
ResSUMO: A Deep Learning Architecture Based on Residual Structure for Prediction of Lysine SUMOylation Sites

Abstract
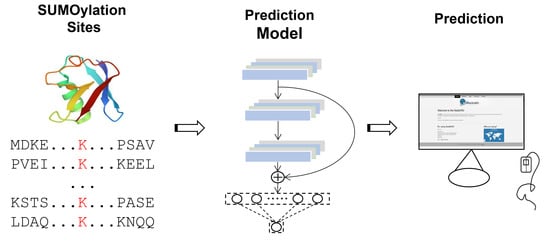
:
1. Introduction
2. Materials and Methods
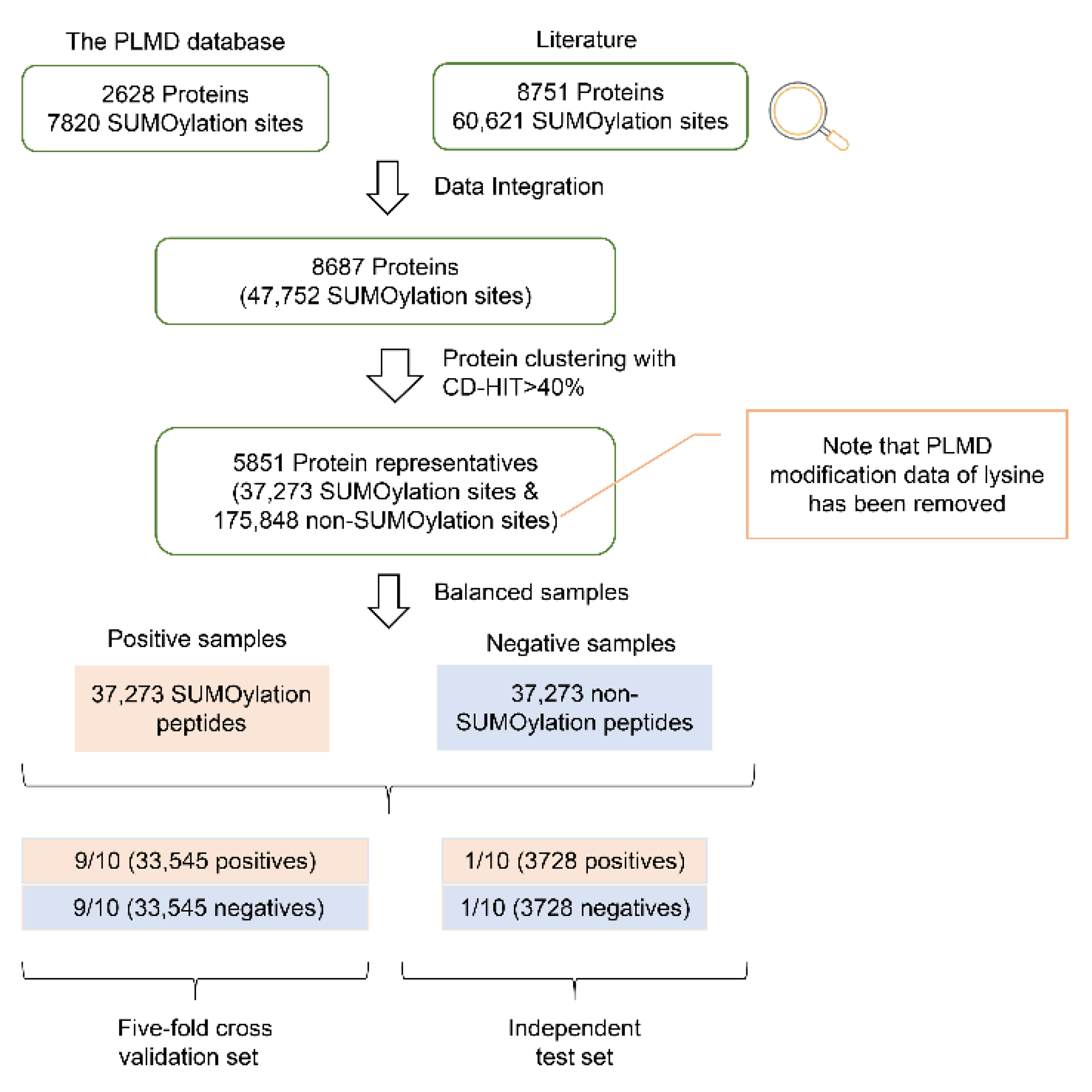
2.1. Data Construction
2.2. Feature Encoding
2.2.1. EAAC Encoding
2.2.2. BLOSUM62 Encoding
2.2.3. AAindex Encoding
2.2.4. ZScale Encoding
3. Machine Learning Algorithms
3.1. Random Forest (RF) and Light Gradient Boosting Machine (LGBM)
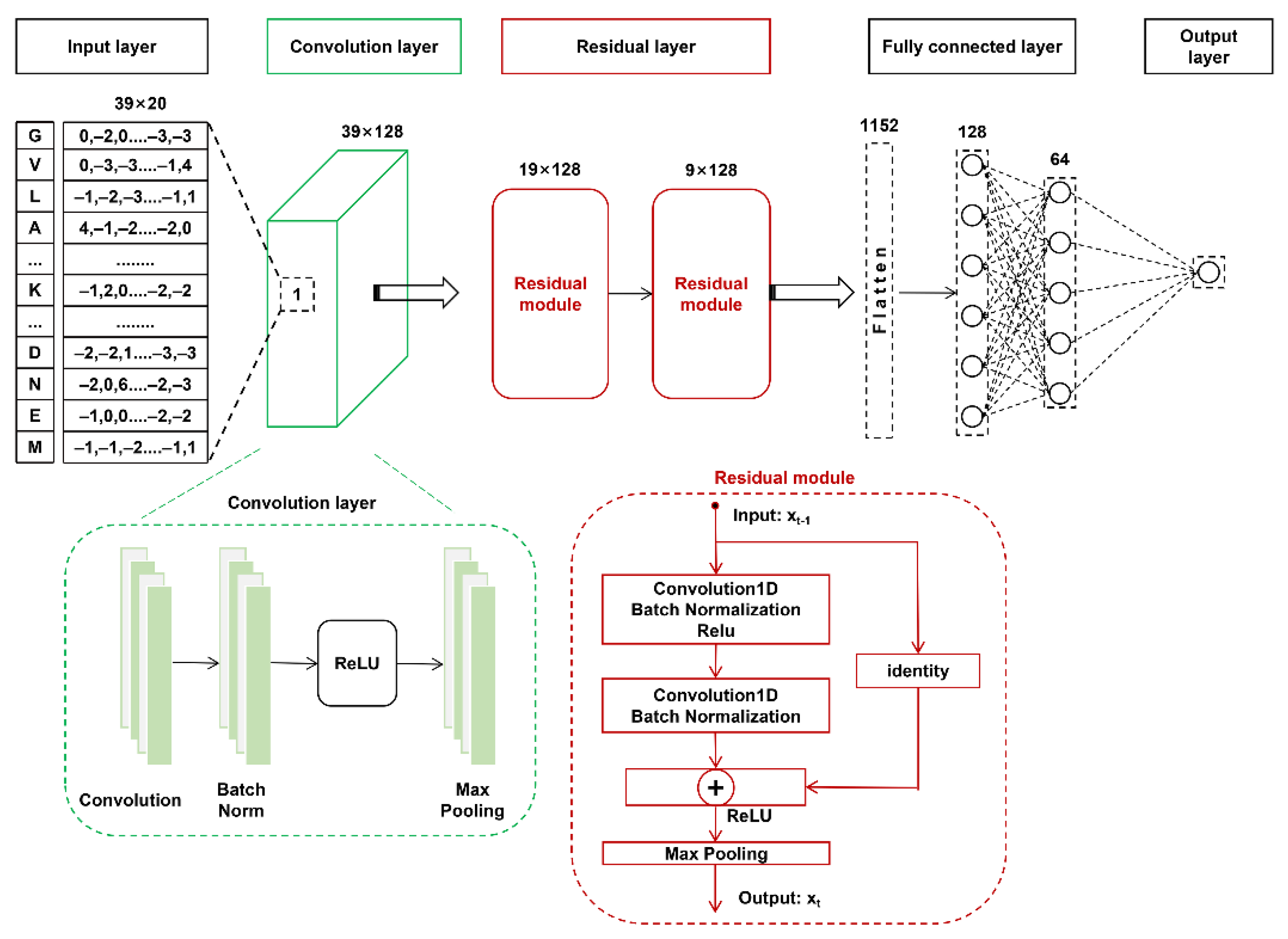
3.2. The Architecture of the DL Models
3.2.1. CNN-Based Classifiers
- Input layer. The peptide segment was converted into a two-dimensional matrix using each encoding feature listed above.
- Convolutional layer. This layer includes two sequentially connected blocks. Each block includes a convolution sublayer, a batch normalization sublayer, a rectified linear unit (ReLU) [40] as the activation function, and a max-pooling sublayer. The number of convolution kernels was set to 128, and each convolution kernel size was set as 3. The max-pooling parameters pool_size, strides, and padding were set as 2, 1, and “same”, respectively.
- Fully connected layer. The output from the above layer was flattened and received by the fully connected layer. This layer contained two fully connected sublayers, one with 128 neurons followed by another with 64 neurons.
- Output layer. This layer contains a single neuron to output the probability score (within the range from 0 to 1), indicating the likelihood of the SUMOylation modification, which was calculated by the “Sigmoid” function as follows:
3.2.2. The Residual Structure Layered CNN Architecture (RSCNN)
- Input layer. This layer is the same as the input layer of the above CNN architecture.
- Convolution layer. This layer includes a single convolution sublayer, a batch normalization sublayer, a ReLU, and a max-pooling sublayer. The number of convolution kernels was 128, and the convolution kernel size was 3. The max-pooling parameters pool_size, strides, and padding were set as 2, 1, and “same”, respectively.
- Residual layer. This layer contains two sequentially connected residual module blocks. Each block includes two convolution sublayers and two batch normalization sublayers. In each convolution sublayer, the number of convolution kernels, convolution kernel size, and the convolution parameter padding were 128, 3, and “same”, respectively. The output of the residual module block t can be calculated using the following formula:where is the input of the t-th residual module block; represents a set of weights of the t-th block. refers to the convolution and batch normalization operation in the t-th block, R is the ReLU activation function, and P is the max-pooling function.
- Fully connected layer. This layer is the same as for the above CNN architecture.
- Output layer. This layer is the same as for the above CNN architecture.
3.3. The Strategy to Avoiding Overfitting for the DL Approaches
3.4. Performance Evaluation Strategies
3.5. Statistical Analysis
4. Results
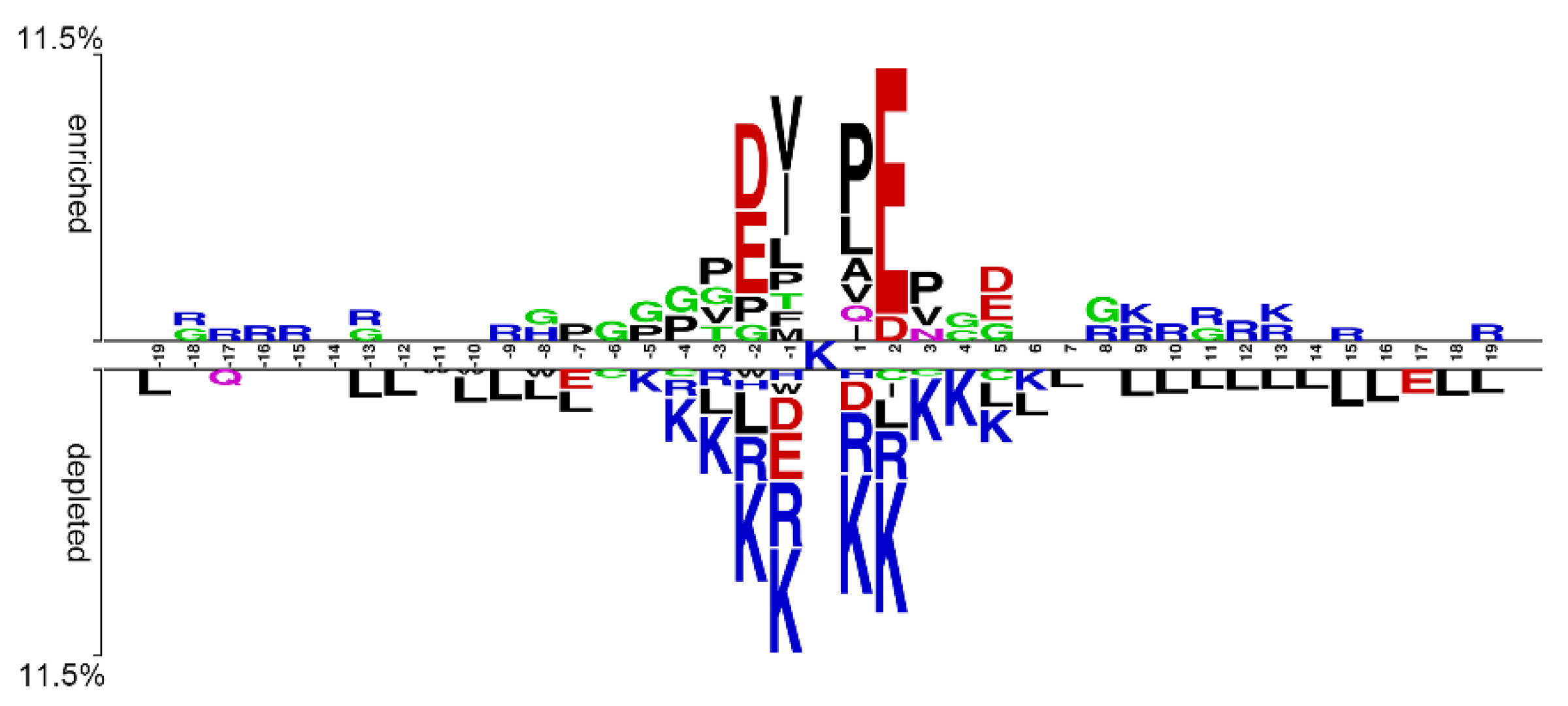
4.1. Construction of the SUMOylation Dataset and Sequence Preference Analysis
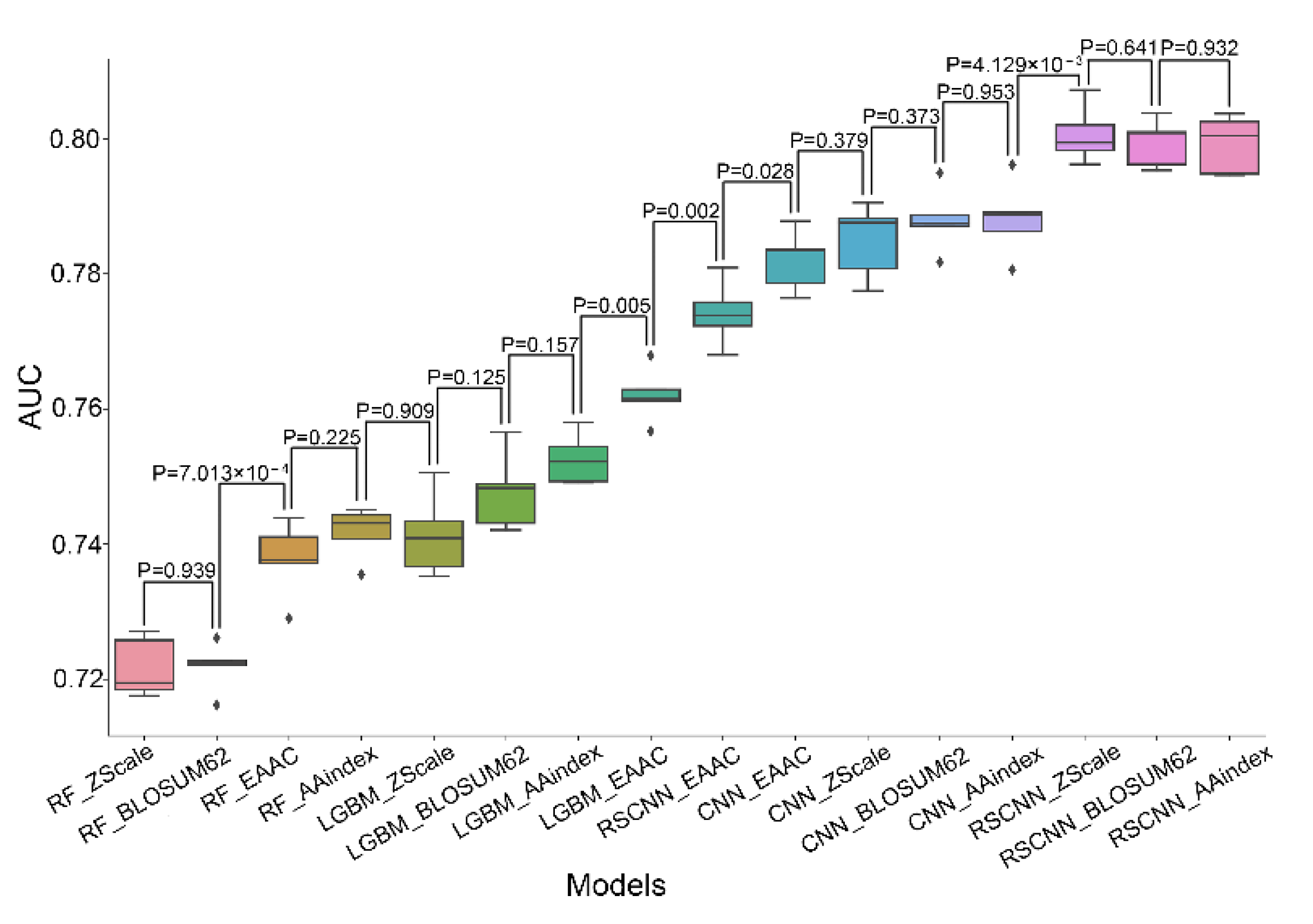
4.2. Performance Evaluation of Various Classifiers Combined with Distinct Features
4.3. Most RSCNN Models Compare Favorably to Other Models
4.4. Our Model ResSUMO Shows Superior Performance to the Reported Classifiers
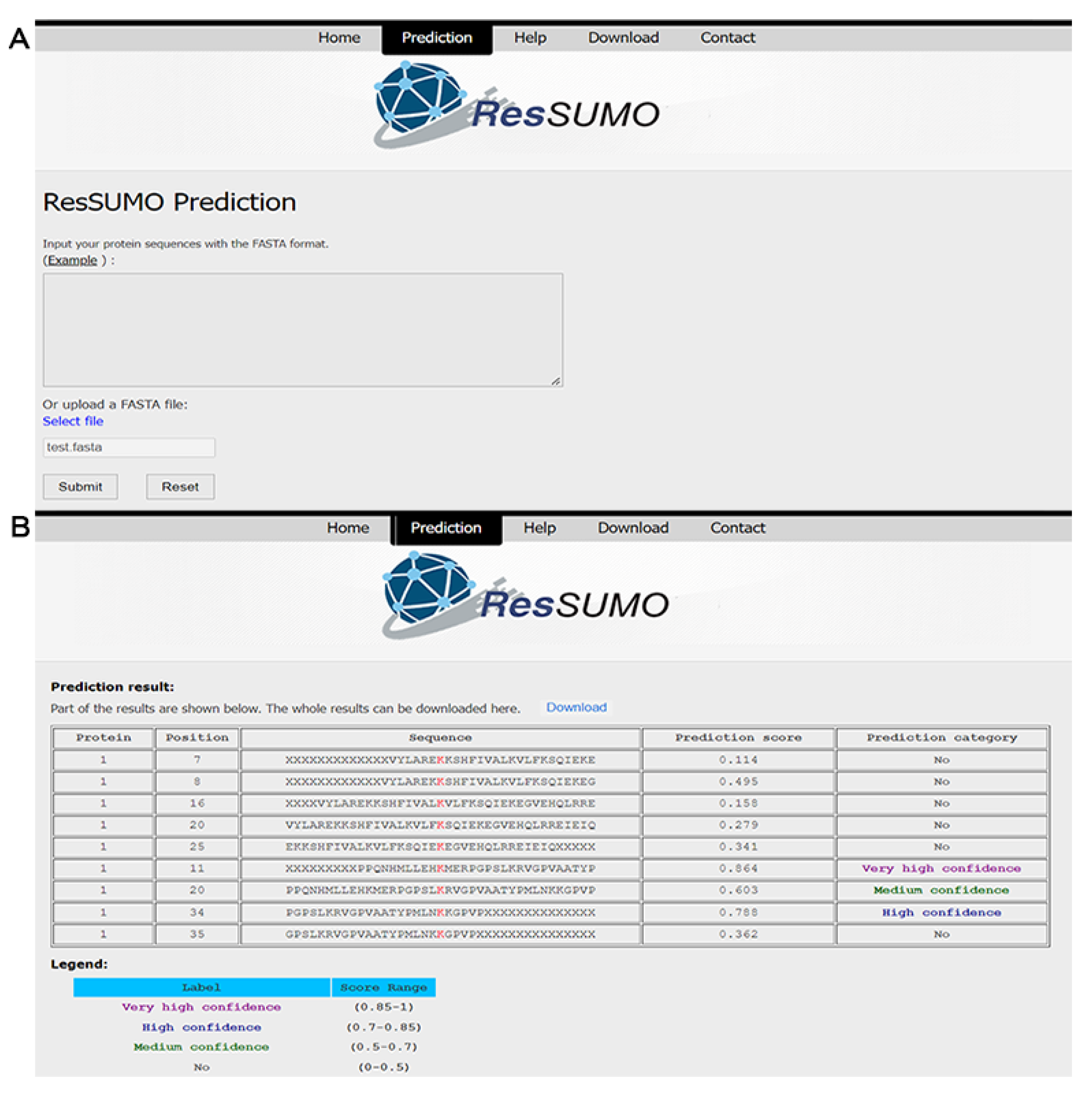
4.5. Construction of the Online SUMOylation Predictor
5. Discussions
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, J.; Sha, M.; Wang, Q.; Ma, Y.; Geng, X.; Gao, Y.; Feng, L.; Shen, Y.; Shen, Y. Small ubiquitin-related modifier 2/3 interacts with p65 and stabilizes it in the cytoplasm in HBV-associated hepatocellular carcinoma. BMC Cancer 2015, 15, 675. [Google Scholar] [CrossRef]
- Seeler, J.S.; Dejean, A. Nuclear and unclear functions of SUMO. Nat. Rev. Mol. Cell Biol. 2003, 4, 690–699. [Google Scholar] [CrossRef]
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947–956. [Google Scholar] [CrossRef]
- Hay, R.T. SUMO: A history of modification. Mol. Cell 2005, 18, 1–12. [Google Scholar] [CrossRef]
- Muller, S.; Hoege, C.; Pyrowolakis, G.; Jentsch, S. SUMO, ubiquitin’s mysterious cousin. Nat. Rev. Mol. Cell Biol. 2001, 2, 202–210. [Google Scholar] [CrossRef]
- Seeler, J.S.; Bischof, O.; Nacerddine, K.; Dejean, A. SUMO, the three Rs and cancer. Curr. Top. Microbiol. Immunol. 2007, 313, 49–71. [Google Scholar] [CrossRef]
- Princz, A.; Tavernarakis, N. SUMOylation in Neurodegenerative Diseases. Gerontology 2020, 66, 122–130. [Google Scholar] [CrossRef]
- Marmor-Kollet, H.; Siany, A.; Kedersha, N.; Knafo, N.; Rivkin, N.; Danino, Y.M.; Moens, T.G.; Olender, T.; Sheban, D.; Cohen, N.; et al. Spatiotemporal Proteomic Analysis of Stress Granule Disassembly Using APEX Reveals Regulation by SUMOylation and Links to ALS Pathogenesis. Mol. Cell 2020, 80, 876–891.e876. [Google Scholar] [CrossRef]
- Hendriks, I.A.; D’Souza, R.C.; Yang, B.; Verlaan-de Vries, M.; Mann, M.; Vertegaal, A.C. Uncovering global SUMOylation signaling networks in a site-specific manner. Nat. Struct. Mol. Biol. 2014, 21, 927–936. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Vertegaal, A.C. A high-yield double-purification proteomics strategy for the identification of SUMO sites. Nat. Protoc. 2016, 11, 1630–1649. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Lyon, D.; Su, D.; Skotte, N.H.; Daniel, J.A.; Jensen, L.J.; Nielsen, M.L. Site-specific characterization of endogenous SUMOylation across species and organs. Nat. Commun. 2018, 9, 2456. [Google Scholar] [CrossRef]
- Matic, I.; Schimmel, J.; Hendriks, I.A.; van Santen, M.A.; van de Rijke, F.; van Dam, H.; Gnad, F.; Mann, M.; Vertegaal, A.C. Site-specific identification of SUMO-2 targets in cells reveals an inverted SUMOylation motif and a hydrophobic cluster SUMOylation motif. Mol. Cell 2010, 39, 641–652. [Google Scholar] [CrossRef]
- Jia, J.; Zhang, L.; Liu, Z.; Xiao, X.; Chou, K.C. pSumo-CD: Predicting sumoylation sites in proteins with covariance discriminant algorithm by incorporating sequence-coupled effects into general PseAAC. Bioinformatics 2016, 32, 3133–3141. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Ding, J.; Xu, Y. iAcet-Sumo: Identification of lysine acetylation and sumoylation sites in proteins by multi-class transformation methods. Comput. Biol. Med. 2018, 100, 144–151. [Google Scholar] [CrossRef]
- Chang, C.C.; Tung, C.H.; Chen, C.W.; Tu, C.H.; Chu, Y.W. SUMOgo: Prediction of sumoylation sites on lysines by motif screening models and the effects of various post-translational modifications. Sci. Rep. 2018, 8, 15512. [Google Scholar] [CrossRef]
- Dehzangi, A.; Lopez, Y.; Taherzadeh, G.; Sharma, A.; Tsunoda, T. SumSec: Accurate Prediction of Sumoylation Sites Using Predicted Secondary Structure. Molecules 2018, 23, 3260. [Google Scholar] [CrossRef]
- Sharma, A.; Lysenko, A.; Lopez, Y.; Dehzangi, A.; Sharma, R.; Reddy, H.; Sattar, A.; Tsunoda, T. HseSUMO: Sumoylation site prediction using half-sphere exposures of amino acids residues. BMC Genom. 2019, 19, 982. [Google Scholar] [CrossRef]
- Qian, Y.; Ye, S.; Zhang, Y.; Zhang, J. SUMO-Forest: A Cascade Forest based method for the prediction of SUMOylation sites on imbalanced data. Gene 2020, 741, 144536. [Google Scholar] [CrossRef]
- Lopez, Y.; Dehzangi, A.; Reddy, H.M.; Sharma, A. C-iSUMO: A sumoylation site predictor that incorporates intrinsic characteristics of amino acid sequences. Comput. Biol. Chem. 2020, 87, 107235. [Google Scholar] [CrossRef]
- Khan, Y.D.; Khan, N.S.; Naseer, S.; Butt, A.H. iSUMOK-PseAAC: Prediction of lysine sumoylation sites using statistical moments and Chou’s PseAAC. PeerJ 2021, 9, e11581. [Google Scholar] [CrossRef]
- Hamelryck, T. An amino acid has two sides: A new 2D measure provides a different view of solvent exposure. Proteins 2005, 59, 38–48. [Google Scholar] [CrossRef]
- Heffernan, R.; Paliwal, K.; Lyons, J.; Dehzangi, A.; Sharma, A.; Wang, J.; Sattar, A.; Yang, Y.; Zhou, Y. Improving prediction of secondary structure, local backbone angles, and solvent accessible surface area of proteins by iterative deep learning. Sci. Rep. 2015, 5, 11476. [Google Scholar] [CrossRef]
- Yang, Y.; Heffernan, R.; Paliwal, K.; Lyons, J.; Dehzangi, A.; Sharma, A.; Wang, J.; Sattar, A.; Zhou, Y. SPIDER2: A Package to Predict Secondary Structure, Accessible Surface Area, and Main-Chain Torsional Angles by Deep Neural Networks. Methods Mol. Biol. 2017, 1484, 55–63. [Google Scholar] [CrossRef]
- Zhou, Z.H.; Feng, J. Deep forest. Natl. Sci. Rev. 2019, 6, 74–86. [Google Scholar] [CrossRef]
- Beauclair, G.; Bridier-Nahmias, A.; Zagury, J.F.; Saib, A.; Zamborlini, A. JASSA: A comprehensive tool for prediction of SUMOylation sites and SIMs. Bioinformatics 2015, 31, 3483–3491. [Google Scholar] [CrossRef]
- Chen, Z.; He, N.; Huang, Y.; Qin, W.T.; Liu, X.; Li, L. Integration of A Deep Learning Classifier with A Random Forest Approach for Predicting Malonylation Sites. Genom. Proteom. Bioinform. 2018, 16, 451–459. [Google Scholar] [CrossRef]
- Huang, Y.; He, N.; Chen, Y.; Chen, Z.; Li, L. BERMP: A cross-species classifier for predicting m(6)A sites by integrating a deep learning algorithm and a random forest approach. Int. J. Biol. Sci. 2018, 14, 1669–1677. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Lyon, D.; Young, C.; Jensen, L.J.; Vertegaal, A.C.; Nielsen, M.L. Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat. Struct. Mol. Biol. 2017, 24, 325–336. [Google Scholar] [CrossRef]
- Xu, H.; Zhou, J.; Lin, S.; Deng, W.; Zhang, Y.; Xue, Y. PLMD: An updated data resource of protein lysine modifications. J. Genet. Genom. 2017, 44, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Sha, Y.; Ma, C.; Wei, X.; Liu, Y.; Chen, Y.; Li, L. DeepSADPr: A Hybrid-learning Architecture for Serine ADP-ribosylation site prediction. Methods 2021, 203, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhao, P.; Li, F.; Leier, A.; Marquez-Lago, T.T.; Wang, Y.; Webb, G.I.; Smith, A.I.; Daly, R.J.; Chou, K.C.; et al. iFeature: A Python package and web server for features extraction and selection from protein and peptide sequences. Bioinformatics 2018, 34, 2499–2502. [Google Scholar] [CrossRef] [PubMed]
- Pearson, W.R. Selecting the Right Similarity-Scoring Matrix. Curr. Protoc. Bioinform. 2013, 43, 3–5. [Google Scholar] [CrossRef]
- Kawashima, S.; Pokarowski, P.; Pokarowska, M.; Kolinski, A.; Katayama, T.; Kanehisa, M. AAindex: Amino acid index database, progress report 2008. Nucleic Acids Res. 2008, 36, D202–D205. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.; Eriksson, L.; Jonsson, J.; Sjostrom, M.; Wold, S. New chemical descriptors relevant for the design of biologically active peptides. A multivariate characterization of 87 amino acids. J. Med. Chem. 1998, 41, 2481–2491. [Google Scholar] [CrossRef]
- Tasmia, S.A.; Kibria, M.K.; Tuly, K.F.; Islam, M.A.; Khatun, M.S.; Hasan, M.M.; Mollah, M.N.H. Prediction of serine phosphorylation sites mapping on Schizosaccharomyces Pombe by fusing three encoding schemes with the random forest classifier. Sci. Rep. 2022, 12, 2632. [Google Scholar] [CrossRef]
- Weng, S.L.; Kao, H.J.; Huang, C.H.; Lee, T.Y. MDD-Palm: Identification of protein S-palmitoylation sites with substrate motifs based on maximal dependence decomposition. PLoS ONE 2017, 12, e0179529. [Google Scholar] [CrossRef]
- Hasan, M.M.; Guo, D.; Kurata, H. Computational identification of protein S-sulfenylation sites by incorporating the multiple sequence features information. Mol. Biosyst. 2017, 13, 2545–2550. [Google Scholar] [CrossRef]
- Deng, L.; Pan, J.; Xu, X.; Yang, W.; Liu, C.; Liu, H. PDRLGB: Precise DNA-binding residue prediction using a light gradient boosting machine. BMC Bioinform. 2018, 19, 522. [Google Scholar] [CrossRef]
- Nair, V.; Hinton, G.E. Rectified linear units improve restricted boltzmann machines. In Proceedings of the 27th International Conference on International Conference on Machine Learning, Haifa, Israel, 21–24 June 2010; pp. 807–814. [Google Scholar]
- Xu, H.D.; Liang, R.P.; Wang, Y.G.; Qiu, J.D. mUSP: A high-accuracy map of the in situ crosstalk of ubiquitylation and SUMOylation proteome predicted via the feature enhancement approach. Brief. Bioinform. 2021, 22, bbaa050. [Google Scholar] [CrossRef]
- Vacic, V.; Iakoucheva, L.M.; Radivojac, P. Two Sample Logo: A graphical representation of the differences between two sets of sequence alignments. Bioinformatics 2006, 22, 1536–1537. [Google Scholar] [CrossRef]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B.; Chou, K.C. iSuc-PseOpt: Identifying lysine succinylation sites in proteins by incorporating sequence-coupling effects into pseudo components and optimizing imbalanced training dataset. Anal. Biochem. 2016, 497, 48–56. [Google Scholar] [CrossRef]
- Yin, S.; Zheng, J.; Jia, C.; Zou, Q.; Lin, Z.; Shi, H. UPFPSR: A ubiquitylation predictor for plant through combining sequence information and random forest. Math. Biosci. Eng. 2022, 19, 775–791. [Google Scholar] [CrossRef]
- Xu, Y.; Ding, Y.X.; Ding, J.; Lei, Y.H.; Wu, L.Y.; Deng, N.Y. iSuc-PseAAC: Predicting lysine succinylation in proteins by incorporating peptide position-specific propensity. Sci. Rep. 2015, 5, 10184. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Shahjaman, M.; Kabir, E.; Kamruzzaman, M. Prediction of Protein Acetylation Sites using Kernel Naive Bayes Classifier Based on Protein Sequences Profiling. Bioinformation 2018, 14, 213–218. [Google Scholar] [CrossRef]
- Xue, Y.; Chen, H.; Jin, C.; Sun, Z.; Yao, X. NBA-Palm: Prediction of palmitoylation site implemented in Naive Bayes algorithm. BMC Bioinform. 2006, 7, 458. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, M.; Qin, X.; Liu, G. Succinylation Site Prediction Based on Protein Sequences Using the IFS-LightGBM (BO) Model. Comput. Math. Methods Med. 2020, 2020, 8858489. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, Z.; Chen, C.; Han, Y.; Yu, B. Prediction of protein crotonylation sites through LightGBM classifier based on SMOTE and elastic net. Anal. Biochem. 2020, 609, 113903. [Google Scholar] [CrossRef]
- Sharma, A.; Singh, B. AE-LGBM: Sequence-based novel approach to detect interacting protein pairs via ensemble of autoencoder and LightGBM. Comput. Biol. Med. 2020, 125, 103964. [Google Scholar] [CrossRef]
- Eraslan, G.; Avsec, Z.; Gagneur, J.; Theis, F.J. Deep learning: New computational modelling techniques for genomics. Nat. Rev. Genet. 2019, 20, 389–403. [Google Scholar] [CrossRef]
- Song, Z.; Huang, D.; Song, B.; Chen, K.; Song, Y.; Liu, G.; Su, J.; Magalhaes, J.P.; Rigden, D.J.; Meng, J. Attention-based multi-label neural networks for integrated prediction and interpretation of twelve widely occurring RNA modifications. Nat. Commun. 2021, 12, 4011. [Google Scholar] [CrossRef]
- Wang, D.; Zeng, S.; Xu, C.; Qiu, W.; Liang, Y.; Joshi, T.; Xu, D. MusiteDeep: A deep-learning framework for general and kinase-specific phosphorylation site prediction. Bioinformatics 2017, 33, 3909–3916. [Google Scholar] [CrossRef]
- Cao, Z.; Chu, Z.; Liu, D.; Chen, Y. A vector-based representation to enhance head pose estimation. In Proceedings of the IEEE/CVF Winter Conference on Applications of Computer Vision, Waikoloa, HI, USA, 3–8 January 2021; pp. 1188–1197. [Google Scholar]
- Cui, Y.; Cao, Z.; Xie, Y.; Jiang, X.; Tao, F.; Chen, Y.V.; Li, L.; Liu, D. Dg-labeler and dgl-mots dataset: Boost the autonomous driving perception. In Proceedings of the IEEE/CVF Winter Conference on Applications of Computer Vision, Waikoloa, HI, USA, 4–8 January 2022; pp. 58–67. [Google Scholar]
- Cui, Y.; Yan, L.; Cao, Z.; Liu, D. Tf-blender: Temporal feature blender for video object detection. In Proceedings of the IEEE/CVF International Conference on Computer Vision, Montreal, QC, Canada, 10–17 October 2021; pp. 8138–8147. [Google Scholar]
- Liu, D.; Cui, Y.; Tan, W.; Chen, Y. Sg-net: Spatial granularity network for one-stage video instance segmentation. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, Nashville, TN, USA, 20–25 June 2021; pp. 9816–9825. [Google Scholar]
- Liu, D.; Cui, Y.; Yan, L.; Mousas, C.; Yang, B.; Chen, Y. Densernet: Weakly supervised visual localization using multi-scale feature aggregation. In Proceedings of the AAAI Conference on Artificial Intelligence, Virtually, 2–9 February 2021; pp. 6101–6109. [Google Scholar]
- Sun, S.; Lopez, J.A.; Xie, Y.; Guo, W.; Liu, D.; Li, L. HIT web server: A hybrid method to improve electrostatic calculations for biomolecules. Comput. Struct. Biotechnol. J. 2022, 20, 1580–1583. [Google Scholar] [CrossRef]
- Wang, Q.; Fang, Y.; Ravula, A.; Feng, F.; Quan, X.; Liu, D. WebFormer: The Web-page Transformer for Structure Information Extraction. In Proceedings of the ACM Web Conference 2022, Lyon, France, 25–29 April 2022; pp. 3124–3133. [Google Scholar]
- Xie, Y.; Karki, C.B.; Chen, J.; Liu, D.; Li, L. Computational study on DNA repair: The roles of electrostatic interactions between uracil-DNA glycosylase (UDG) and DNA. Front. Mol. Biosci. 2021, 8, 718587. [Google Scholar] [CrossRef]
- Yan, L.; Ma, S.; Wang, Q.; Chen, Y.; Zhang, X.; Savakis, A.; Liu, D. Video Captioning Using Global-Local Representation. IEEE Trans. Circuits Syst. Video Technol. 2022, 1. [Google Scholar] [CrossRef]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep Residual Learning for Image Recognition. In Proceedings of the 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Feng, F.; Wang, S.; Wang, C.; Zhang, J. Learning Deep Hierarchical Spatial-Spectral Features for Hyperspectral Image Classification Based on Residual 3D-2D CNN. Sensors 2019, 19, 5276. [Google Scholar] [CrossRef]
- Li, K.; Wang, B. DAR-Net: Dense Attentional Residual Network for Vehicle Detection in Aerial Images. Comput. Intell. Neurosci. 2021, 2021, 6340823. [Google Scholar] [CrossRef]
- Si, Y.; Yan, C. Improved protein contact prediction using dimensional hybrid residual networks and singularity enhanced loss function. Brief. Bioinform. 2021, 22, bbab341. [Google Scholar] [CrossRef]
- Li, Y.; Hu, J.; Zhang, C.; Yu, D.J.; Zhang, Y. ResPRE: High-accuracy protein contact prediction by coupling precision matrix with deep residual neural networks. Bioinformatics 2019, 35, 4647–4655. [Google Scholar] [CrossRef]
- Shen, L.C.; Liu, Y.; Song, J.; Yu, D.J. SAResNet: Self-attention residual network for predicting DNA-protein binding. Brief. Bioinform. 2021, 22, bbab101. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, P.; Li, C.; Li, F.; Xiang, D.; Chen, Y.Z.; Akutsu, T.; Daly, R.J.; Webb, G.I.; Zhao, Q.; et al. iLearnPlus: A comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res. 2021, 49, e60. [Google Scholar] [CrossRef]
- Lyu, X.; Li, S.; Jiang, C.; He, N.; Chen, Z.; Zou, Y.; Li, L. DeepCSO: A Deep-Learning Network Approach to Predicting Cysteine S-Sulphenylation Sites. Front. Cell Dev. Biol. 2020, 8, 594587. [Google Scholar] [CrossRef]
- Wei, X.; Sha, Y.; Zhao, Y.; He, N.; Li, L. DeepKcrot: A deep-learning architecture for general and species-specific lysine crotonylation site prediction. IEEE Access 2021, 9, 49504–49513. [Google Scholar] [CrossRef]
- Zhang, L.; Zou, Y.; He, N.; Chen, Y.; Chen, Z.; Li, L. DeepKhib: A Deep-Learning Framework for Lysine 2-Hydroxyisobutyrylation Sites Prediction. Front. Cell Dev. Biol. 2020, 8, 580217. [Google Scholar] [CrossRef]
- Wang, M.; Cui, X.; Yu, B.; Chen, C.; Ma, Q.; Zhou, H. SulSite-GTB: Identification of protein S-sulfenylation sites by fusing multiple feature information and gradient tree boosting. Neural Comput. Appl. 2020, 32, 13843–13862. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Sn | Sp | MCC | ACC | AUC |
|---|---|---|---|---|---|
| RF_AAindex [15] | 0.697 ± 0.004 | 0.650 ± 0.000 | 0.348 ± 0.004 | 0.674 ± 0.002 | 0.742 ± 0.003 |
| RF_BLOSUM62 [15] | 0.664 ± 0.004 | 0.650 ± 0.000 | 0.314 ± 0.004 | 0.657 ± 0.002 | 0.722 ± 0.003 |
| RF_EAAC [15] | 0.693 ± 0.008 | 0.650 ± 0.000 | 0.344 ± 0.008 | 0.672 ± 0.004 | 0.738 ± 0.005 |
| RF_ZScale [15] | 0.661 ± 0.006 | 0.650 ± 0.000 | 0.311 ± 0.006 | 0.655 ± 0.003 | 0.721 ± 0.004 |
| LGBM_AAindex [39] | 0.709 ± 0.011 | 0.650 ± 0.000 | 0.360 ± 0.011 | 0.680 ± 0.005 | 0.752 ± 0.003 |
| LGBM_BLOSUM62 [39] | 0.708 ± 0.013 | 0.650 ± 0.000 | 0.358 ± 0.013 | 0.679 ± 0.007 | 0.748 ± 0.005 |
| LGBM_EAAC [39] | 0.734 ± 0.008 | 0.650 ± 0.000 | 0.385 ± 0.008 | 0.692 ± 0.004 | 0.762 ± 0.004 |
| LGBM_ZScale [39] | 0.694 ± 0.012 | 0.650 ± 0.000 | 0.344 ± 0.012 | 0.672 ± 0.006 | 0.741 ± 0.005 |
| CNN_AAindex [31] | 0.784 ± 0.007 | 0.650 ± 0.000 | 0.438 ± 0.008 | 0.717 ± 0.004 | 0.788 ± 0.005 |
| CNN_BLOSUM62 [31] | 0.784 ± 0.006 | 0.650 ± 0.000 | 0.438 ± 0.007 | 0.717 ± 0.003 | 0.788 ± 0.004 |
| CNN_EAAC [31] | 0.771 ± 0.009 | 0.650 ± 0.000 | 0.424 ± 0.010 | 0.711 ± 0.004 | 0.782 ± 0.004 |
| CNN_ZScale [31] | 0.779 ± 0.008 | 0.650 ± 0.000 | 0.433 ± 0.009 | 0.714 ± 0.004 | 0.785 ± 0.005 |
| RSCNN_AAindex [63] | 0.803 ± 0.008 | 0.650 ± 0.000 | 0.458 ± 0.009 | 0.726 ± 0.004 | 0.799 ± 0.004 |
| RSCNN_BLOSUM62 [63] | 0.803 ± 0.007 | 0.650 ± 0.000 | 0.458 ± 0.008 | 0.726 ± 0.004 | 0.799 ± 0.003 |
| RSCNN_EAAC [63] | 0.763 ± 0.007 | 0.650 ± 0.000 | 0.416 ± 0.008 | 0.706 ± 0.004 | 0.774 ± 0.004 |
| RSCNN_ZScale [63] | 0.802 ± 0.007 | 0.650 ± 0.000 | 0.457 ± 0.007 | 0.726 ± 0.003 | 0.800 ± 0.004 |
| Model | Sn | Sp | MCC | ACC | AUC |
|---|---|---|---|---|---|
| Five-fold cross-validation | |||||
| SUMO-Forest | 0.729 ± 0.006 | 0.650 ± 0.000 | 0.380 ± 0.006 | 0.689 ± 0.003 | 0.760 ± 0.003 |
| iSUMOK-PseAAC | 0.506 ± 0.023 | 0.650 ± 0.000 | 0.158 ± 0.023 | 0.578 ± 0.012 | 0.620 ± 0.016 |
| ResSUMO | 0.802 ± 0.007 | 0.650 ±0.000 | 0.457 ± 0.007 | 0.726 ± 0.003 | 0.800 ± 0.004 |
| Independent test | |||||
| SUMO-Forest | 0.745 ± 0.002 | 0.650 ± 0.000 | 0.397 ± 0.002 | 0.698 ± 0.001 | 0.769 ± 0.002 |
| iSUMOK-PseAAC | 0.524 ± 0.006 | 0.650 ± 0.000 | 0.176 ± 0.006 | 0.587 ± 0.003 | 0.628 ± 0.006 |
| ResSUMO | 0.795 ± 0.007 | 0.650 ±0.000 | 0.450 ± 0.008 | 0.722 ± 0.003 | 0.801 ± 0.003 |
| Model * | Sn | Sp | MCC | ACC | AUC |
|---|---|---|---|---|---|
| CNN-1 | 0.716 ± 0.015 | 0.650 ± 0.000 | 0.367 ± 0.015 | 0.683 ± 0.007 | 0.747 ± 0.009 |
| CNN-2 | 0.779 ± 0.008 | 0.650 ± 0.000 | 0.433 ± 0.009 | 0.714 ± 0.004 | 0.785 ± 0.005 |
| CNN-4 | 0.773 ± 0.005 | 0.650 ± 0.000 | 0.426 ± 0.006 | 0.711 ± 0.003 | 0.783 ± 0.002 |
| CNN-6 | 0.765 ± 0.009 | 0.650 ± 0.000 | 0.418 ± 0.009 | 0.708 ± 0.004 | 0.779 ± 0.007 |
| CNN-8 | 0.764 ± 0.013 | 0.650 ± 0.000 | 0.417 ± 0.014 | 0.707 ± 0.006 | 0.778 ± 0.005 |
| ResSUMO | 0.802 ± 0.007 | 0.650 ± 0.000 | 0.457 ± 0.007 | 0.726 ± 0.003 | 0.800 ± 0.004 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Liu, Y.; Chen, Y.; Li, L. ResSUMO: A Deep Learning Architecture Based on Residual Structure for Prediction of Lysine SUMOylation Sites. Cells 2022, 11, 2646. https://doi.org/10.3390/cells11172646
Zhu Y, Liu Y, Chen Y, Li L. ResSUMO: A Deep Learning Architecture Based on Residual Structure for Prediction of Lysine SUMOylation Sites. Cells. 2022; 11(17):2646. https://doi.org/10.3390/cells11172646
Chicago/Turabian StyleZhu, Yafei, Yuhai Liu, Yu Chen, and Lei Li. 2022. "ResSUMO: A Deep Learning Architecture Based on Residual Structure for Prediction of Lysine SUMOylation Sites" Cells 11, no. 17: 2646. https://doi.org/10.3390/cells11172646