
Autophagy in Rat Müller Glial Cells Is Modulated by the Sirtuin 4/AMPK/mTOR Pathway and Induces Apoptosis under Oxidative Stress

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Transient Retinal I/R Model and Drug Administration
2.3. Cell Culture
2.4. Cell Coculture
2.5. Transfection of Lentivirus Vectors
2.6. Construction and Transfection of Plasmid
2.7. Western Blot Analysis
2.8. Immunofluorescence Staining
2.9. Cell Viability Assay
2.10. TUNEL Staining
2.11. ROS Detection
2.12. Dead/Live Cell Staining
2.13. Mitochondrial Membrane Potential
2.14. Mitochondrial Morphology Assessment
2.15. ATP Content
2.16. DNA Extraction and Quantitative PCR Assay
2.17. Statistics
3. Results
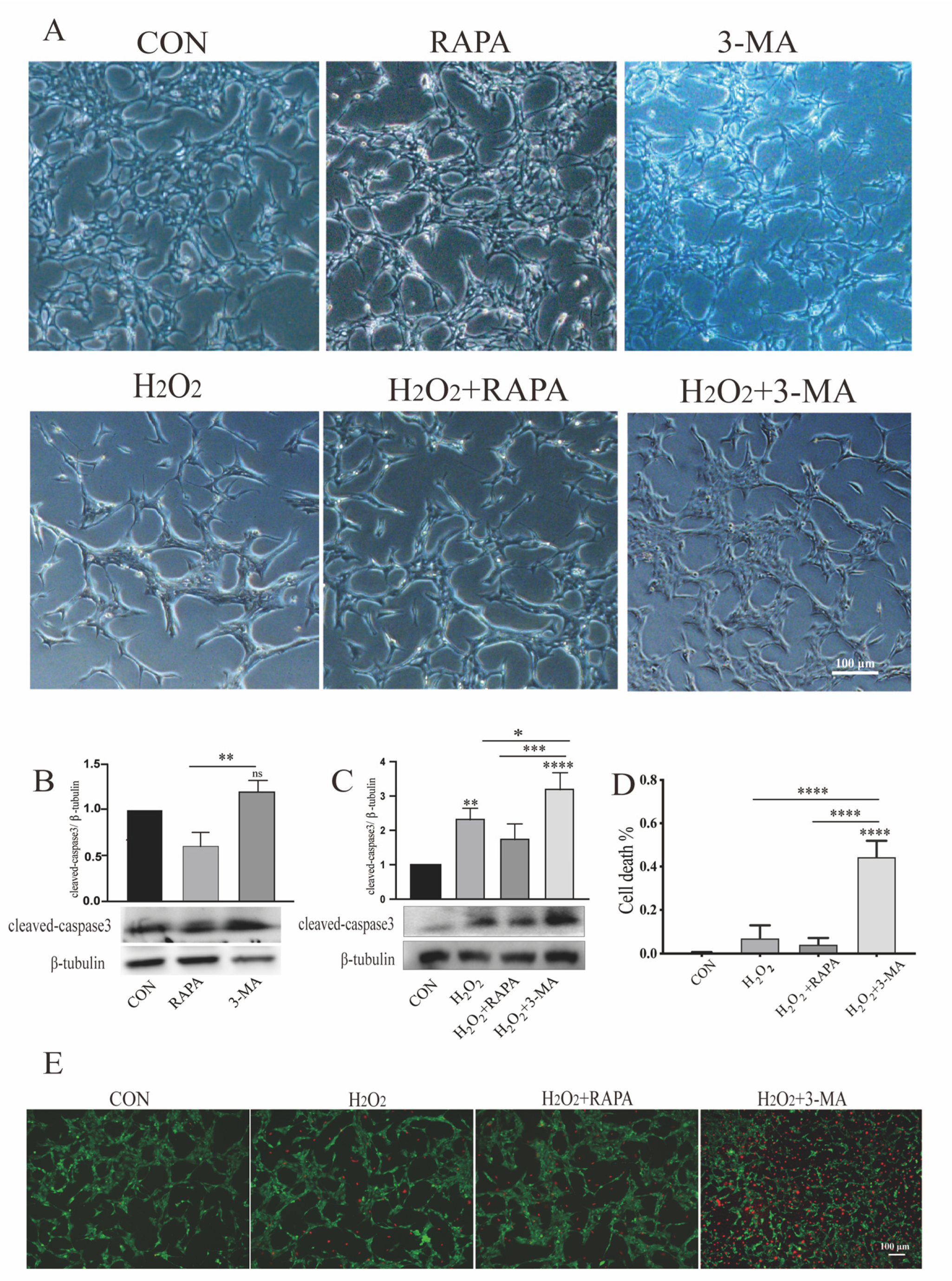
3.1. Regulation of Autophagy Can Modulate Morphologic Alterations and Apoptosis of Müller Glial Cells under Oxidative Stress
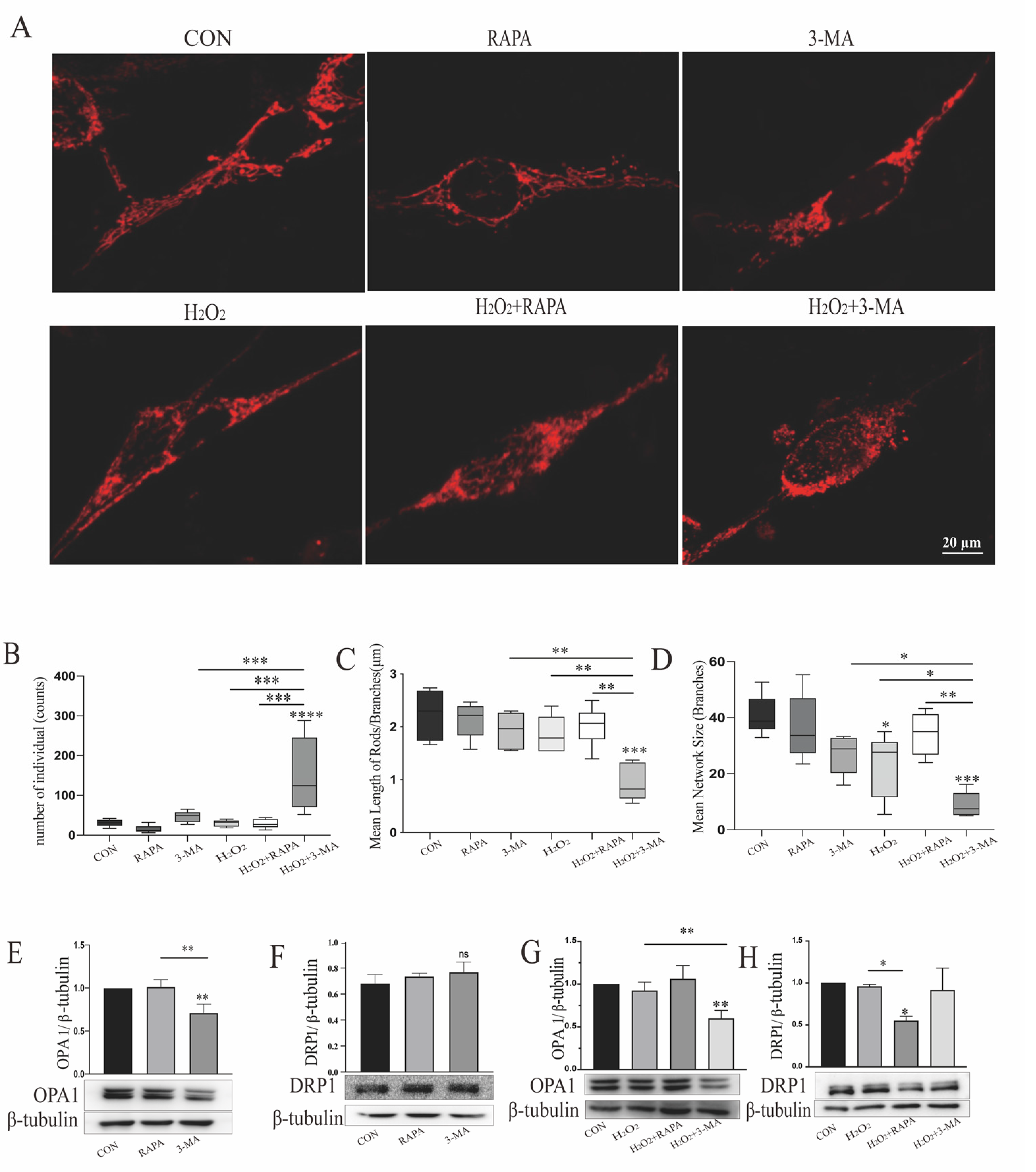
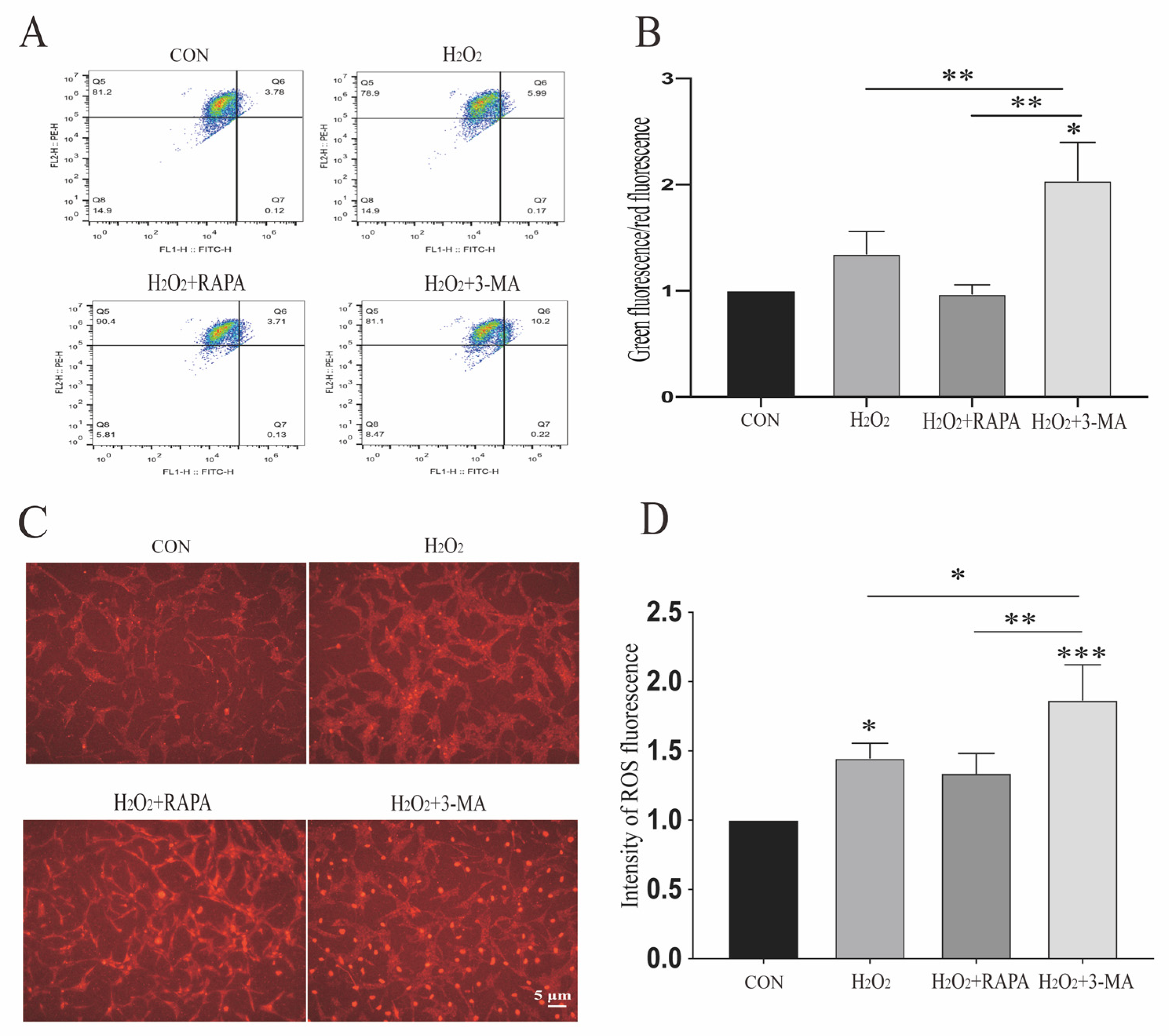
3.2. Regulation of Autophagy Can Impact Mitochondrial Function of r-MCs under Oxidative Stress
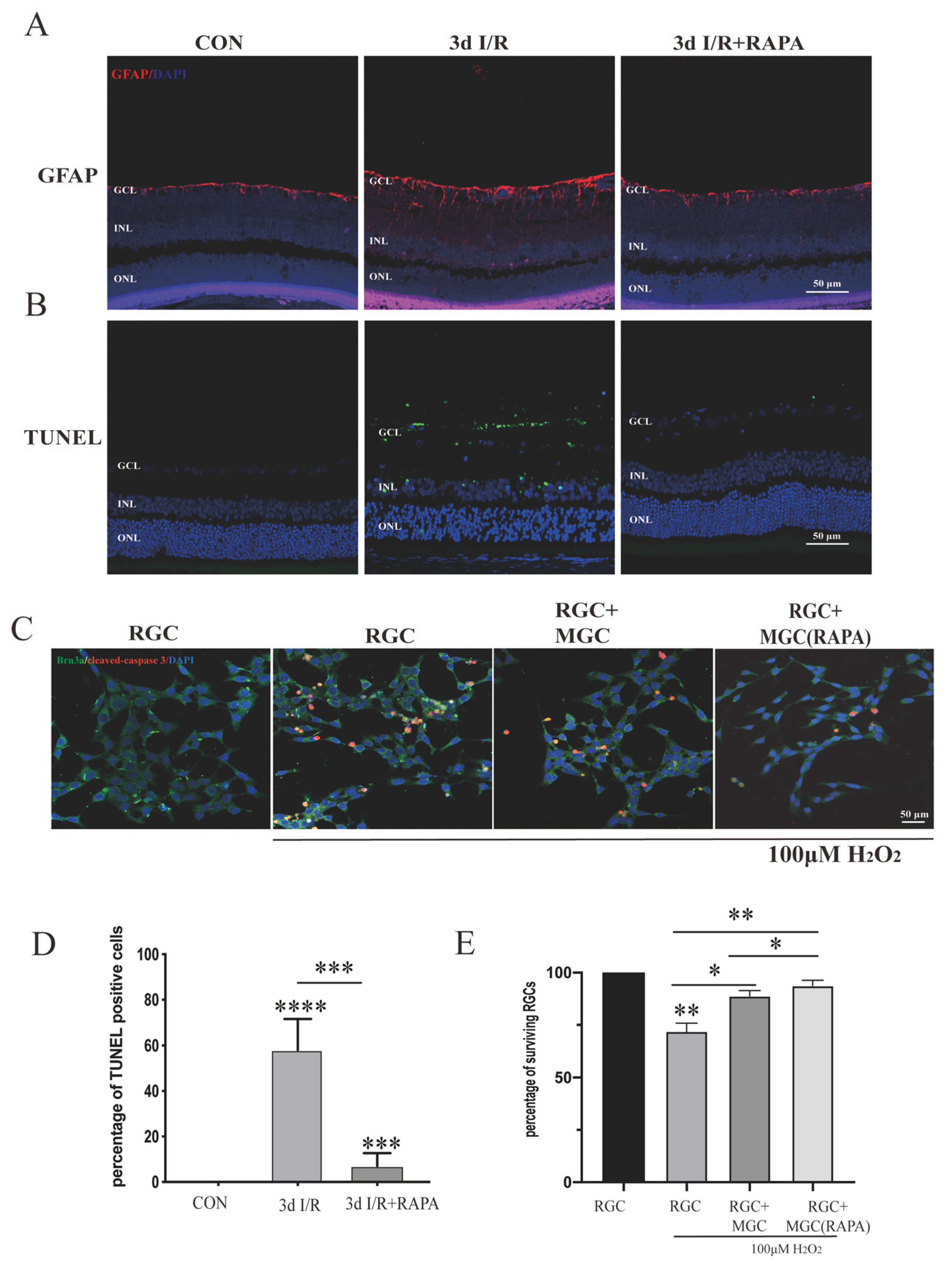
3.3. Activation of Autophagy by Rapamycin Can Help Retinal Neurons to Survive under Ischemia/Reperfusion Injury and Oxidative Stress
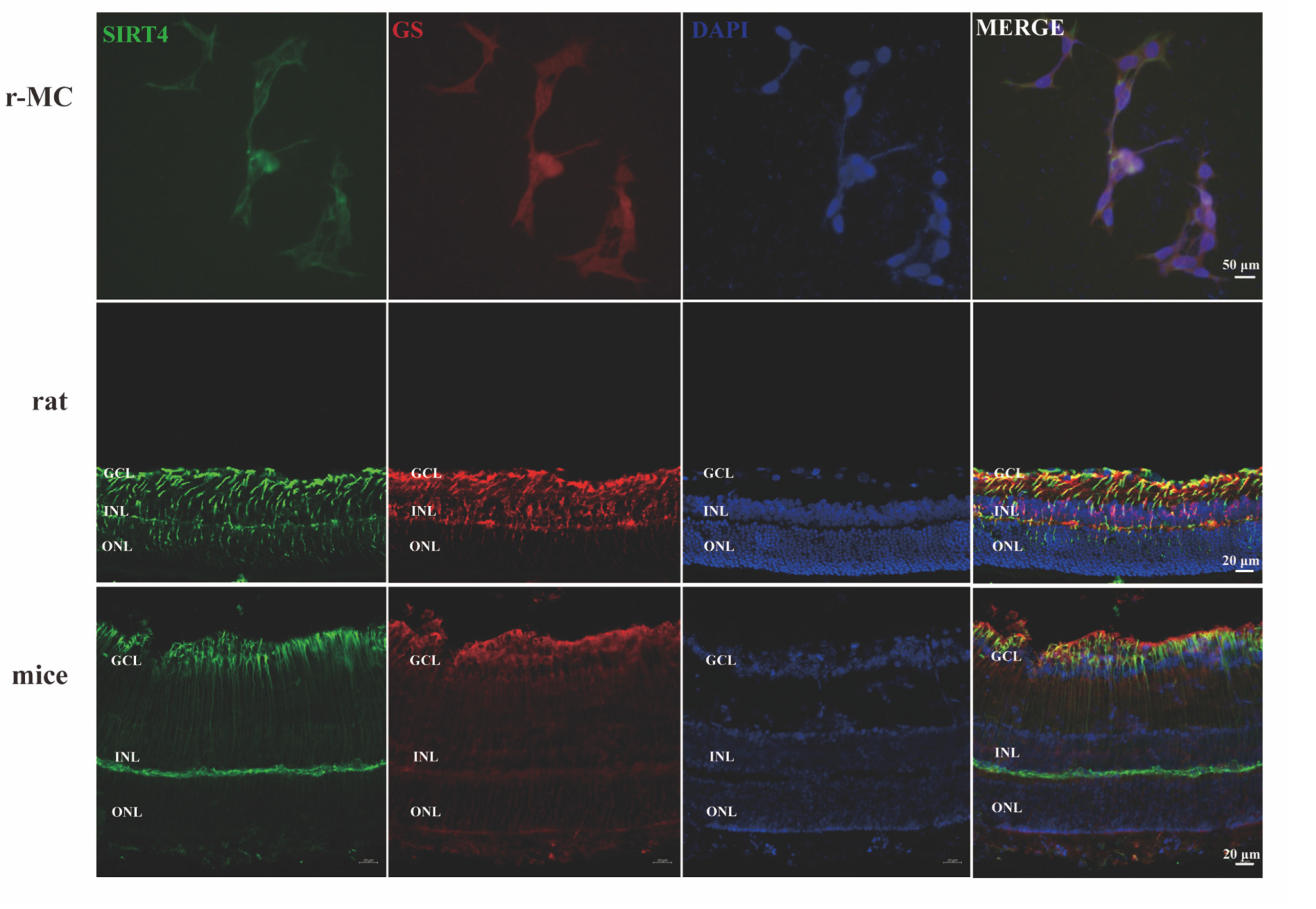
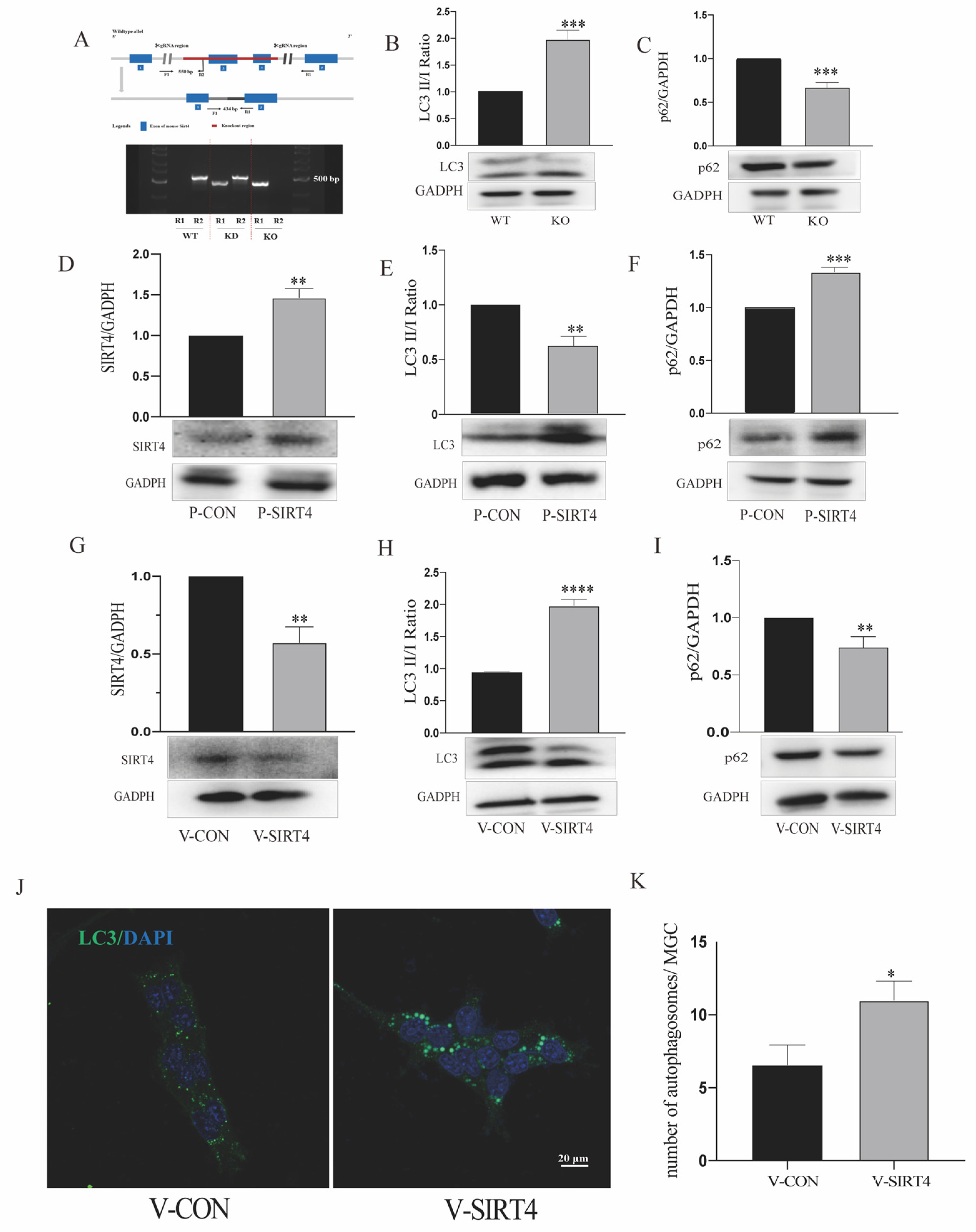
3.4. Depletion of Sirtuin 4 Can Increase Autophagic Function In Vitro and In Vivo
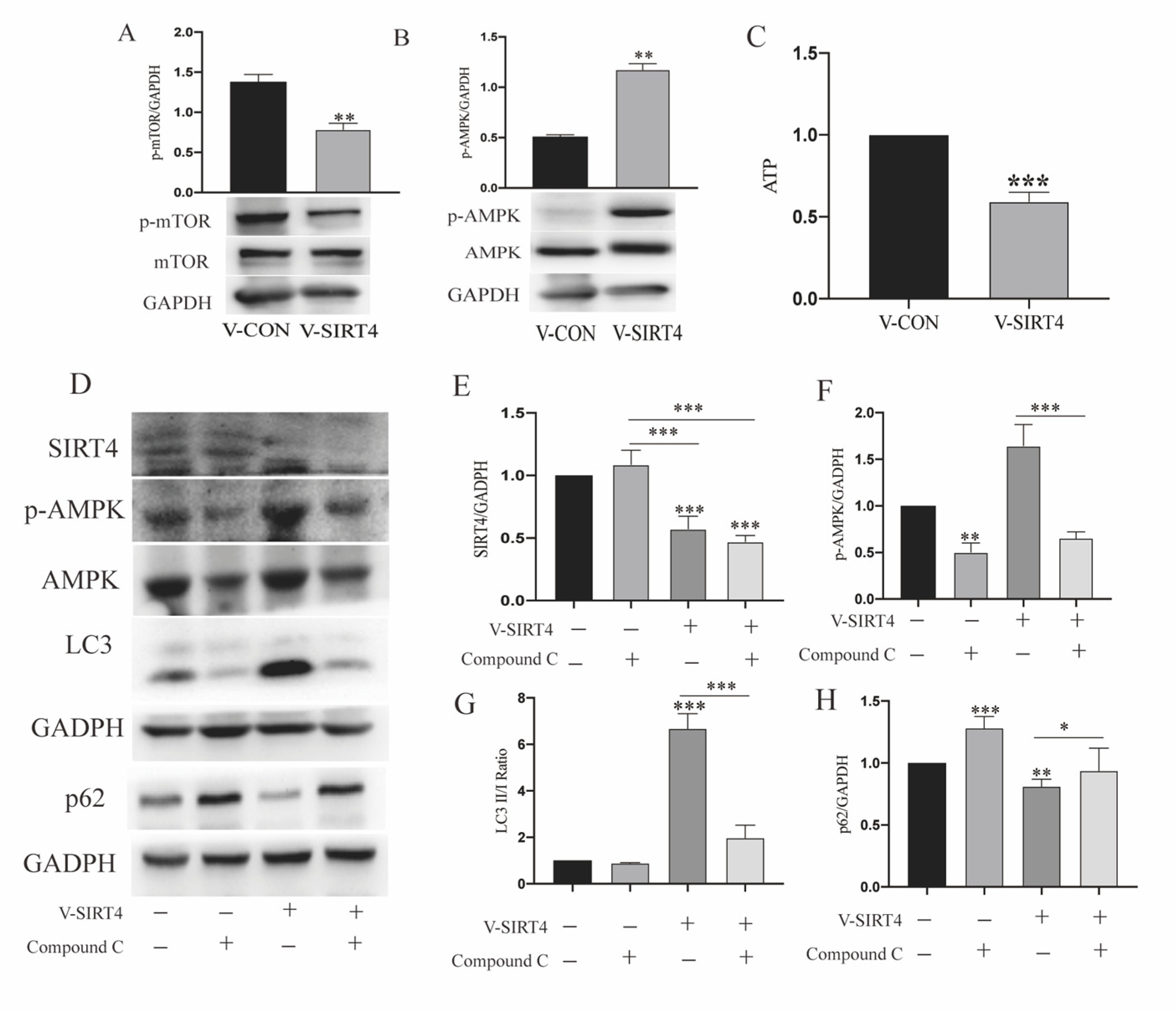
3.5. SIRT4 Participates in Modulating r-MC Autophagic Function through AMPK/mTOR Signaling Pathway
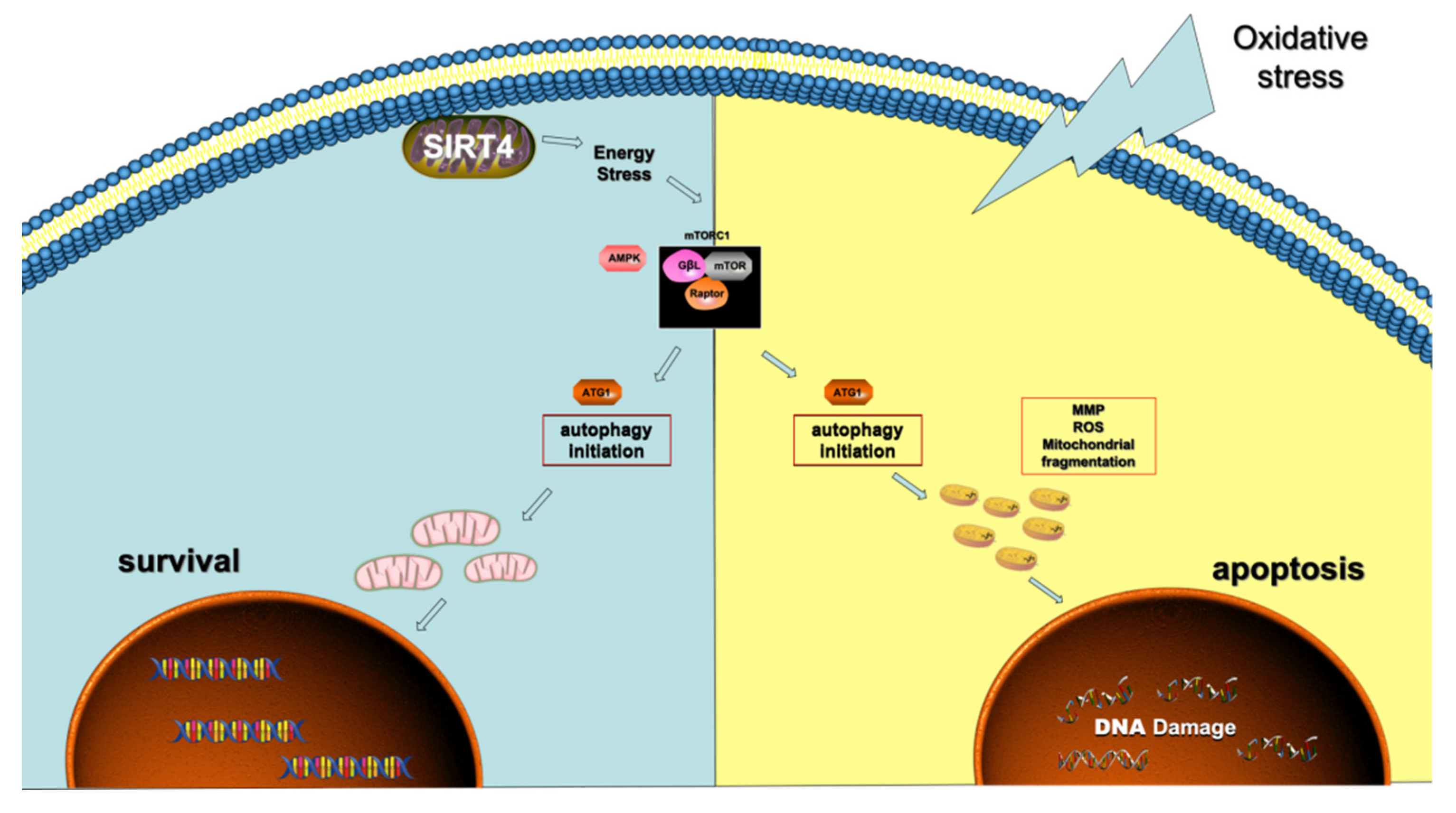
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bringmann, A.; Pannicke, T.; Biedermann, B.; Francke, M.; Iandiev, I.; Grosche, J.; Wiedemann, P.; Albrecht, J.; Reichenbach, A. Role of Retinal Glial Cells in Neurotransmitter Uptake and Metabolism. Neurochem. Int. 2009, 54, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Dauphin, M.; Poitry-Yamate, C.; de Bilbao, F.; Julliard, A.K.; Jourdan, F.; Donati, G. Early Postnatal Müller Cell Death Leads to Retinal but Not Optic Nerve Degeneration in NSE-Hu-Bcl-2 Transgenic Mice. Neuroscience 1999, 95, 9–21. [Google Scholar] [CrossRef]
- Byrne, L.C.; Khalid, F.; Lee, T.; Zin, E.A.; Greenberg, K.P.; Visel, M.; Schaffer, D.V.; Flannery, J.G. AAV-Mediated, Optogenetic Ablation of Müller Glia Leads to Structural and Functional Changes in the Mouse Retina. PLoS ONE 2013, 8, e76075. [Google Scholar] [CrossRef]
- Lindsey, A.E.; Powers, M.K. Visual Behavior of Adult Goldfish with Regenerating Retina. Vis. Neurosci. 2007, 24, 247–255. [Google Scholar] [CrossRef]
- Mensinger, A.F.; Powers, M.K. Visual Function in Regenerating Teleost Retina Following Cytotoxic Lesioning. Vis. Neurosci. 1999, 16, 241–251. [Google Scholar] [CrossRef]
- Sherpa, T.; Fimbel, S.M.; Mallory, D.E.; Maaswinkel, H.; Spritzer, S.D.; Sand, J.A.; Li, L.; Hyde, D.R.; Stenkamp, D.L. Ganglion Cell Regeneration Following Whole-Retina Destruction in Zebrafish. Dev. Neurobiol. 2008, 68, 166–181. [Google Scholar] [CrossRef]
- Das, A.V.; Mallya, K.B.; Zhao, X.; Ahmad, F.; Bhattacharya, S.; Thoreson, W.B.; Hegde, G.V.; Ahmad, I. Neural Stem Cell Properties of Müller Glia in the Mammalian Retina: Regulation by Notch and Wnt Signaling. Dev. Biol. 2006, 299, 283–302. [Google Scholar] [CrossRef]
- Lawrence, J.M.; Singhal, S.; Bhatia, B.; Keegan, D.J.; Reh, T.A.; Luthert, P.J.; Khaw, P.T.; Limb, G.A. MIO-M1 Cells and Similar Müller Glial Cell Lines Derived from Adult Human Retina Exhibit Neural Stem Cell Characteristics. Stem Cells 2007, 25, 2033–2043. [Google Scholar] [CrossRef]
- Scrivo, A.; Bourdenx, M.; Pampliega, O.; Cuervo, A.M. Selective Autophagy as a Potential Therapeutic Target for Neurodegenerative Disorders. Lancet Neurol. 2018, 17, 802–815. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, C.; Zhang, H.; Wu, R. Autophagy in Glaucoma: Crosstalk with Apoptosis and Its Implications. Brain Res. Bull. 2015, 117, 1–9. [Google Scholar] [CrossRef]
- Deng, S.; Wang, M.; Yan, Z.; Tian, Z.; Chen, H.; Yang, X.; Zhuo, Y. Autophagy in Retinal Ganglion Cells in a Rhesus Monkey Chronic Hypertensive Glaucoma Model. PLoS ONE 2013, 8, e77100. [Google Scholar] [CrossRef]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W.; Ding, J.; et al. Dysregulated Autophagy in the RPE Is Associated with Increased Susceptibility to Oxidative Stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [PubMed]
- Subirada, P.V.; Paz, M.C.; Ridano, M.E.; Lorenc, V.E.; Fader, C.M.; Chiabrando, G.A.; Sánchez, M.C. Effect of Autophagy Modulators on Vascular, Glial, and Neuronal Alterations in the Oxygen-Induced Retinopathy Mouse Model. Front. Cell. Neurosci. 2019, 13, 279. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef]
- Han, Y.; Zhou, S.; Coetzee, S.; Chen, A. SIRT4 and Its Roles in Energy and Redox Metabolism in Health, Disease and during Exercise. Front. Physiol. 2019, 10, 1006. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Du, L.; Liang, X.; Meng, P.; Bi, L.; Wang, Y.; Wang, C.; Tang, B. Sirtuin 4 Depletion Promotes Hepatocellular Carcinoma Tumorigenesis Through Regulating Adenosine-Monophosphate–Activated Protein Kinase Alpha/Mammalian Target of Rapamycin Axis in Mice. Hepatology 2019, 69, 1614–1631. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, F.; Zhang, Q.; Jin, M.; Lu, Y.; Chen, S.; Guo, C.; Zhang, X. Rapamycin Mediates MTOR Signaling in Reactive Astrocytes and Reduces Retinal Ganglion Cell Loss. Exp. Eye Res. 2018, 176, 10–19. [Google Scholar] [CrossRef]
- Su, W.; Li, Z.; Jia, Y.; Zhuo, Y. Rapamycin is neuroprotective in a rat chronic hypertensive glaucoma model. PLoS ONE 2014, 9, e99719. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-J.; Liu, C.; Shan, K.; Liu, B.-H.; Li, X.-M.; Zhang, S.-J.; Zhou, R.-M.; Dong, R.; Yan, B.; Sun, X.-H. Circular rna-znf609 regulates retinal neurodegeneration by acting as mir-615 sponge. Theranostics 2018, 8, 3408–3415. [Google Scholar] [CrossRef]
- Wang, R.; Peng, L.; Zhao, J.; Zhang, L.; Guo, C.; Zheng, W.; Chen, H. Gardenamide A Protects RGC-5 Cells from H2O2-Induced Oxidative Stress Insults by Activating PI3K/Akt/eNOS Signaling Pathway. Int. J. Mol. Sci. 2015, 16, 22350–22367. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Hu, P.; Qin, M.; Chen, G.; Wang, F.; Yao, S.; Jin, M.; Xie, Z.; Zhang, X. SIRT4 Is Highly Expressed in Retinal Müller Glial Cells. Front. Neurosci. 2022, 16, 840443. [Google Scholar] [CrossRef] [PubMed]
- Valente, A.J.; Maddalena, L.A.; Robb, E.L.; Moradi, F.; Stuart, J.A. A Simple ImageJ Macro Tool for Analyzing Mitochondrial Network Morphology in Mammalian Cell Culture. Acta Histochem. 2017, 119, 315–326. [Google Scholar] [CrossRef]
- Lopes de Faria, J.M.; Duarte, D.A.; Montemurro, C.; Papadimitriou, A.; Consonni, S.R.; Lopes de Faria, J.B. Defective Autophagy in Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4356–4366. [Google Scholar] [CrossRef]
- Yang, F.; Liao, J.; Yu, W.; Qiao, N.; Guo, J.; Han, Q.; Li, Y.; Hu, L.; Pan, J.; Tang, Z. Exposure to Copper Induces Mitochondria-Mediated Apoptosis by Inhibiting Mitophagy and the PINK1/Parkin Pathway in Chicken (Gallus Gallus) Livers. J. Hazard. Mater. 2021, 408, 124888. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Luo, Z.; Jin, M.; Sheng, W.; Wang, H.-T.; Long, X.; Wu, Y.; Hu, P.; Xu, H.; Zhang, X. Exploration of Age-Related Mitochondrial Dysfunction and the Anti-Aging Effects of Resveratrol in Zebrafish Retina. Aging 2019, 11, 3117–3137. [Google Scholar] [CrossRef]
- Hasnat, M.; Yuan, Z.; Ullah, A.; Naveed, M.; Raza, F.; Baig, M.M.F.A.; Khan, A.; Xu, D.; Su, Y.; Sun, L.; et al. Mitochondria-Dependent Apoptosis in Triptolide-Induced Hepatotoxicity Is Associated with the Drp1 Activation. Toxicol. Mech. Methods 2020, 30, 124–133. [Google Scholar] [CrossRef]
- Chen, L.B. Mitochondrial Membrane Potential in Living Cells. Annu. Rev. Cell Biol. 1988, 4, 155–181. [Google Scholar] [CrossRef] [PubMed]
- Shirozu, R.; Yashiroda, H.; Murata, S. Proteasome Impairment Induces Recovery of Mitochondrial Membrane Potential and an Alternative Pathway of Mitochondrial Fusion. Mol. Cell. Biol. 2016, 36, 347–362. [Google Scholar] [CrossRef]
- Sakthivel, R.; Malar, D.S.; Devi, K.P. Phytol Shows Anti-Angiogenic Activity and Induces Apoptosis in A549 Cells by Depolarizing the Mitochondrial Membrane Potential. Biomed. Pharmacother. 2018, 105, 742–752. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, J.; Tian, H.; Fu, G.-H. Mitochondrial ROS Accumulation Inhibiting JAK2/STAT3 Pathway Is a Critical Modulator of CYT997-Induced Autophagy and Apoptosis in Gastric Cancer. J. Exp. Clin. Cancer Res. 2020, 39, 119. [Google Scholar] [CrossRef]
- Fontana, F.; Marzagalli, M.; Raimondi, M.; Zuco, V.; Zaffaroni, N.; Limonta, P. Δ-Tocotrienol Sensitizes and Re-sensitizes Ovarian Cancer Cells to Cisplatin via Induction of G1 Phase Cell Cycle Arrest and ROS/MAPK-mediated Apoptosis. Cell Prolif. 2021, 54, e13111. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Yan, F.; Wei, W.; Wang, F.; Wang, Z.; Nie, J.; Jin, M.; Pang, Y.; Qin, M.; Wang, L. CD38 Deficiency Protects the Retina from Ischaemia/Reperfusion Injury Partly via Suppression of TLR4/MyD88/NF-ΚB Signalling. Exp. Eye Res. 2022, 219, 109058. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ji, Y.; Yan, X.; Su, G.; Chen, L.; Xiao, J. Berberine Attenuates Apoptosis in Rat Retinal Müller Cells Stimulated with High Glucose via Enhancing Autophagy and the AMPK/MTOR Signaling. Biomed. Pharmacother. 2018, 108, 1201–1207. [Google Scholar] [CrossRef]
- Kwakye, G.F.; Jiménez, J.A.; Thomas, M.G.; Kingsley, B.A.; McIIvin, M.; Saito, M.A.; Korley, E.M. Heterozygous Huntingtin Promotes Cadmium Neurotoxicity and Neurodegeneration in Striatal Cells via Altered Metal Transport and Protein Kinase C Delta Dependent Oxidative Stress and Apoptosis Signaling Mechanisms. NeuroToxicology 2019, 70, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wu, W.; Gabbi, C.; Dai, Y.; So, M.; Chaurasiya, S.P.; Wang, L.; Warner, M.; Gustafsson, J.-Å. Retinal and Optic Nerve Degeneration in Liver X Receptor β Knockout Mice. Proc. Natl. Acad. Sci. USA 2019, 116, 16507–16512. [Google Scholar] [CrossRef]
- Basu, S.; Rajakaruna, S.; Reyes, B.; Van Bockstaele, E.; Menko, A.S. Suppression of MAPK/JNK-MTORC1 Signaling Leads to Premature Loss of Organelles and Nuclei by Autophagy during Terminal Differentiation of Lens Fiber Cells. Autophagy 2014, 10, 1193–1211. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Esteban-Martínez, L.; Serrano-Puebla, A.; Gómez-Sintes, R.; Villarejo-Zori, B. Autophagy in the Eye: Development, Degeneration, and Aging. Prog. Retin. Eye Res. 2016, 55, 206–245. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. Age-Related Decline in Chaperone-Mediated Autophagy. J. Biol. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef]
- Dehay, B.; Bove, J.; Rodriguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic Lysosomal Depletion in Parkinson’s Disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef]
- Rodríguez-Muela, N.; Germain, F.; Mariño, G.; Fitze, P.S.; Boya, P. Autophagy Promotes Survival of Retinal Ganglion Cells after Optic Nerve Axotomy in Mice. Cell Death Differ. 2012, 19, 162–169. [Google Scholar] [CrossRef]
- Chen, Y.; Sawada, O.; Kohno, H.; Le, Y.-Z.; Subauste, C.; Maeda, T.; Maeda, A. Autophagy Protects the Retina from Light-Induced Degeneration. J. Biol. Chem. 2013, 288, 7506–7518. [Google Scholar] [CrossRef]
- Wohl, S.G.; Jorstad, N.L.; Levine, E.M.; Reh, T.A. SMüller Glial MicroRNAs Are Required for the Maintenance of Glial Homeostasis and Retinal Architecture. Nat. Commun. 2017, 8, 1603. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.E.; Grosser, J.A.; Fehrman, R.L.; Schlamp, C.L.; Nickells, R.W. Completion of BAX Recruitment Correlates with Mitochondrial Fission during Apoptosis. Sci. Rep. 2019, 9, 16565. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Liu, W.; Zhang, Q.; Huang, J.; Hu, C.; Liu, Y.; Wang, Q.; Zhou, M.; Lai, W.; Sheng, F.; et al. Dynamin-related Protein 1-mediated Mitochondrial Fission Contributes to IR -783-induced Apoptosis in Human Breast Cancer Cells. J. Cell. Mol. Med. 2018, 22, 4474–4485. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Pawlowska, E.; Szczepanska, J.; Jablkowska, A.; Blasiak, J. Role of Mitochondrial DNA Damage in ROS-Mediated Pathogenesis of Age-Related Macular Degeneration (AMD). Int. J. Mol. Sci. 2019, 20, 2374. [Google Scholar] [CrossRef]
- Zhang, S.; Rao, S.; Yang, M.; Ma, C.; Hong, F.; Yang, S. Role of Mitochondrial Pathways in Cell Apoptosis during He-Patic Ischemia/Reperfusion Injury. Int. J. Mol. Sci. 2022, 23, 2357. [Google Scholar] [CrossRef]
- Singh, C.; Tran, V.; McCollum, L.; Bolok, Y.; Allan, K.; Yuan, A.; Hoppe, G.; Brunengraber, H.; Sears, J.E. Hyperoxia Induces Glutamine-Fuelled Anaplerosis in Retinal Müller Cells. Nat. Commun. 2020, 11, 1277. [Google Scholar] [CrossRef]
- Ho, L.; Titus, A.S.; Banerjee, K.K.; George, S.; Lin, W.; Deota, S.; Saha, A.K.; Nakamura, K.; Gut, P.; Verdin, E.; et al. SIRT4 Regulates ATP Homeostasis and Mediates a Retrograde Signaling via AMPK. Aging 2013, 5, 835–849. [Google Scholar] [CrossRef]
- Gatica, D.; Klionsky, D.J. New Tricks of an Old Autophagy Regulator: AMPK-Dependent Regulation of Autophagy through CCNY (Cyclin Y)-CDK16. Autophagy 2020, 16, 973–974. [Google Scholar] [CrossRef]
- Lang, A.; Anand, R.; Altinoluk-Hambüchen, S.; Ezzahoini, H.; Stefanski, A.; Iram, A.; Bergmann, L.; Urbach, J.; Böhler, P.; Hänsel, J.; et al. SIRT4 Interacts with OPA1 and Regulates Mitochondrial Quality Control and Mitophagy. Aging 2017, 9, 2163–2189. [Google Scholar] [CrossRef] [PubMed]
- Shaw, E.; Talwadekar, M.; Rashida, Z.; Mohan, N.; Acharya, A.; Khatri, S.; Laxman, S.; Kolthur-Seetharam, U. Anabolic SIRT4 Exerts Retrograde Control over TORC1 Signaling by Glutamine Sparing in the Mitochondria. Mol. Cell. Biol. 2020, 40, e00212–e00219. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wang, J.; Yang, Y.; Zou, P.; Xia, Z.; Li, J. SIRT4 Suppresses Doxorubicin-Induced Cardiotoxicity by Regulating the AKT/MTOR/Autophagy Pathway. Toxicology 2022, 469, 153119. [Google Scholar] [CrossRef] [PubMed]
- Choubey, S.K.; Prabhu, D.; Nachiappan, M.; Biswal, J.; Jeyakanthan, J. Molecular Modeling, Dynamics Studies and Density Functional Theory Approaches to Identify Potential Inhibitors of SIRT4 Protein from Homo Sapiens: A Novel Target for the Treatment of Type 2 Diabetes. J. Biomol. Struct. Dyn. 2017, 35, 3316–3329. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, M.; Xie, Z.; Cao, T.; Wang, Z.; Zhang, X.; Wang, F.; Wei, W.; Jin, M.; Ma, J.; Zeng, L.; et al. Autophagy in Rat Müller Glial Cells Is Modulated by the Sirtuin 4/AMPK/mTOR Pathway and Induces Apoptosis under Oxidative Stress. Cells 2022, 11, 2645. https://doi.org/10.3390/cells11172645
Qin M, Xie Z, Cao T, Wang Z, Zhang X, Wang F, Wei W, Jin M, Ma J, Zeng L, et al. Autophagy in Rat Müller Glial Cells Is Modulated by the Sirtuin 4/AMPK/mTOR Pathway and Induces Apoptosis under Oxidative Stress. Cells. 2022; 11(17):2645. https://doi.org/10.3390/cells11172645
Chicago/Turabian StyleQin, Mengqi, Zhi Xie, Ting Cao, Zhiruo Wang, Xiaoyu Zhang, Feifei Wang, Wei Wei, Ming Jin, Jingyuan Ma, Ling Zeng, and et al. 2022. "Autophagy in Rat Müller Glial Cells Is Modulated by the Sirtuin 4/AMPK/mTOR Pathway and Induces Apoptosis under Oxidative Stress" Cells 11, no. 17: 2645. https://doi.org/10.3390/cells11172645