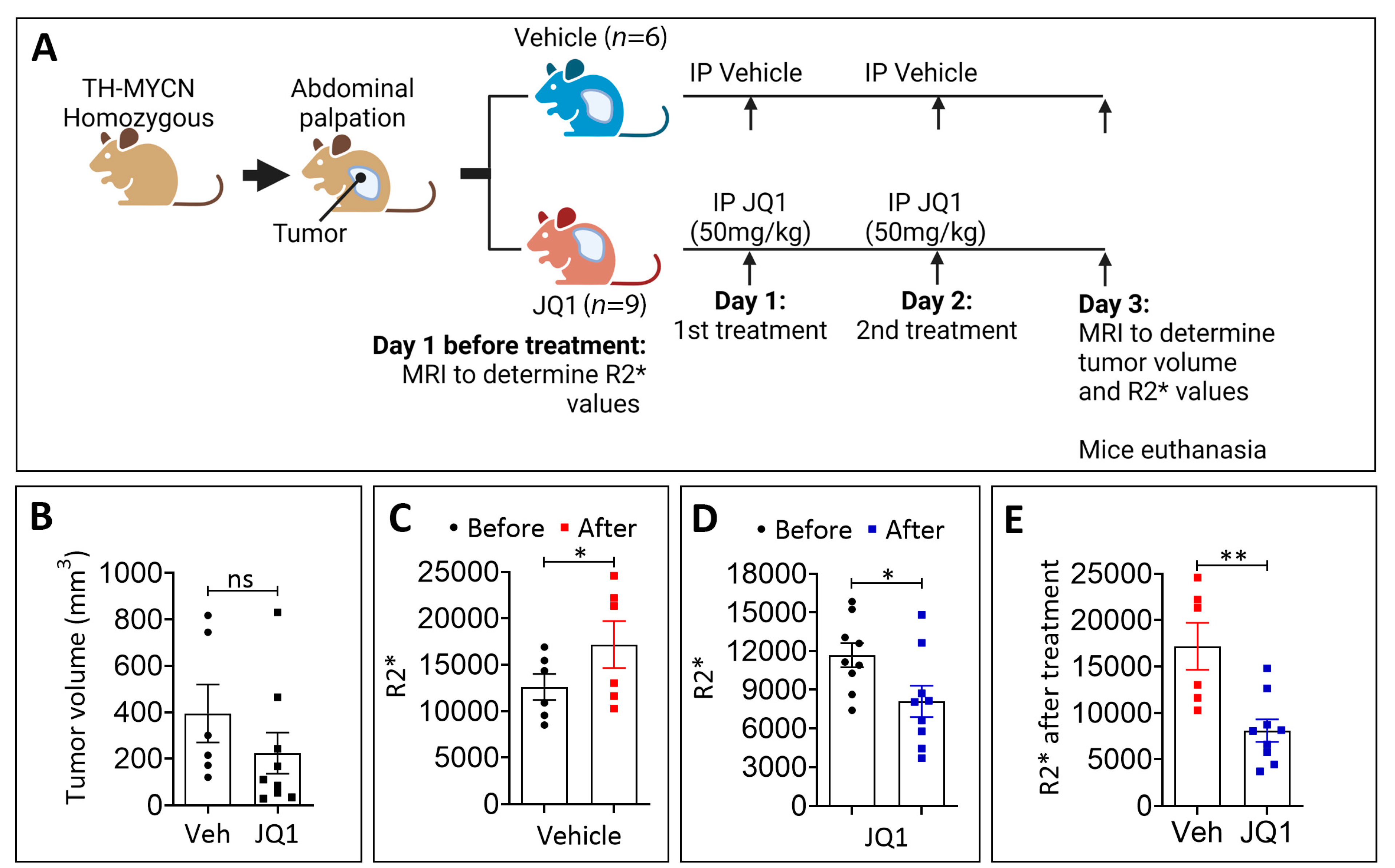
3.2. JQ1 Treatment Reduces Hypoxia in TH-MYCN Tumors
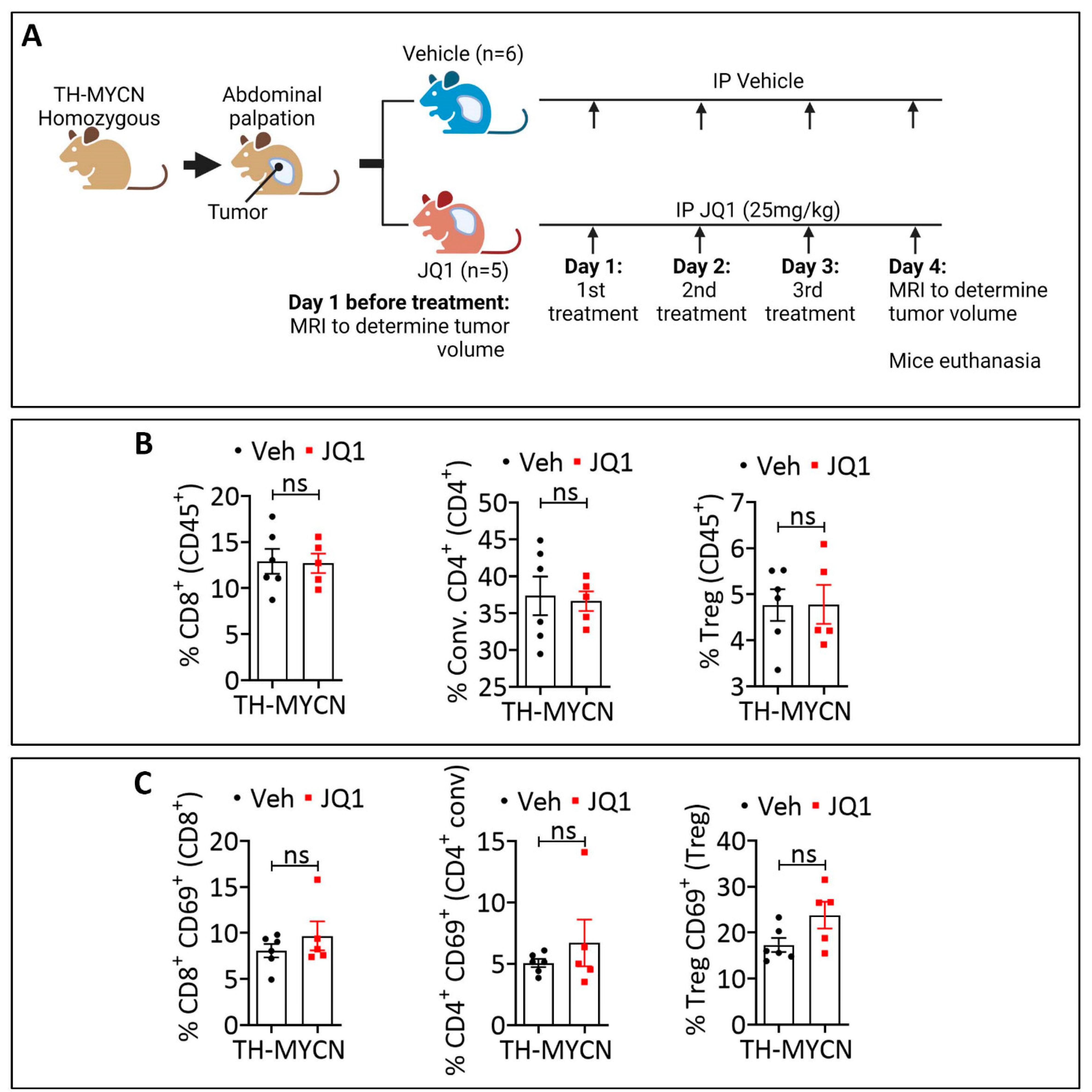
3.3. JQ1-Treated Tumors Displayed a Higher Infiltration Level of CD8+ PD-1+, Conventional CD4+ PD-1+, and Treg PD-1+ Cells Compared to the Control
3.4. Combining JQ1 Improves the Therapeutic Benefit of PD-1 in TH-MYCN Tumor-Bearing Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayat, M.A. Neuroblastoma: Diagnosis, Therapy and Prognosis; Springer: Dordrecht, The Netherlands, 2012. [Google Scholar]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef]
- Pinto, N.R.; Applebaum, M.A.; Volchenboum, S.L.; Matthay, K.K.; London, W.B.; Ambros, P.F.; Nakagawara, A.; Berthold, F.; Schleiermacher, G.; Park, J.R.; et al. Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J. Clin. Oncol. 2015, 33, 3008–3017. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.K.; Dyer, M.A. Neuroblastoma: Developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.U.; Shohet, J.M. Neuroblastoma: Molecular pathogenesis and therapy. Annu. Rev. Med. 2015, 66, 49–63. [Google Scholar] [CrossRef]
- Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O. Recent insights into the biology of neuroblastoma. Int. J. Cancer 2014, 135, 2249–2261. [Google Scholar] [CrossRef] [PubMed]
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116. [Google Scholar] [CrossRef]
- Bagatell, R.; Beck-Popovic, M.; London, W.B.; Zhang, Y.; Pearson, A.D.; Matthay, K.K.; Monclair, T.; Ambros, P.F.; Cohn, S.L.; International Neuroblastoma Risk, G. Significance of MYCN amplification in international neuroblastoma staging system stage 1 and 2 neuroblastoma: A report from the International Neuroblastoma Risk Group database. J. Clin. Oncol. 2009, 27, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Mosse, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Janoueix-Lerosey, I.; Lequin, D.; Brugieres, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Bresler, S.C.; Weiser, D.A.; Huwe, P.J.; Park, J.H.; Krytska, K.; Ryles, H.; Laudenslager, M.; Rappaport, E.F.; Wood, A.C.; McGrady, P.W.; et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 2014, 26, 682–694. [Google Scholar] [CrossRef] [Green Version]
- Valentijn, L.J.; Koster, J.; Zwijnenburg, D.A.; Hasselt, N.E.; van Sluis, P.; Volckmann, R.; van Noesel, M.M.; George, R.E.; Tytgat, G.A.; Molenaar, J.J.; et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat. Genet. 2015, 47, 1411–1414. [Google Scholar] [CrossRef]
- Peifer, M.; Hertwig, F.; Roels, F.; Dreidax, D.; Gartlgruber, M.; Menon, R.; Kramer, A.; Roncaioli, J.L.; Sand, F.; Heuckmann, J.M.; et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015, 526, 700–704. [Google Scholar] [CrossRef]
- Yang, L.; Ke, X.X.; Xuan, F.; Tan, J.; Hou, J.; Wang, M.; Cui, H.; Zhang, Y. PHOX2B Is Associated with Neuroblastoma Cell Differentiation. Cancer Biother. Radiopharm. 2016, 31, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Pritchard, J.; Berthold, F.; Carlsen, N.L.; Castel, V.; Castelberry, R.P.; De Bernardi, B.; Evans, A.E.; Favrot, M.; Hedborg, F.; et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J. Clin. Oncol. 1993, 11, 1466–1477. [Google Scholar] [CrossRef]
- Sokol, E.; Desai, A.V. The Evolution of Risk Classification for Neuroblastoma. Children 2019, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Smith, V.; Foster, J. High-Risk Neuroblastoma Treatment Review. Children 2018, 5, 114. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1 and mechanisms of hypoxia sensing. Curr. Opin. Cell Biol. 2001, 13, 167–171. [Google Scholar] [CrossRef]
- Liu, Y.V.; Baek, J.H.; Zhang, H.; Diez, R.; Cole, R.N.; Semenza, G.L. RACK1 competes with HSP90 for binding to HIF-1alpha and is required for O(2)-independent and HSP90 inhibitor-induced degradation of HIF-1alpha. Mol. Cell 2007, 25, 207–217. [Google Scholar] [CrossRef]
- Qing, G.; Skuli, N.; Mayes, P.A.; Pawel, B.; Martinez, D.; Maris, J.M.; Simon, M.C. Combinatorial regulation of neuroblastoma tumor progression by N-Myc and hypoxia inducible factor HIF-1alpha. Cancer Res. 2010, 70, 10351–10361. [Google Scholar] [CrossRef] [Green Version]
- Pahlman, S.; Mohlin, S. Hypoxia and hypoxia-inducible factors in neuroblastoma. Cell Tissue Res. 2018, 372, 269–275. [Google Scholar] [CrossRef]
- Romano, N.; Veronese, M.; Manfrini, N.; Zolla, L.; Ceci, M. Ribosomal RACK1 promotes proliferation of neuroblastoma cells independently of global translation upregulation. Cell Signal 2019, 53, 102–110. [Google Scholar] [CrossRef]
- Da Motta, L.L.; Ledaki, I.; Purshouse, K.; Haider, S.; De Bastiani, M.A.; Baban, D.; Morotti, M.; Steers, G.; Wigfield, S.; Bridges, E.; et al. The BET inhibitor JQ1 selectively impairs tumour response to hypoxia and downregulates CA9 and angiogenesis in triple negative breast cancer. Oncogene 2017, 36, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Weiss, W.A.; Aldape, K.; Mohapatra, G.; Feuerstein, B.G.; Bishop, J.M. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997, 16, 2985–2995. [Google Scholar] [CrossRef]
- Little, R.A.; Jamin, Y.; Boult, J.K.R.; Naish, J.H.; Watsoon, Y.; Cheung, S.; Holliday, K.F.; Lu, H.; McHugh, D.J.; Irlam, J.; et al. Mapping Hypoxia in Renal Carcinoma with Oxygen-enhanced MRI: Comparison with Intrinsic Susceptibility MRI and Pathology. Radiology 2018, 288, 739–747. [Google Scholar] [CrossRef]
- Ma, M.; Liang, J.; Zhang, D.; Xu, X.; Cheng, Q.; Xiao, Z.; Shi, C.; Luo, L. Monitoring Treatment Efficacy of Antiangiogenic Therapy Combined With Hypoxia-Activated Prodrugs Online Using Functional MRI. Front. Oncol. 2021, 11, 672047. [Google Scholar] [CrossRef] [PubMed]
- Jamin, Y.; Glass, L.; Hallsworth, A.; George, R.; Koh, D.M.; Pearson, A.D.; Chesler, L.; Robinson, S.P. Intrinsic susceptibility MRI identifies tumors with ALKF1174L mutation in genetically-engineered murine models of high-risk neuroblastoma. PLoS ONE 2014, 9, e92886. [Google Scholar] [CrossRef]
- Schaaf, M.B.; Garg, A.D.; Agostinis, P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018, 9, 115. [Google Scholar] [CrossRef]
- Chouaib, S.; Messai, Y.; Couve, S.; Escudier, B.; Hasmim, M.; Noman, M.Z. Hypoxia promotes tumor growth in linking angiogenesis to immune escape. Front. Immunol. 2012, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Baginska, J.; Viry, E.; Paggetti, J.; Medves, S.; Berchem, G.; Moussay, E.; Janji, B. The critical role of the tumor microenvironment in shaping natural killer cell-mediated anti-tumor immunity. Front. Immunol. 2013, 4, 490. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Deng, Y.; Cheng, J.; Wei, S.; Luo, H.; Liu, L. Tumor-Infiltrating PD-1(hi)CD8(+)-T-Cell Signature as an Effective Biomarker for Immune Checkpoint Inhibitor Therapy Response Across Multiple Cancers. Front. Oncol. 2021, 11, 695006. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, I.; Baruchel, A.; Doz, F.; Schulte, J. Bromodomain and extraterminal protein inhibitors in pediatrics: A review of the literature. Pediatr. Blood Cancer 2017, 64, e26334. [Google Scholar] [CrossRef]
- Bid, H.K.; Kerk, S. BET bromodomain inhibitor (JQ1) and tumor angiogenesis. Oncoscience 2016, 3, 316–317. [Google Scholar] [CrossRef] [PubMed]
- Benaron, D.A.; Benitz, W.E.; Ariagno, R.L.; Stevenson, D.K. Noninvasive methods for estimating in vivo oxygenation. Clin. Pediatr. 1992, 31, 258–273. [Google Scholar] [CrossRef]
- Thommen, D.S.; Koelzer, V.H.; Herzig, P.; Roller, A.; Trefny, M.; Dimeloe, S.; Kiialainen, A.; Hanhart, J.; Schill, C.; Hess, C.; et al. A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med. 2018, 24, 994–1004. [Google Scholar] [CrossRef]
- Duchemann, B.; Naigeon, M.; Auclin, E.; Ferrara, R.; Cassard, L.; Jouniaux, J.M.; Boselli, L.; Grivel, J.; Desnoyer, A.; Danlos, F.X.; et al. CD8(+)PD-1(+) to CD4(+)PD-1(+) ratio (PERLS) is associated with prognosis of patients with advanced NSCLC treated with PD-(L)1 blockers. J. Immunother. Cancer 2022, 10, e004012. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Nishina, H.; Takimoto, H.; Marengere, L.E.; Wakeham, A.C.; Bouchard, D.; Kong, Y.Y.; Ohteki, T.; Shahinian, A.; Bachmann, M.; et al. The transcription factor NF-ATc1 regulates lymphocyte proliferation and Th2 cytokine production. Immunity 1998, 8, 115–124. [Google Scholar] [CrossRef]
- Klein-Hessling, S.; Muhammad, K.; Klein, M.; Pusch, T.; Rudolf, R.; Floter, J.; Qureischi, M.; Beilhack, A.; Vaeth, M.; Kummerow, C.; et al. NFATc1 controls the cytotoxicity of CD8(+) T cells. Nat. Commun. 2017, 8, 511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Fei, Q.; Xiong, P.; Yang, J.; Zhang, Z.; Lin, X.; Pan, M.; Lu, F.; Huang, H. Synergistic inhibition of pancreatic cancer with anti-PD-L1 and c-Myc inhibitor JQ1. Oncoimmunology 2019, 8, e1581529. [Google Scholar] [CrossRef] [PubMed]
- Adeegbe, D.O.; Liu, S.; Hattersley, M.M.; Bowden, M.; Zhou, C.W.; Li, S.; Vlahos, R.; Grondine, M.; Dolgalev, I.; Ivanova, E.V.; et al. BET Bromodomain Inhibition Cooperates with PD-1 Blockade to Facilitate Antitumor Response in Kras-Mutant Non-Small Cell Lung Cancer. Cancer Immunol. Res. 2018, 6, 1234–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence | Purpose | Parameters | Number of Mice in Group Treated with | |
|---|---|---|---|---|
| JQ1 | Vehicle | |||
| FSE T2w | Tumor detection, delineation and volume measurement | SR: 156 µm × 161 µm × 1500 µm, Sl: 16, TE: 68 ms, TR: 5000 ms, ET: 8, AVG: 3, SD: 7 min 45 s, RG ON | 9 | 6 |
| FSE T1w | Tumor detection, delineation and volume measurement | SR: 156 µm × 159 µm × 1500 µm, Sl: 16, TE: 11 ms, TR: 1000 ms, ET: 4, AVG: 4, SD: 4 min 12 s, RG ON | ||
| MEMS | T2 relaxometry map | SR: 156 µm × 208 µm × 1500 µm, Sl: 16, fTE: 15 ms, NOE: 10, TR: 3000 ms, AVG: 1, SD: 9 min 36 s, RG ON | ||
| MGE | T2* relaxometry map | FLASH, SR: 313 µm × 313 µm × 1500 µm, Sl: 16, fTE: 4 ms, TR: 600 ms, NOE: 10, AVG: 1, SD: 1 min 16 s, RG ON | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sauvage, D.; Bosseler, M.; Viry, E.; Kanli, G.; Oudin, A.; Berchem, G.; Keunen, O.; Janji, B. The BET Protein Inhibitor JQ1 Decreases Hypoxia and Improves the Therapeutic Benefit of Anti-PD-1 in a High-Risk Neuroblastoma Mouse Model. Cells 2022, 11, 2783. https://doi.org/10.3390/cells11182783
Sauvage D, Bosseler M, Viry E, Kanli G, Oudin A, Berchem G, Keunen O, Janji B. The BET Protein Inhibitor JQ1 Decreases Hypoxia and Improves the Therapeutic Benefit of Anti-PD-1 in a High-Risk Neuroblastoma Mouse Model. Cells. 2022; 11(18):2783. https://doi.org/10.3390/cells11182783
Chicago/Turabian StyleSauvage, Delphine, Manon Bosseler, Elodie Viry, Georgia Kanli, Anais Oudin, Guy Berchem, Olivier Keunen, and Bassam Janji. 2022. "The BET Protein Inhibitor JQ1 Decreases Hypoxia and Improves the Therapeutic Benefit of Anti-PD-1 in a High-Risk Neuroblastoma Mouse Model" Cells 11, no. 18: 2783. https://doi.org/10.3390/cells11182783