
The ATG8 Family Proteins GABARAP and GABARAPL1 Target Antigen to Dendritic Cells to Prime CD4+ and CD8+ T Cells
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid Constructs
2.2. Cell Culture
2.3. Reagents, Peptides and Antibodies
2.4. Dendritic Cell Generation
2.5. Cell Transfections
2.6. T-Cell Proliferation Assay
2.7. Western Blotting
2.8. Mice
2.9. Statistical Analyses
3. Results
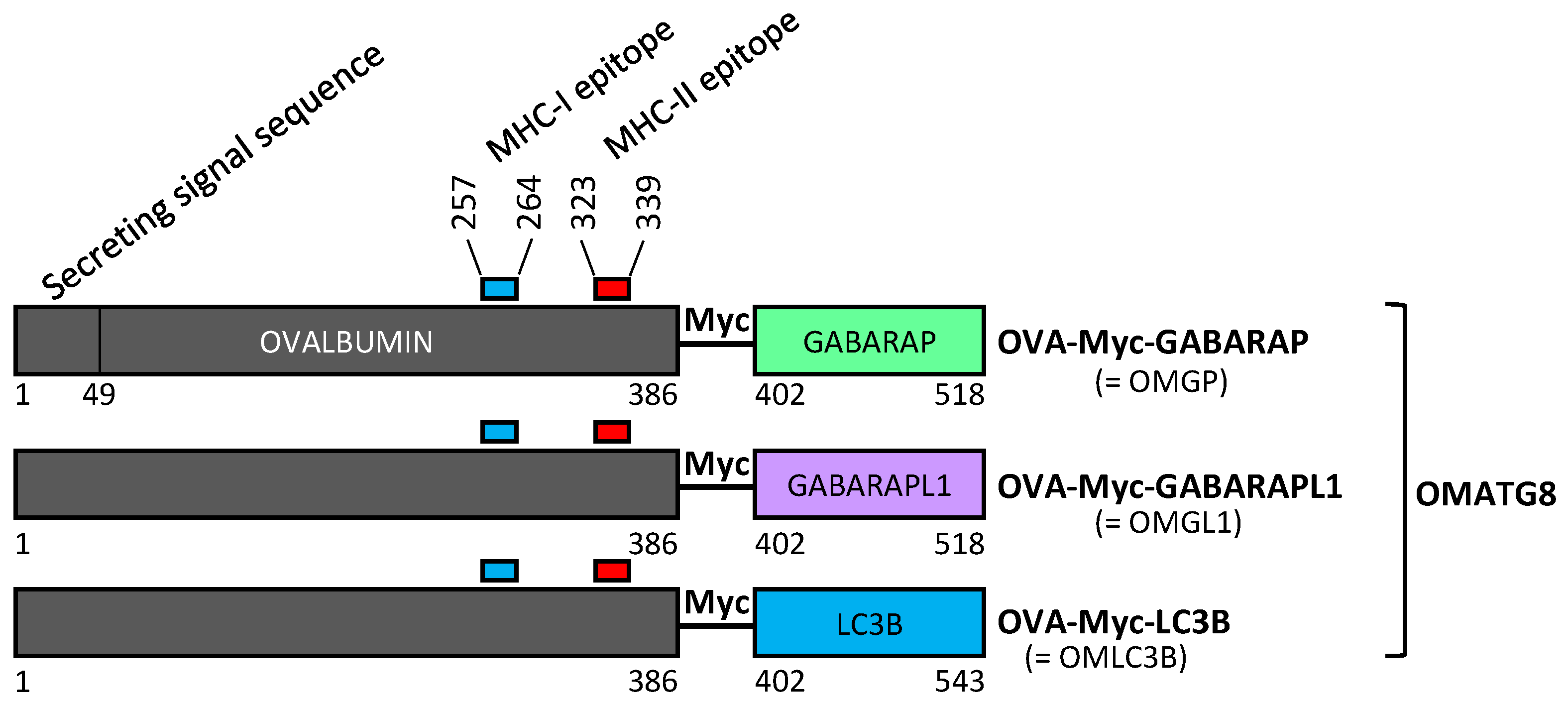
3.1. Design of Plasmid Constructions Expressing OVALBUMIN Fused with the ATG8 Autophagy Proteins GABARAP and GABARAPL1
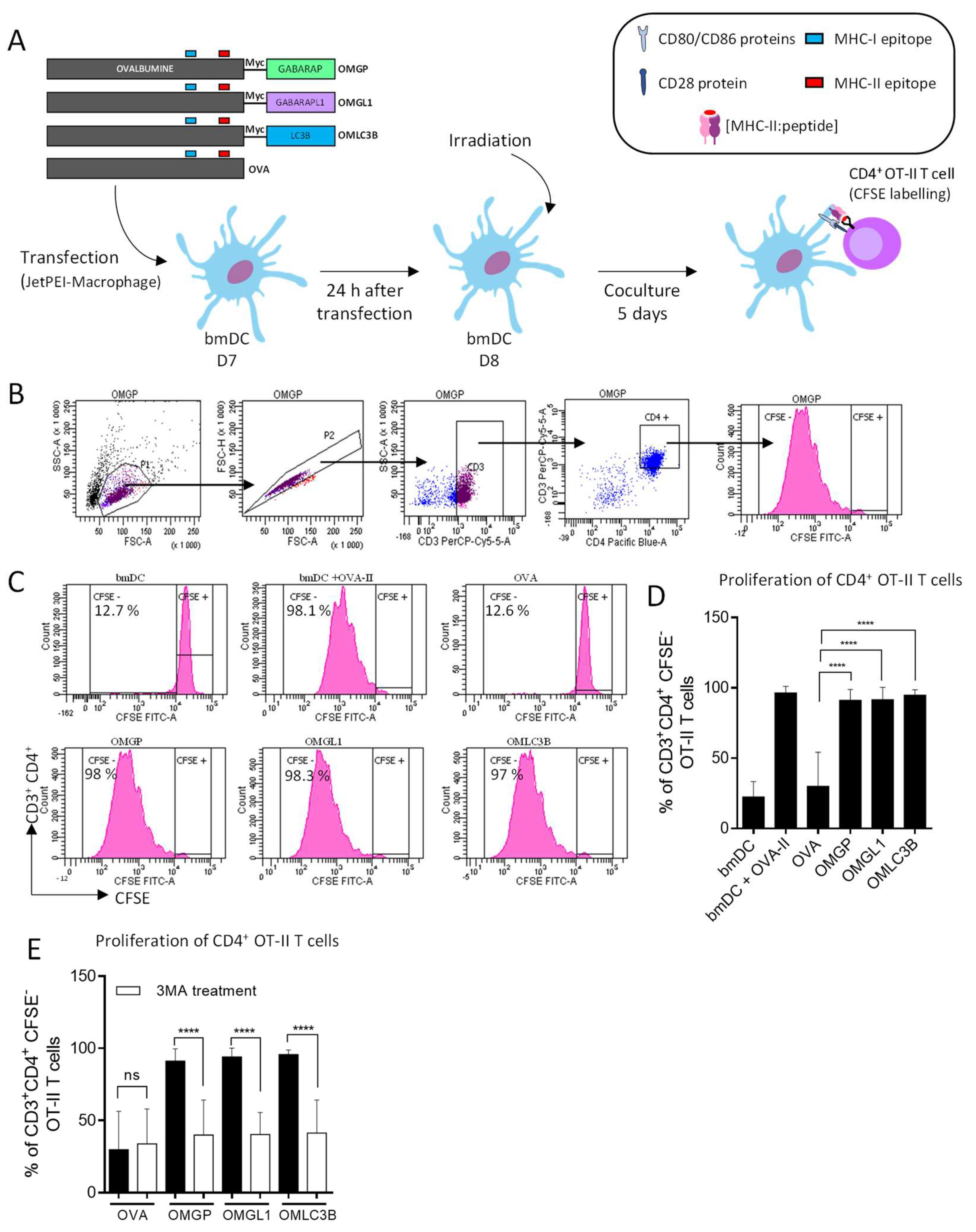
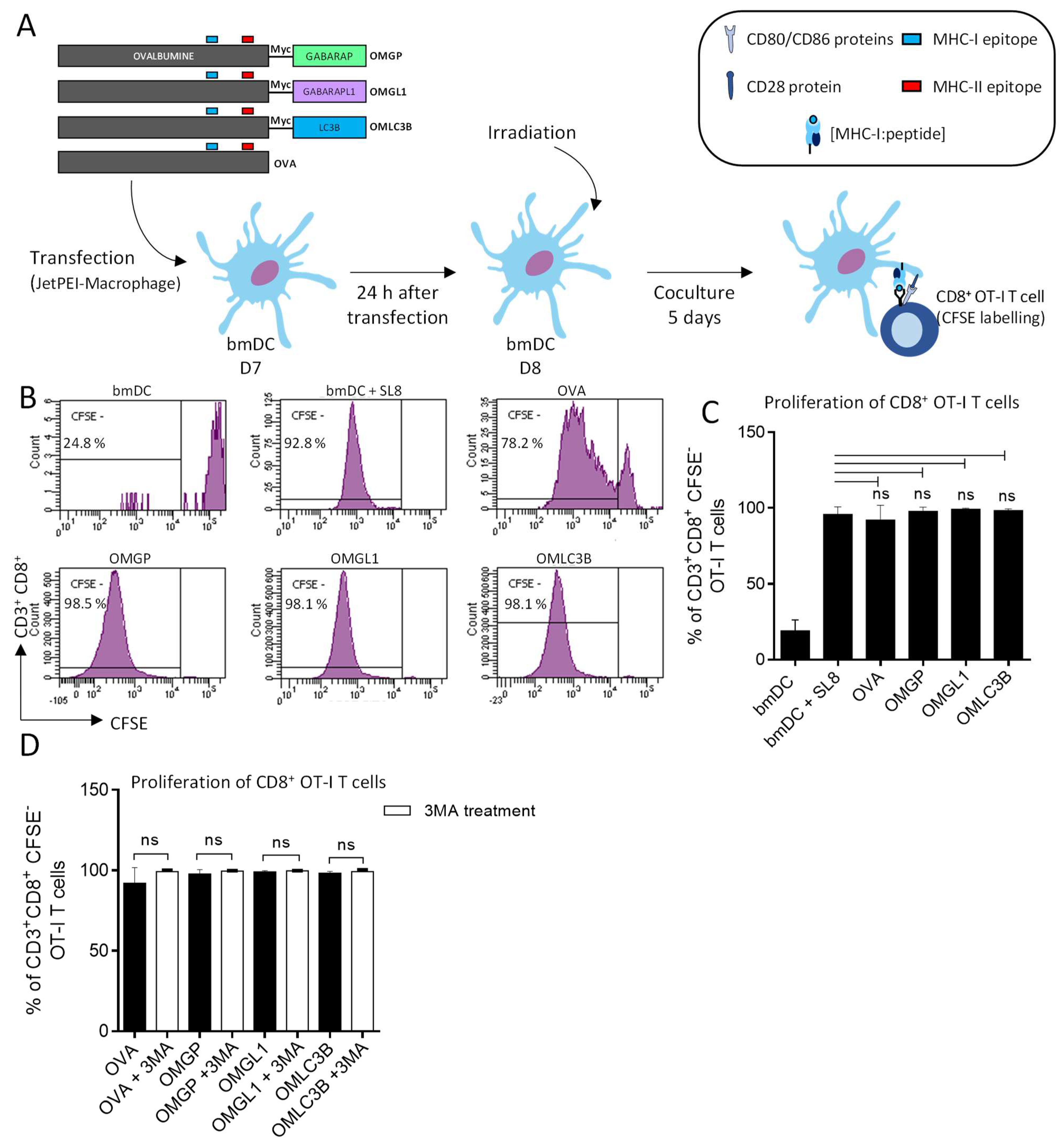
3.2. The Overexpression of OVA-Myc-GABARAP and OVA-Myc-GABARAPL1 Proteins in Dendritic Cells Induce Efficient MHC-II-Restricted Peptide Presentation
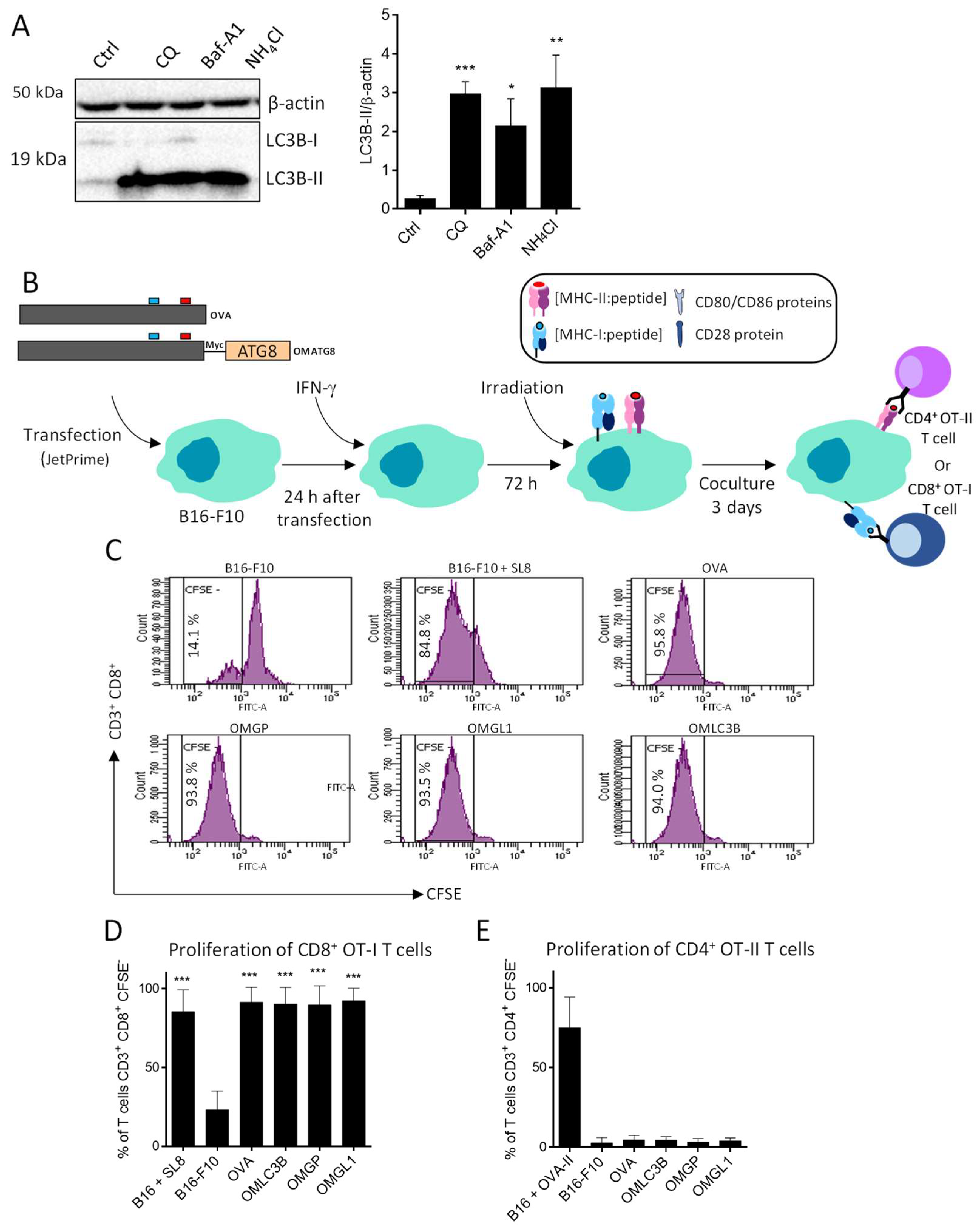
3.3. B16-F10 Genetically Modified to Expressed OMGP or OMGL1 Fusion Proteins Did Not Enhance T Cell Recognition
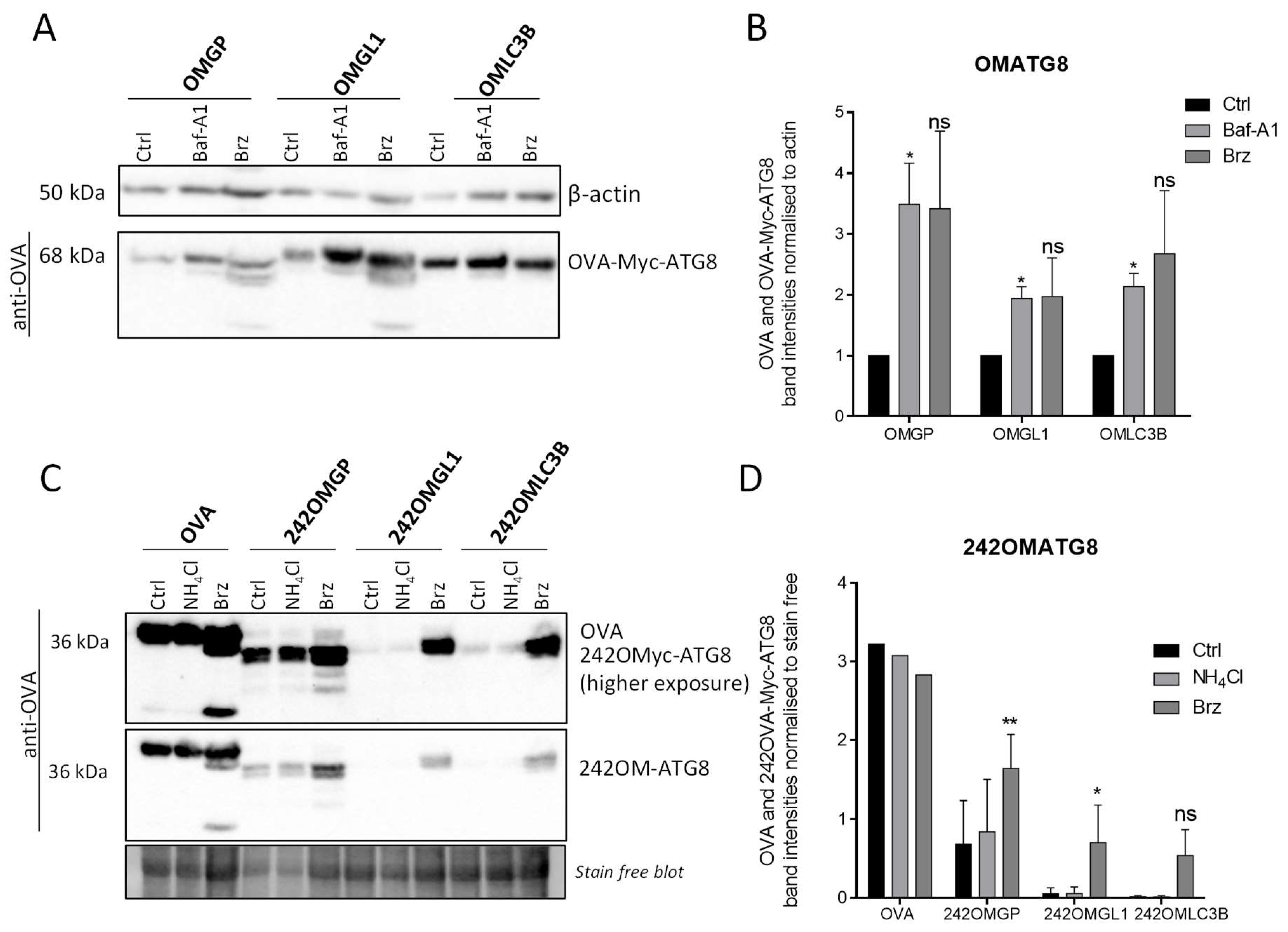
3.4. OVA-Myc-ATG8 Fusion Proteins Are Highly Degraded by Proteasome Pathway in B16-F10 Cell Lines
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adotévi, O.; Dosset, M.; Galaine, J.; Beziaud, L.; Godet, Y.; Borg, C. Targeting Antitumor CD4 Helper T Cells with Universal Tumor-Reactive Helper Peptides Derived from Telomerase for Cancer Vaccine. Hum. Vaccines Immunother. 2013, 9, 1073–1077. [Google Scholar] [CrossRef] [PubMed]
- Hunder, N.N.; Wallen, H.; Cao, J.; Hendricks, D.W.; Reilly, J.Z.; Rodmyre, R.; Jungbluth, A.; Gnjatic, S.; Thompson, J.A.; Yee, C. Treatment of Metastatic Melanoma with Autologous CD4+ T Cells against NY-ESO-1. N. Engl. J. Med. 2008, 358, 2698–2703. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Chhabra, A.; Chakraborty, N.G.; Hegde, U.; Dorsky, D.I.; Chodon, T.; von Euw, E.; Comin-Anduix, B.; Koya, R.C.; Ribas, A.; et al. MHC-I-Restricted Melanoma Antigen Specific TCR-Engineered Human CD4+ T Cells Exhibit Multifunctional Effector and Helper Responses, in Vitro. Clin. Immunol. 2010, 136, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Haabeth, O.A.W.; Tveita, A.A.; Fauskanger, M.; Schjesvold, F.; Lorvik, K.B.; Hofgaard, P.O.; Omholt, H.; Munthe, L.A.; Dembic, Z.; Corthay, A.; et al. How Do CD4(+) T Cells Detect and Eliminate Tumor Cells That Either Lack or Express MHC Class II Molecules? Front. Immunol. 2014, 5, 174. [Google Scholar] [CrossRef] [PubMed]
- Janssen, E.M.; Lemmens, E.E.; Wolfe, T.; Christen, U.; von Herrath, M.G.; Schoenberger, S.P. CD4+ T Cells Are Required for Secondary Expansion and Memory in CD8+ T Lymphocytes. Nature 2003, 421, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Andersen, A.N.; Landsverk, O.J.; Simonsen, A.; Bogen, B.; Corthay, A.; Øynebråten, I. Coupling of HIV-1 Antigen to the Selective Autophagy Receptor SQSTM1/P62 Promotes T-Cell-Mediated Immunity. Front. Immunol. 2016, 7, 167. [Google Scholar] [CrossRef]
- Fonteneau, J.F.; Brilot, F.; Münz, C.; Gannagé, M. The Tumor Antigen NY-ESO-1 Mediates Direct Recognition of Melanoma Cells by CD4+ T Cells after Intercellular Antigen Transfer. J. Immunol. 2016, 196, 64–71. [Google Scholar] [CrossRef]
- Li, Y.; Wang, L.-X.; Pang, P.; Cui, Z.; Aung, S.; Haley, D.; Fox, B.A.; Urba, W.J.; Hu, H.-M. Tumor-Derived Autophagosome Vaccine: Mechanism of Cross-Presentation and Therapeutic Efficacy. Clin. Cancer Res. 2011, 17, 7047–7057. [Google Scholar] [CrossRef]
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of Antigen Processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef]
- Trombetta, E.S.; Mellman, I. Cell Biology of Antigen Processing in Vitro and in Vivo. Annu. Rev. Immunol. 2004, 23, 975–1028. [Google Scholar] [CrossRef]
- Chemali, M.; Radtke, K.; Desjardins, M.; English, L. Alternative Pathways for MHC Class I Presentation: A New Function for Autophagy. Cell. Mol. Life Sci. 2011, 68, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Munz, C. Antigen Processing for MHC Class II Presentation via Autophagy. Front. Immunol. 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Baehrecke, E.H.; Brumell, J.H.; Chu, C.T.; Codogno, P.; Cuervo, A.M.; Debnath, J.; Deretic, V.; Elazar, Z.; Eskelinen, E.-L.; et al. A Comprehensive Glossary of Autophagy-Related Molecules and Processes (2nd Edition). Autophagy 2011, 7, 1273–1294. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, I.; Vuillermoz, C.; Jouvenot, M.; Ordener, C.; Royez, M.; Adessi, G.L. Identification and Characterization of an Early Estrogen-Regulated RNA in Cultured Guinea-Pig Endometrial Cells. Mol. Cell. Endocrinol. 1993, 90, R17–R21. [Google Scholar] [CrossRef]
- Le Grand, J.N.; Chakrama, F.Z.; Seguin-Py, S.; Fraichard, A.; Delage-Mourroux, R.; Jouvenot, M.; Boyer-Guittaut, M. GABARAPL1 (GEC1): Original or Copycat? Autophagy 2011, 7, 1098–1107. [Google Scholar] [CrossRef]
- Jacquet, M.; Guittaut, M.; Fraichard, A.; Despouy, G. The Functions of Atg8-Family Proteins in Autophagy and Cancer: Linked or Unrelated? Autophagy 2020, 17, 599–611. [Google Scholar] [CrossRef]
- Schmid, D.; Pypaert, M.; Münz, C. MHC Class II Antigen Loading Compartments Continuously Receive Input from Autophagosomes. Immunity 2007, 26, 79–92. [Google Scholar] [CrossRef]
- Dengjel, J.; Schoor, O.; Fischer, R.; Reich, M.; Kraus, M.; Müller, M.; Kreymborg, K.; Altenberend, F.; Brandenburg, J.; Kalbacher, H.; et al. Autophagy Promotes MHC Class II Presentation of Peptides from Intracellular Source Proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 7922–7927. [Google Scholar] [CrossRef]
- Jagannath, C.; Lindsey, D.R.; Dhandayuthapani, S.; Xu, Y.; Hunter, R.L.; Eissa, N.T. Autophagy Enhances the Efficacy of BCG Vaccine by Increasing Peptide Presentation in Mouse Dendritic Cells. Nat. Med. 2009, 15, 267–276. [Google Scholar] [CrossRef]
- Romao, S.; Gasser, N.; Becker, A.C.; Guhl, B.; Bajagic, M.; Vanoaica, D.; Ziegler, U.; Roesler, J.; Dengjel, J.; Reichenbach, J.; et al. Autophagy Proteins Stabilize Pathogen-Containing Phagosomes for Prolonged MHC II Antigen Processing. J. Cell Biol. 2013, 203, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Joubert, P.-E.; Albert, M.L. Antigen Cross-Priming of Cell-Associated Proteins Is Enhanced by Macroautophagy within the Antigen Donor Cell. Front. Immunol. 2012, 3, 61. [Google Scholar] [CrossRef]
- Crotzer, V.L.; Blum, J.S. Autophagy and Adaptive Immunity. Immunology 2010, 131, 9–17. [Google Scholar] [CrossRef]
- Jin, Y.; Sun, C.; Feng, L.; Li, P.; Xiao, L.; Ren, Y.; Wang, D.; Li, C.; Chen, L. Regulation of SIV Antigen-Specific CD4+ T Cellular Immunity via Autophagosome-Mediated MHC II Molecule-Targeting Antigen Presentation in Mice. PLoS ONE 2014, 9, e93143. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The Machinery of Macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Li, H.; Li, Y.; Jiao, J.; Hu, H.-M. Alpha-Alumina Nanoparticles Induce Efficient Autophagy-Dependent Cross-Presentation and Potent Antitumour Response. Nat. Nanotechnol. 2011, 6, 645–650. [Google Scholar] [CrossRef]
- Nakatogawa, H. Two Ubiquitin-like Conjugation Systems That Mediate Membrane Formation during Autophagy. Essays Biochem. 2013, 55, 39–50. [Google Scholar] [CrossRef]
- Coulon, P.-G.; Richetta, C.; Rouers, A.; Blanchet, F.P.; Urrutia, A.; Guerbois, M.; Piguet, V.; Theodorou, I.; Bet, A.; Schwartz, O.; et al. HIV-Infected Dendritic Cells Present Endogenous MHC Class II–Restricted Antigens to HIV-Specific CD4+ T Cells. J. Immunol. 2016, 197, 517–532. [Google Scholar] [CrossRef]
- Seglen, P.O.; Gordon, P.B. 3-Methyladenine: Specific Inhibitor of Autophagic/Lysosomal Protein Degradation in Isolated Rat Hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Milosevic, S.; Behrends, U.; Jaffee, E.M.; Pardoll, D.M.; Bornkamm, G.W.; Mautner, J. Major Histocompatibility Complex Class II-Restricted Presentation of a Cytosolic Antigen by Autophagy. Eur. J. Immunol. 2003, 33, 1250–1259. [Google Scholar] [CrossRef]
- Riedel, A.; Nimmerjahn, F.; Burdach, S.; Behrends, U.; Bornkamm, G.W.; Mautner, J. Endogenous Presentation of a Nuclear Antigen on MHC Class II by Autophagy in the Absence of CRM1-Mediated Nuclear Export. Eur. J. Immunol. 2008, 38, 2090–2095. [Google Scholar] [CrossRef]
- Brazil, M.I.; Weiss, S.; Stockinger, B. Excessive Degradation of Intracellular Protein in Macrophages Prevents Presentation in the Context of Major Histocompatibility Complex Class II Molecules. Eur. J. Immunol. 1997, 27, 1506–1514. [Google Scholar] [CrossRef]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Münz, C. Endogenous MHC Class II Processing of a Viral Nuclear Antigen After Autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef]
- Dörfel, D.; Appel, S.; Grünebach, F.; Weck, M.M.; Müller, M.R.; Heine, A.; Brossart, P. Processing and Presentation of HLA Class I and II Epitopes by Dendritic Cells after Transfection with in Vitro-Transcribed MUC1 RNA. Blood 2005, 105, 3199–3205. [Google Scholar] [CrossRef]
- Lee, H.K.; Mattei, L.M.; Steinberg, B.E.; Alberts, P.; Lee, Y.H.; Chervonsky, A.; Mizushima, N.; Grinstein, S.; Iwasaki, A. In Vivo Requirement for Atg5 in Antigen Presentation by Dendritic Cells. Immunity 2010, 32, 227–239. [Google Scholar] [CrossRef]
- Borst, J.; Ahrends, T.; Bąbała, N.; Melief, C.J.M.; Kastenmüller, W. CD4+ T Cell Help in Cancer Immunology and Immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic Cancer Vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef]
- Renaude, E.; Kroemer, M.; Borg, C.; Peixoto, P.; Hervouet, E.; Loyon, R.; Adotévi, O. Epigenetic Reprogramming of CD4+ Helper T Cells as a Strategy to Improve Anticancer Immunotherapy. Front. Immunol. 2021, 12, 669992. [Google Scholar] [CrossRef]
- Basu, A.; Ramamoorthi, G.; Albert, G.; Gallen, C.; Beyer, A.; Snyder, C.; Koski, G.; Disis, M.L.; Czerniecki, B.J.; Kodumudi, K. Differentiation and Regulation of TH Cells: A Balancing Act for Cancer Immunotherapy. Front. Immunol. 2021, 12, 669474. [Google Scholar] [CrossRef]
- Diebold, S.S.; Cotten, M.; Koch, N.; Zenke, M. MHC Class II Presentation of Endogenously Expressed Antigens by Transfected Dendritic Cells. Gene Ther. 2001, 8, 487–493. [Google Scholar] [CrossRef]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Löhr, F.; Popovic, D.; Occhipinti, A.; et al. Nix Is a Selective Autophagy Receptor for Mitochondrial Clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [Green Version]
- von Muhlinen, N.; Thurston, T.; Ryzhakov, G.; Bloor, S.; Randow, F. NDP52, a Novel Autophagy Receptor for Ubiquitin-Decorated Cytosolic Bacteria. Autophagy 2010, 6, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent Activation of Nrf2 through P62 in Hepatocellular Carcinoma Cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The Selective Autophagy Substrate P62 Activates the Stress Responsive Transcription Factor Nrf2 through Inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Böhm, W.; Thoma, S.; Leithäuser, F.; Möller, P.; Schirmbeck, R.; Reimann, J. T Cell-Mediated, IFN-Gamma-Facilitated Rejection of Murine B16 Melanomas. J. Immunol. 1998, 161, 897–908. [Google Scholar]
- Pieper, R.; Christian, R.E.; Gonzales, M.I.; Nishimura, M.I.; Gupta, G.; Settlage, R.E.; Shabanowitz, J.; Rosenberg, S.A.; Hunt, D.F.; Topalian, S.L. Biochemical Identification of a Mutated Human Melanoma Antigen Recognized by CD4+ T Cells. J. Exp. Med. 1999, 189, 757–766. [Google Scholar] [CrossRef]
- Robbins, P.F.; El-Gamil, M.; Li, Y.F.; Zeng, G.; Dudley, M.; Rosenberg, S.A. Multiple HLA Class II-Restricted Melanocyte Differentiation Antigens Are Recognized by Tumor-Infiltrating Lymphocytes from a Patient with Melanoma. J. Immunol. 2002, 169, 6036–6047. [Google Scholar] [CrossRef]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the Role of CD4+ T Cells in Cancer Immunotherapy—New Insights into Old Paradigms. Cancer Gene Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Sequence |
|---|---|
| NheI-OVA-F | 5′-GCTAGCCACCATGGGCTCTATCG-3′ |
| BamHI-OVA-R | 5′- AAGGATCCAGGGGGACACGCATCTG-3′ |
| BamHI-GABARAP-F | 5′-CGGGATCCTTATGAAGTTCGTGTACAAAGAAG-3′ |
| XhoI-GABARAP-R | 5′-CCCTCGAGGGTCACAGACCGTAGACACTTTC-3′ |
| BamHI-GABARAPL1-F | 5′-GGATCCTTATGAAGTTCCAGTACAAGGAGG-3′ |
| XhoI-GABARAPL1-R | 5′-CTCGAGTCACTTCCCATAGACACTCTCACTCAC-3′ |
| BamHI-LC3B-F | 5′- GGATCCTTATGCCGTCCGAGAAGACCTTC-3′ |
| XhoI-LC3B-R | 5′- CTCGAGCTGTCACAAGCATGGCTCTC-3′ |
| myc 1 | 5′-GATCGAGCAAAAGCTCATTTCTGAAGAGGACTTGTC-3′OH |
| myc 2 | 5′-GATCCACAAGTCCTCTTCAGAAATGAGCTTTTGCTC-3′OH |
| NheI-242OVA-F | 5′-CTAGCTAGCTAGATGCTGGTGCTGCTG-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fonderflick, L.; Baudu, T.; Adotévi, O.; Guittaut, M.; Adami, P.; Delage-Mourroux, R. The ATG8 Family Proteins GABARAP and GABARAPL1 Target Antigen to Dendritic Cells to Prime CD4+ and CD8+ T Cells. Cells 2022, 11, 2782. https://doi.org/10.3390/cells11182782
Fonderflick L, Baudu T, Adotévi O, Guittaut M, Adami P, Delage-Mourroux R. The ATG8 Family Proteins GABARAP and GABARAPL1 Target Antigen to Dendritic Cells to Prime CD4+ and CD8+ T Cells. Cells. 2022; 11(18):2782. https://doi.org/10.3390/cells11182782
Chicago/Turabian StyleFonderflick, Leïla, Timothée Baudu, Olivier Adotévi, Michaël Guittaut, Pascale Adami, and Régis Delage-Mourroux. 2022. "The ATG8 Family Proteins GABARAP and GABARAPL1 Target Antigen to Dendritic Cells to Prime CD4+ and CD8+ T Cells" Cells 11, no. 18: 2782. https://doi.org/10.3390/cells11182782