
Integration of Human Protein Sequence and Protein-Protein Interaction Data by Graph Autoencoder to Identify Novel Protein-Abnormal Phenotype Associations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Gold Standard Data Sets
2.2. Comparison Dataset
2.3. Protein–Protein Interaction Network and Sequence Evidence for Prediction
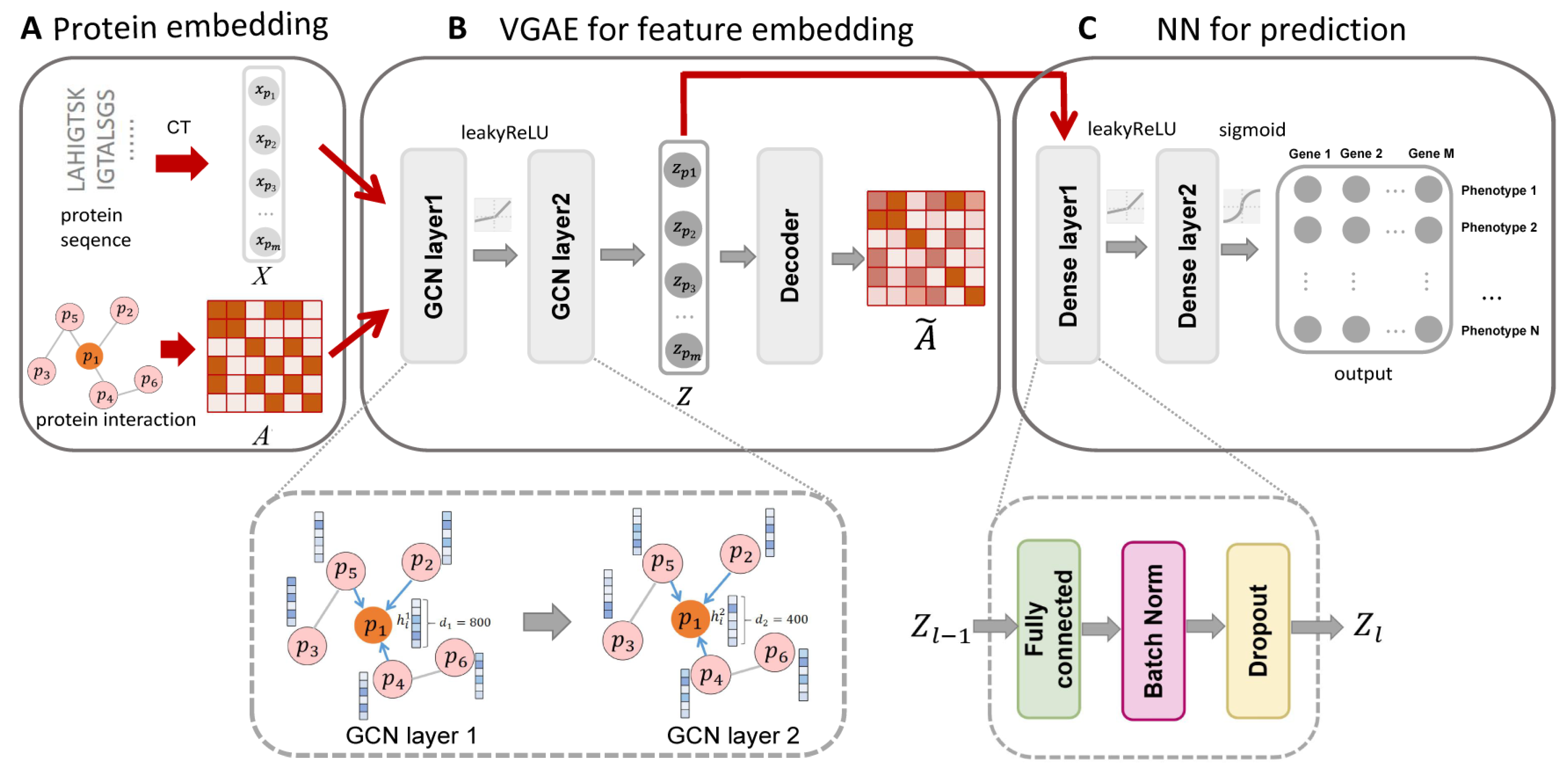
2.4. Variational Graph Autoencoder Model
2.5. Neural Network Model
2.6. Cross-Validation
2.7. F-Max for Protein-Centric Evaluation
2.8. AUC for Term-Centric Evaluation
3. Results
3.1. Overview of Our Prediction Protocol
3.2. Performance Evaluation for GraphPheno
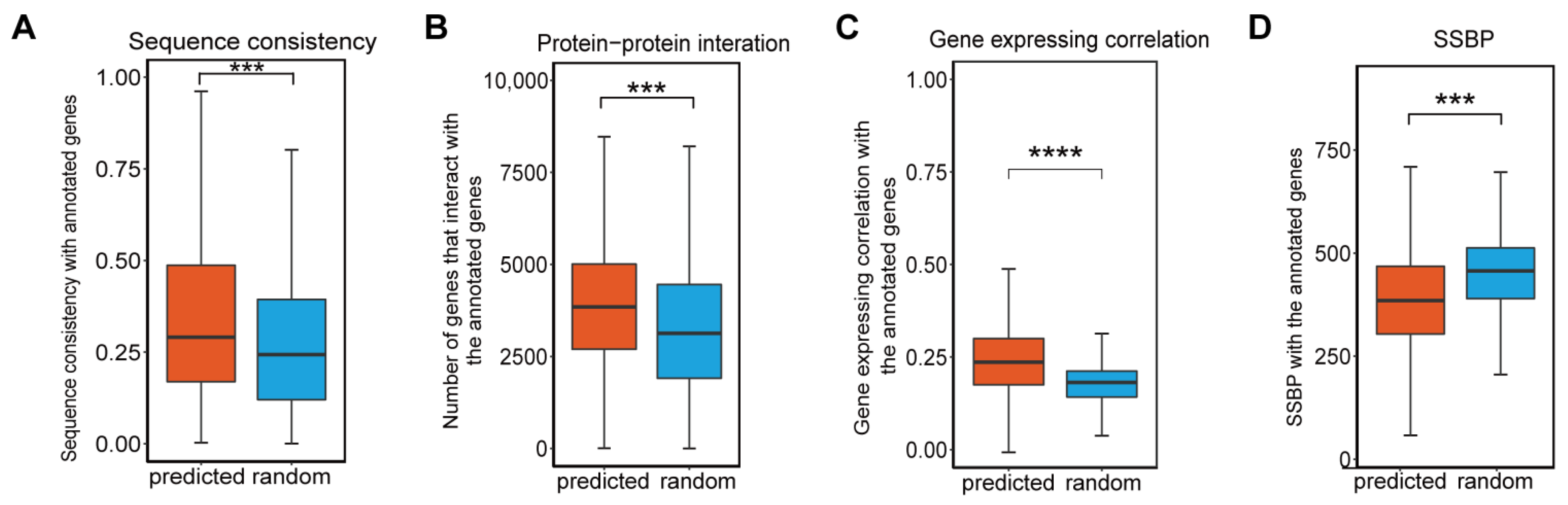
3.3. Predicted and Known Phenotype-Associated Genes Share Similar Biological Properties
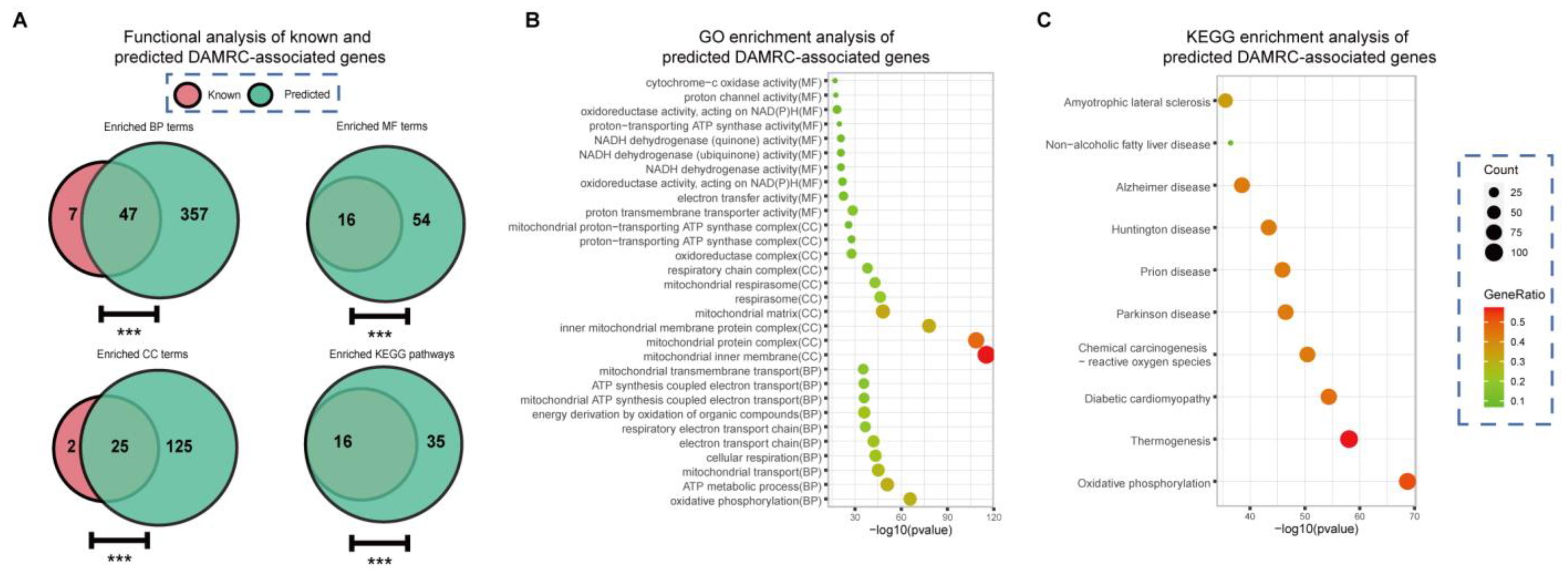
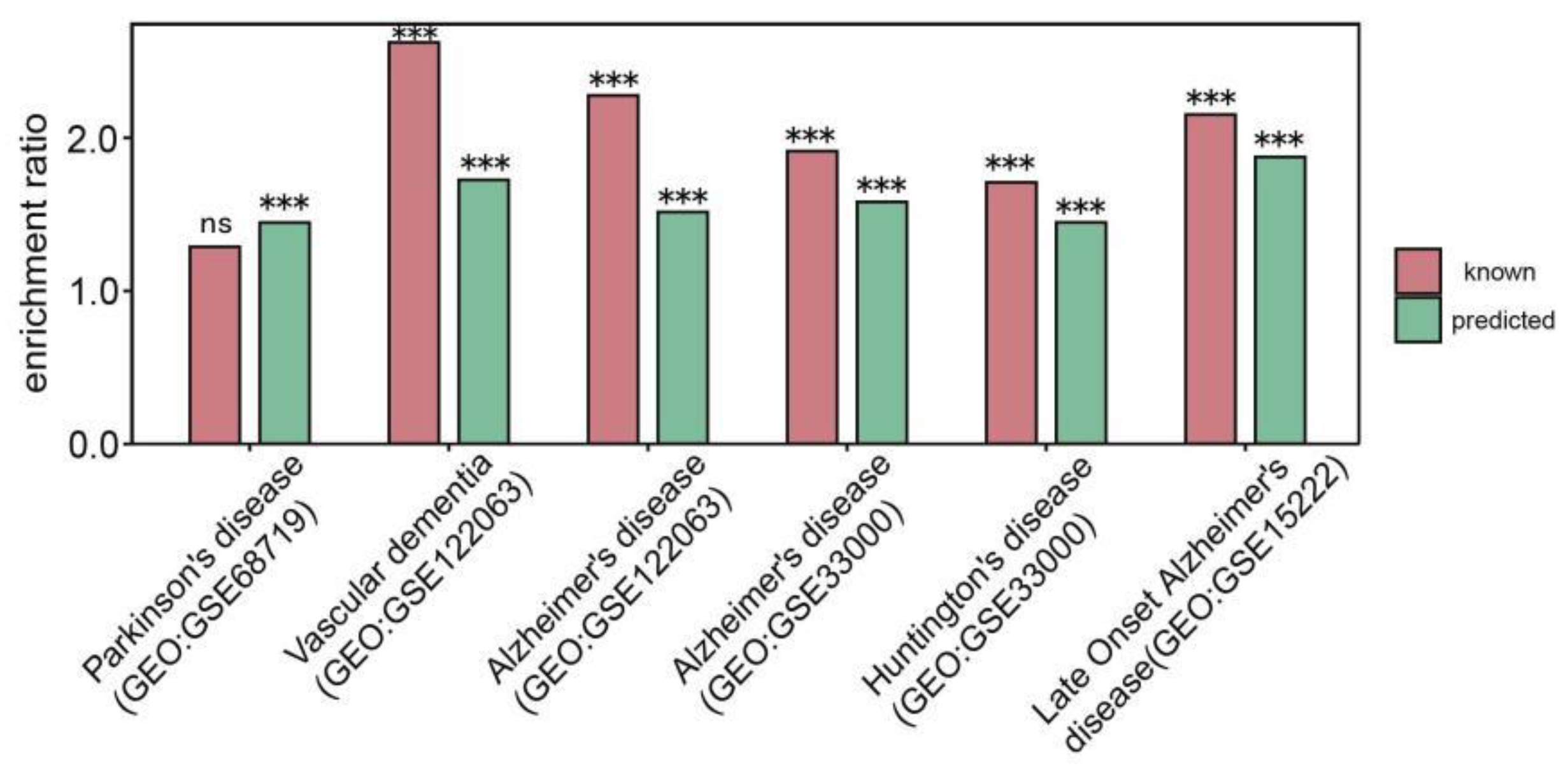
3.4. Discovery of Genes Associated with Abnormality of Mitochondrial Respiratory Chain
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kohler, S.; Doelken, S.C.; Mungall, C.J.; Bauer, S.; Firth, H.V.; Bailleul-Forestier, I.; Black, G.C.; Brown, D.L.; Brudno, M.; Campbell, J. The Human Phenotype Ontology project: Linking molecular biology and disease through phenotype data. Nucleic Acids Res. 2014, 42, D966–D974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef] [PubMed]
- Radivojac, P.; Clark, W.T.; Oron, T.R.; Schnoes, A.M.; Wittkop, T.; Sokolov, A.; Graim, K.; Funk, C.; Verspoor, K.; Ben-Hur, A. A large-scale evaluation of computational protein function prediction. Nat. Methods 2013, 10, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Oron, T.R.; Clark, W.T.; Bankapur, A.R.; D’Andrea, D.; Lepore, R.; Funk, C.S.; Kahanda, I.; Verspoor, K.M.; Ben-Hur, A. An expanded evaluation of protein function prediction methods shows an improvement in accuracy. Genome Biol. 2016, 17, 184. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Jiang, Y.; Bergquist, T.R.; Lee, A.J.; Kacsoh, B.Z.; Crocker, A.W.; Lewis, K.A.; Georghiou, G.; Nguyen, H.N.; Hamid, M.N. The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens. Genome Biol. 2019, 20, 244. [Google Scholar] [CrossRef] [Green Version]
- Gong, Q.; Ning, W.; Tian, W. GoFDR: A sequence alignment based method for predicting protein functions. Methods 2016, 93, 3–14. [Google Scholar] [CrossRef]
- Scheibenreif, L.; Littmann, M.; Orengo, C.; Rost, B. FunFam protein families improve residue level molecular function prediction. BMC Bioinform. 2019, 20, 400. [Google Scholar] [CrossRef] [Green Version]
- Valentini, G.; Armano, G.; Frasca, M.; Lin, J.; Mesiti, M.; Re, M. RANKS: A flexible tool for node label ranking and classification in biological networks. Bioinformatics 2016, 32, 2872–2874. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Xu, Y.; Zhang, Y.; Hwang, T.; Kuang, R. Network-based Phenome-Genome Association Prediction by Bi-Random Walk. PLoS ONE 2015, 10, e0125138. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Liu, L.; Mamitsuka, H.; Zhu, S. HPODNets: Deep graph convolutional networks for predicting human protein-phenotype associations. Bioinformatics 2021, 38, 799–808. [Google Scholar] [CrossRef]
- Lan, L.; Djuric, N.; Guo, Y.; Vucetic, S. MS-kNN: Protein function prediction by integrating multiple data sources. BMC Bioinform. 2013, 14 (Suppl. 3), S8. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Huang, X.; Mamitsuka, H.; Zhu, S. HPOLabeler: Improving prediction of human protein-phenotype associations by learning to rank. Bioinformatics 2020, 36, 4180–4188. [Google Scholar] [CrossRef]
- You, R.; Zhang, Z.; Xiong, Y.; Sun, F.; Mamitsuka, H.; Zhu, S. GOLabeler: Improving sequence-based large-scale protein function prediction by learning to rank. Bioinformatics 2018, 34, 2465–2473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahanda, I.; Funk, C.; Verspoor, K.; Ben-Hur, A. PHENOstruct: Prediction of human phenotype ontology terms using heterogeneous data sources. F1000research 2015, 4, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulmanov, M.; Khan, M.A.; Hoehndorf, R. DeepGO: Predicting protein functions from sequence and interactions using a deep ontology-aware classifier. Bioinformatics 2018, 34, 660–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulmanov, M.; Hoehndorf, R. DeepGOPlus: Improved protein function prediction from sequence. Bioinformatics 2020, 36, 422–429. [Google Scholar] [CrossRef]
- Valentini, G. True path rule hierarchical ensembles for genome-wide gene function prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 8, 832–847. [Google Scholar] [CrossRef]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bansal, P.; Bridge, A.J.; Poux, S.; Bougueleret, L.; Xenarios, I. UniProtKB/Swiss-Prot, the Manually Annotated Section of the UniProt KnowledgeBase: How to Use the Entry View. Methods Mol. Biol. 2016, 1374, 23–54. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, J.; Luo, X.; Zhu, W.; Yu, K.; Chen, K.; Li, Y.; Jiang, H. Predicting protein-protein interactions based only on sequences information. Proc. Natl. Acad. Sci. USA 2007, 104, 4337–4341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, K.; Guan, Y.; Zhang, Y. Graph2GO: A multi-modal attributed network embedding method for inferring protein functions. GigaScience 2020, 9, giaa081. [Google Scholar] [CrossRef] [PubMed]
- Kipf, T.N.; Welling, M. Variational Graph Auto-Encoders. arXiv 2016, arXiv:1611.07308. [Google Scholar]
- Zhang, X.; Zou, Y.; Shi, W. Dilated convolution neural network with LeakyReLU for environmental sound classification. In Proceedings of the 2017 22nd International Conference on Digital Signal Processing (DSP), London, UK, 23–25 August 2017; pp. 1–5. [Google Scholar] [CrossRef]
- Kingma, D.P.; Welling, M. Auto-Encoding Variational Bayes. arXiv 2014, arXiv:1312.6114. [Google Scholar]
- Hershey, J.R.; Olsen, P.A. Approximating the Kullback Leibler Divergence between Gaussian Mixture Models. In Proceedings of the 2007 IEEE International Conference on Acoustics, Speech and Signal Processing—ICASSP ’07, Honolulu, HI, USA, 15–20 April 2007; pp. 317–320. [Google Scholar] [CrossRef] [Green Version]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Clark, W.T.; Radivojac, P. Analysis of protein function and its prediction from amino acid sequence. Proteins 2011, 79, 2086–2096. [Google Scholar] [CrossRef]
- Piovesan, D.; Giollo, M.; Leonardi, E.; Ferrari, C.; Tosatto, S.C. INGA: Protein function prediction combining interaction networks, domain assignments and sequence similarity. Nucleic Acids Res. 2015, 43, W134–W140. [Google Scholar] [CrossRef] [Green Version]
- Gene Ontology Consortium. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017, 45, D331–D338. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.Y.; Dosztanyi, Z.; El-Gebali, S.; Fraser, M. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef]
- Ruopp, M.D.; Perkins, N.J.; Whitcomb, B.W.; Schisterman, E.F. Youden Index and optimal cut-point estimated from observations affected by a lower limit of detection. Biom. J. 2008, 50, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, W.; Liu, Z.; Wang, J.; Liu, Q.; Zhu, Y.; He, F. PRINCESS, a protein interaction confidence evaluation system with multiple data sources. Mol. Cell Proteom. 2008, 7, 1043–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vercellino, I.; Sazanov, L.A. The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell Biol. 2022, 23, 141–161. [Google Scholar] [CrossRef] [PubMed]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef]
- Larsen, S.B.; Hanss, Z.; Kruger, R. The genetic architecture of mitochondrial dysfunction in Parkinson’s disease. Cell Tissue Res. 2018, 373, 21–37. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C.; Fan, W.; Procaccio, V. Mitochondrial energetics and therapeutics. Annu. Rev. Pathol. 2010, 5, 297–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jassal, B.; Matthews, L.; Viteri, G. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [Green Version]
- Connolly, N.M.C.; Theurey, P.; Adam-Vizi, V.; Bazan, N.G.; Bernardi, P.; Bolanos, J.P.; Culmsee, C.; Dawson, V.L.; Deshmukh, M.; Duchen, M.R. Guidelines on experimental methods to assess mitochondrial dysfunction in cellular models of neurodegenerative diseases. Cell Death Differ. 2018, 25, 542–572. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Vidali, S.; Gerlini, R.; Thompson, K.; Urquhart, J.E.; Meisterknecht, J.; Aguilar-Pimentel, J.A.; Amarie, O.V.; Becker, L.; Breen, C.; Calzada-Wack, J. Characterising a homozygous two-exon deletion in UQCRH: Comparing human and mouse phenotypes. EMBO Mol. Med. 2021, 13, e14397. [Google Scholar] [CrossRef]
- Neilson, D.E.; Zech, M.; Hufnagel, R.B.; Slone, J.; Wang, X.; Homan, S.; Gutzwiller, L.M.; Leslie, E.J.; Leslie, N.D.; Xiao, J. A Novel Variant of ATP5MC3 Associated with Both Dystonia and Spastic Paraplegia. Mov. Disord. 2022, 37, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Barel, O.; Shorer, Z.; Flusser, H.; Ofir, R.; Narkis, G.; Finer, G.; Shalev, H.; Nasasra, A.; Saada, A.; Birk, O.S. Mitochondrial complex III deficiency associated with a homozygous mutation in UQCRQ. Am. J. Hum. Genet. 2008, 82, 1211–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, H.; Yu, X.; He, H.; Zhou, L.; Li, Q.; Song, C.; Wang, D.; Ren, T.; Chen, Z.; Huang, H. Hepatocyte miR-33a mediates mitochondrial dysfunction and hepatosteatosis by suppressing NDUFA5. J. Cell. Mol. Med. 2018, 22, 6285–6293. [Google Scholar] [CrossRef] [Green Version]
- Piekutowska-Abramczuk, D.; Assouline, Z.; Matakovic, L.; Feichtinger, R.G.; Konarikova, E.; Jurkiewicz, E.; Stawinski, P.; Gusic, M.; Koller, A.; Pollak, A. NDUFB8 Mutations Cause Mitochondrial Complex I Deficiency in Individuals with Leigh-like Encephalomyopathy. Am. J. Hum. Genet. 2018, 102, 460–467. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.P.; Sun, H.F.; Jiang, H.L.; Li, L.D.; Hu, X.; Xu, X.E.; Jin, W. Loss of COX5B inhibits proliferation and promotes senescence via mitochondrial dysfunction in breast cancer. Oncotarget 2015, 6, 43363–43374. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.F.; He, S.; Zeng, Z.; Gu, X.; Cai, L.; Qi, G. PMPCB Silencing Sensitizes HCC Tumor Cells to Sorafenib Therapy. Mol. Ther. 2019, 27, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Sun, A.; Zhao, Y. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 2019, 567, 257–261. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; He, R.; Qu, Y.; Zhu, Y.; Li, D.; Ling, X.; Xia, S.; Li, Z.; Li, D. Integration of Human Protein Sequence and Protein-Protein Interaction Data by Graph Autoencoder to Identify Novel Protein-Abnormal Phenotype Associations. Cells 2022, 11, 2485. https://doi.org/10.3390/cells11162485
Liu Y, He R, Qu Y, Zhu Y, Li D, Ling X, Xia S, Li Z, Li D. Integration of Human Protein Sequence and Protein-Protein Interaction Data by Graph Autoencoder to Identify Novel Protein-Abnormal Phenotype Associations. Cells. 2022; 11(16):2485. https://doi.org/10.3390/cells11162485
Chicago/Turabian StyleLiu, Yuan, Ruirui He, Yingjie Qu, Yuan Zhu, Dianke Li, Xinping Ling, Simin Xia, Zhenqiu Li, and Dong Li. 2022. "Integration of Human Protein Sequence and Protein-Protein Interaction Data by Graph Autoencoder to Identify Novel Protein-Abnormal Phenotype Associations" Cells 11, no. 16: 2485. https://doi.org/10.3390/cells11162485