
Pharmacological Activation of Potassium Channel Kv11.1 with NS1643 Attenuates Triple Negative Breast Cancer Cell Migration by Promoting the Dephosphorylation of Caveolin-1

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents
2.3. Transfection and Infection
2.4. Western Blot Analysis and Immunoprecipitation
2.5. Immunostaining and Microscopy
2.6. Scratch-Wound Assay
2.7. Single Cell Tracking
2.8. Spheroid Generation and Growth
2.9. TMT Labeling and Mass Spectrometry
2.10. Statistical Analysis
3. Results
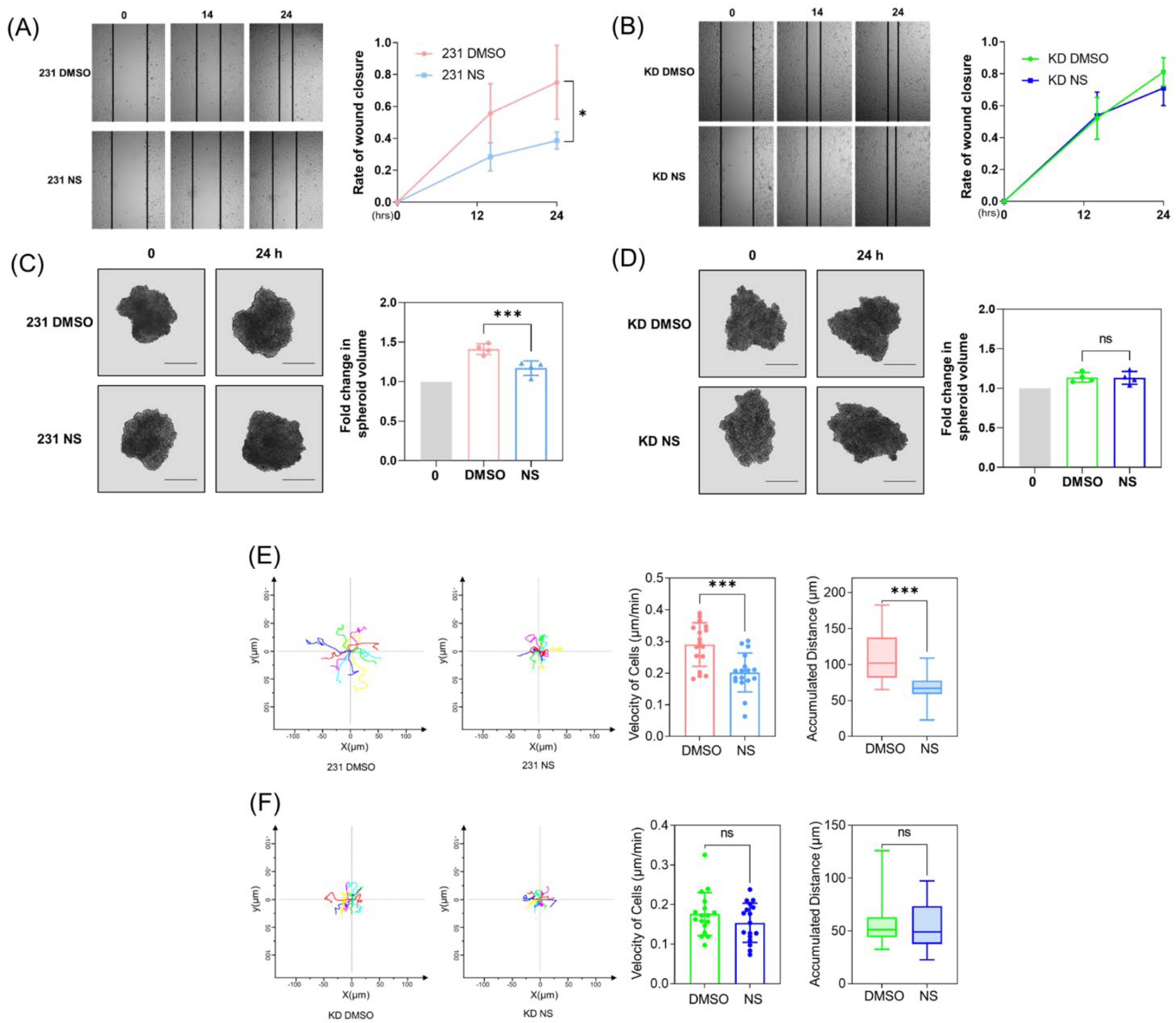
3.1. Cav1 Mediates the Kv11.1 Activation-Dependent Inhibition of Cancer Cell Migration

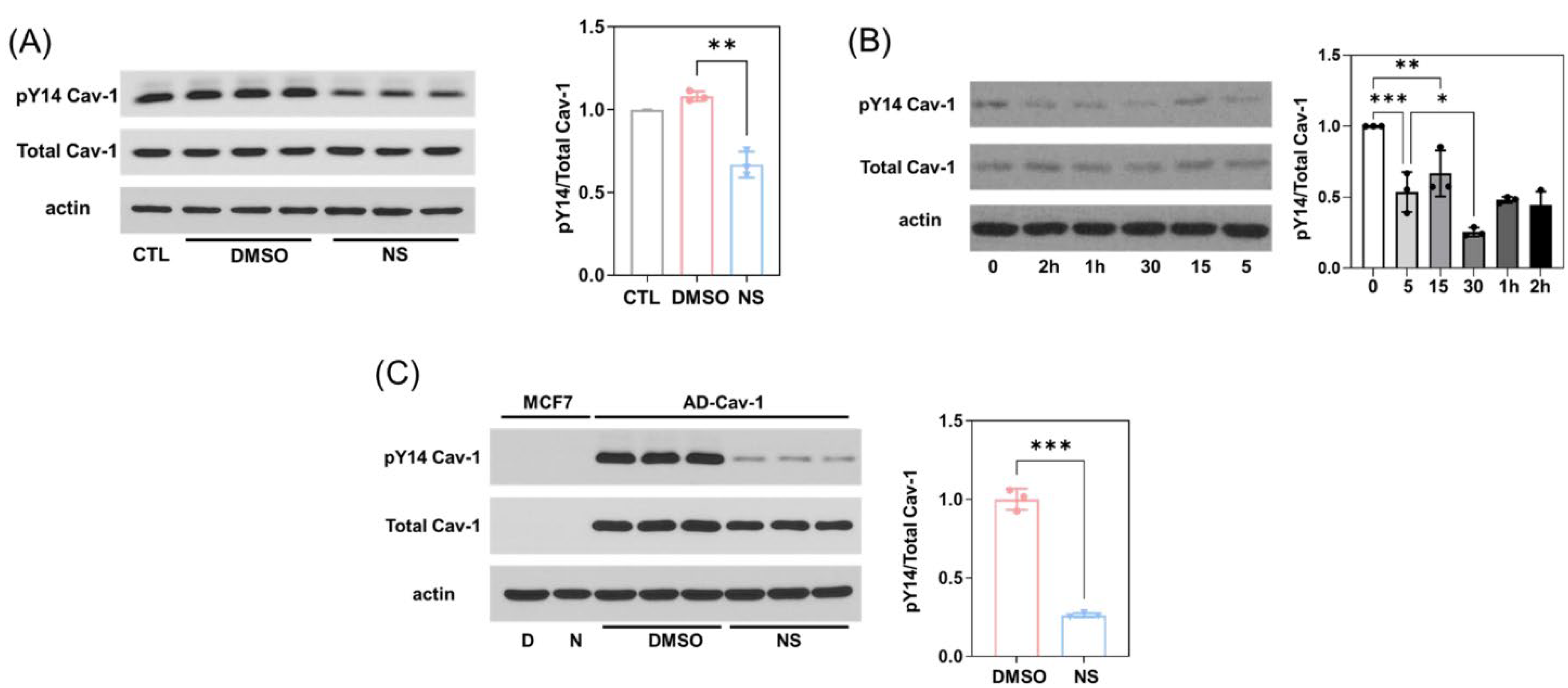
3.2. Kv11.1 Activation Decreases Cav1 Phosphorylation Independent of Src

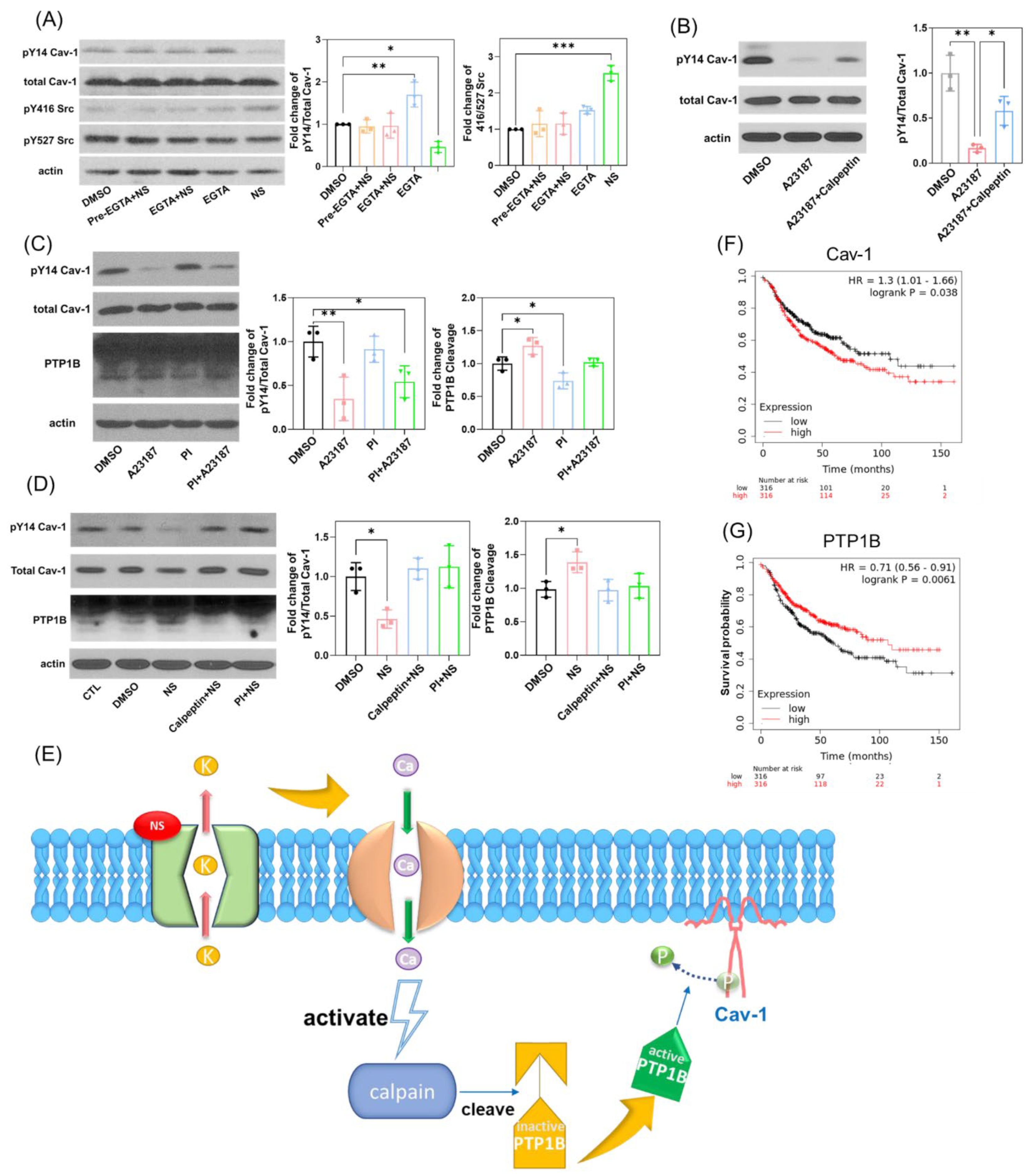
3.3. Kv11.1 Activation of PTP1B Leads to the Dephosphorylation of Cav1 Y14
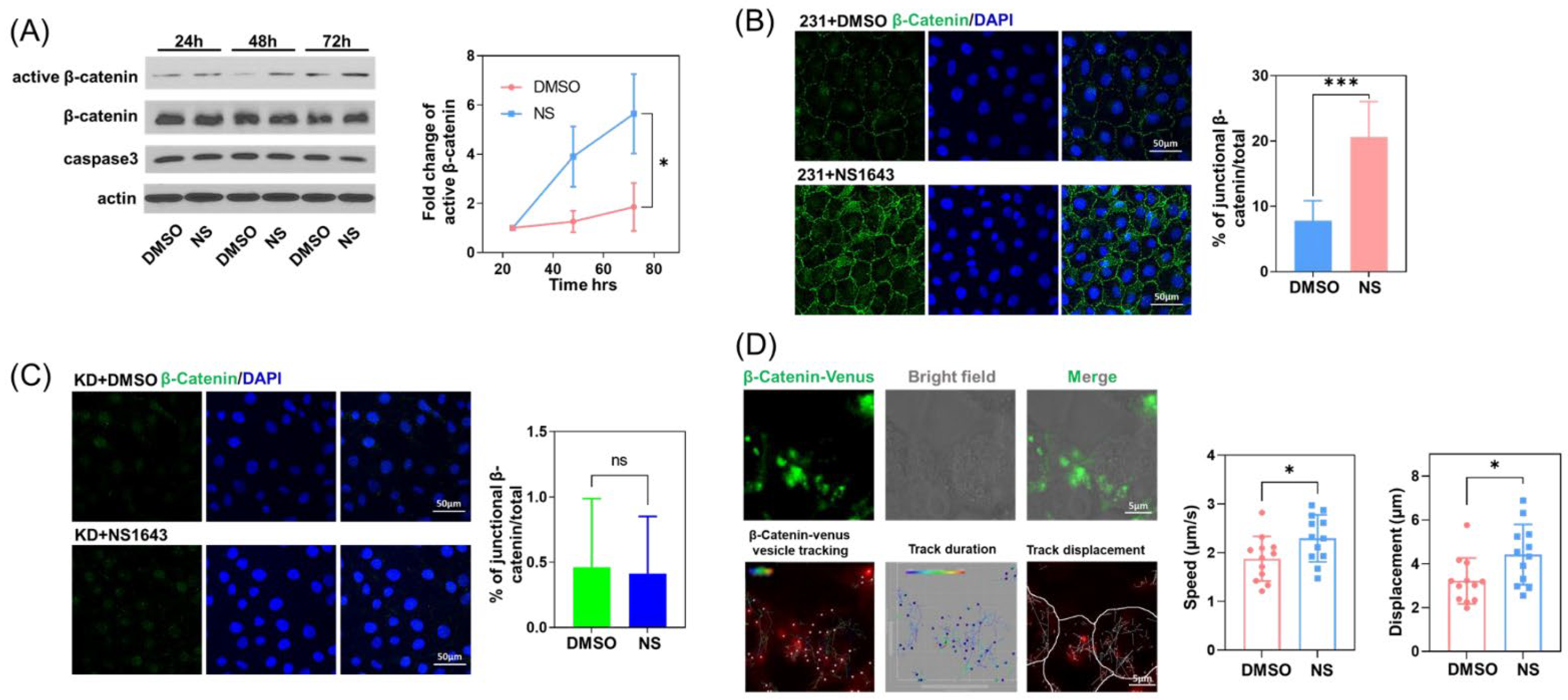
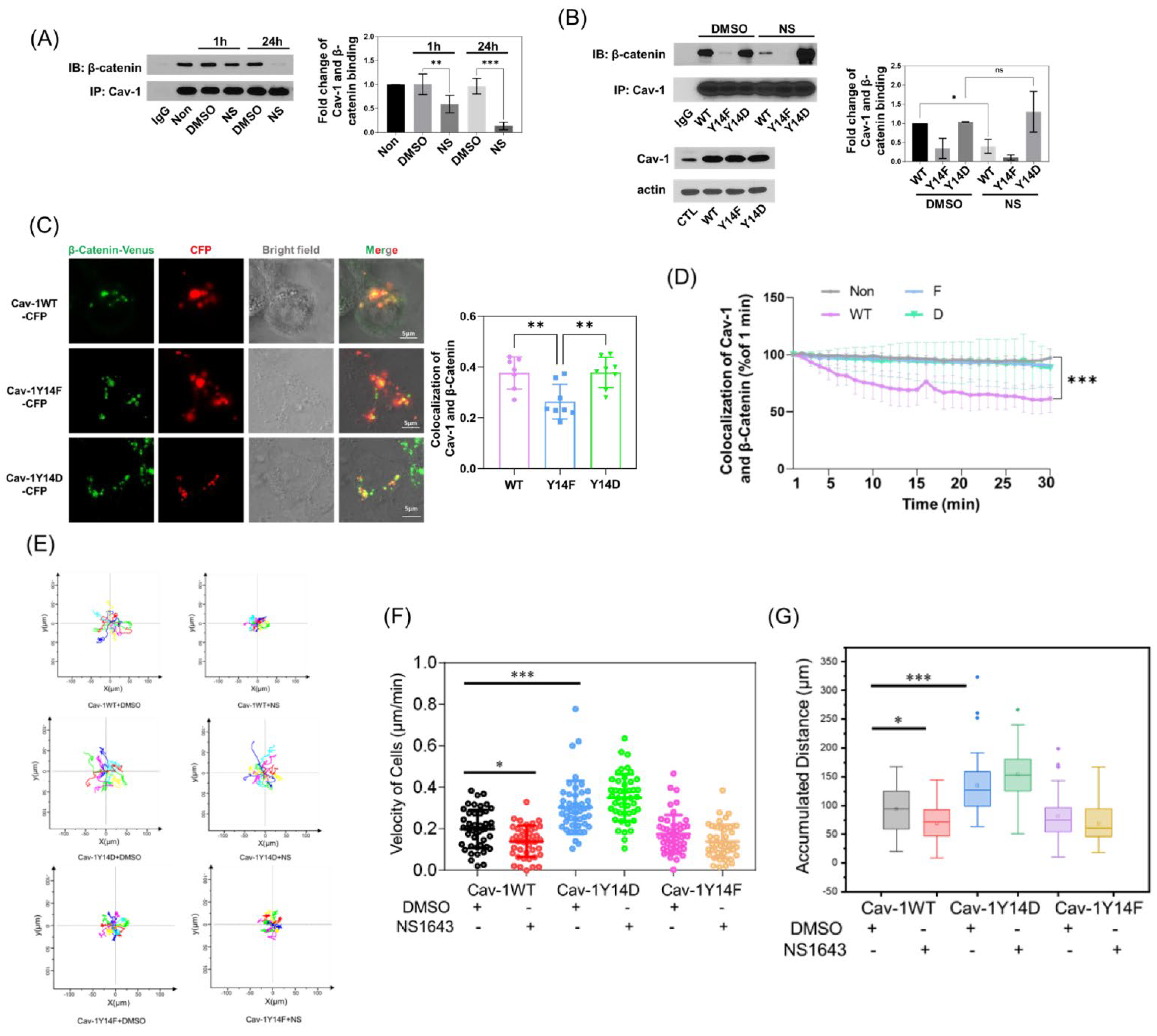
3.4. Kv11.1 Channel Activation Promotes the Accumulation of β-catenin at Cellular Junctions that Are Dependent on Cav-1

3.5. Cav-1 Binds to β-Catenin in a Phosphorylation-Dependent Manner and Prevents Its Junctional Accumulation
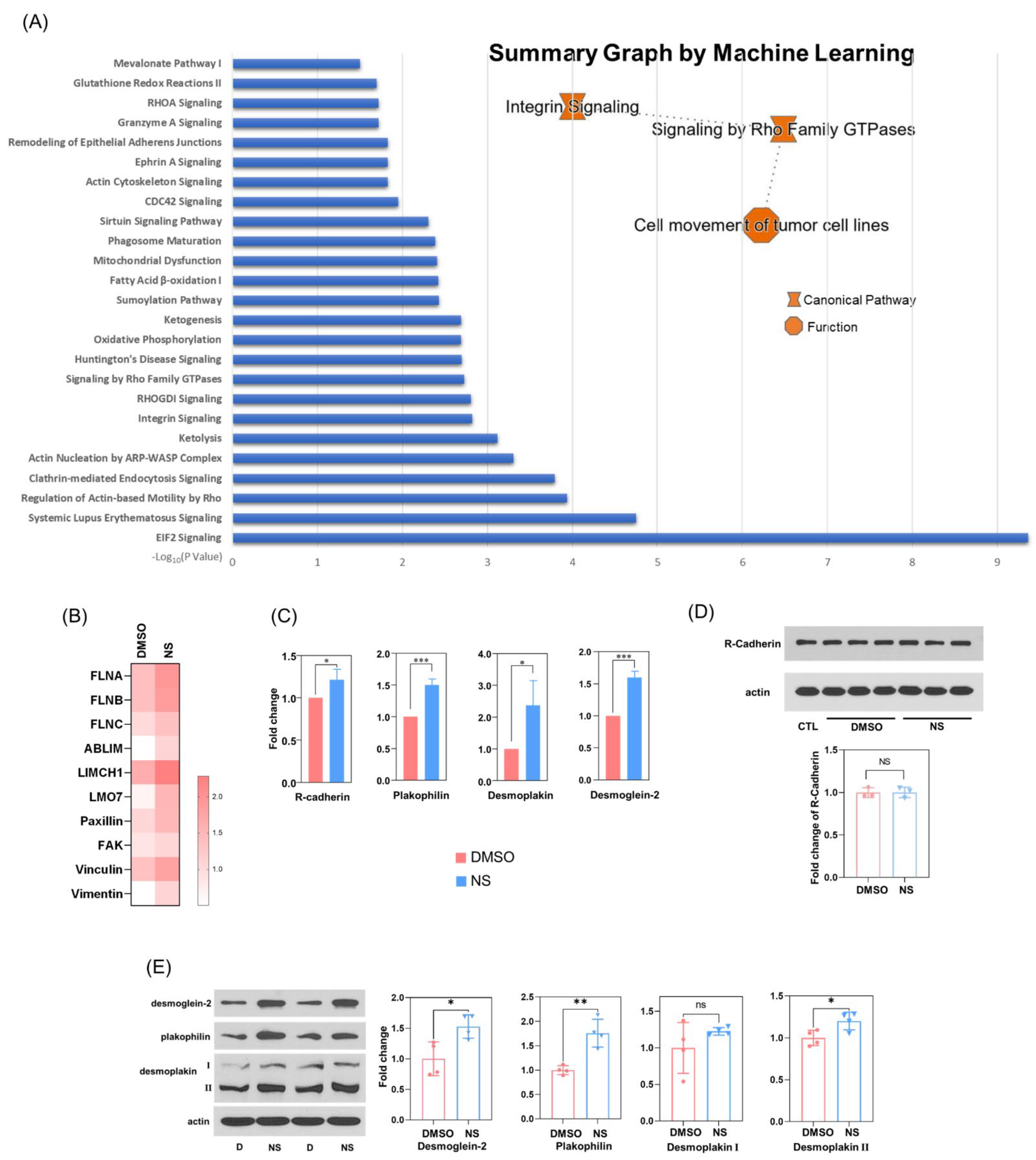
3.6. Kv11.1 Activation Enhances β-catenin Interaction with Adhesion Complexes to Promote contact Inhibition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lastraioli, E.; Iorio, J.; Arcangeli, A. Ion channel expression as promising cancer biomarker. Biochim. Biophys. Acta 2015, 1848, 2685–2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Cheng, G.; Miao, Y.; Qiu, F.; Bai, L.; Gao, Z.; Huang, Y.; Dong, L.; Niu, X.; Wang, X.; et al. Piezo type mechanosensitive ion channel component 1 facilitates gastric cancer omentum metastasis. J. Cell. Mol. Med. 2021, 25, 2238–2253. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Robitaille, M.; Armitage, K.; So, C.L.; Milevskiy, M.J.G.; Northwood, K.; Lim, H.F.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Activation of the Ion Channel TRPV4 Induces Epithelial to Mesenchymal Transition in Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 49417. [Google Scholar] [CrossRef]
- Zhou, Z.H.; Song, J.W.; Li, W.; Liu, X.; Cao, L.; Wan, L.M.; Tan, Y.X.; Ji, S.P.; Liang, Y.M.; Gong, F. The acid-sensing ion channel, ASIC2, promotes invasion and metastasis of colorectal cancer under acidosis by activating the calcineurin/NFAT1 axis. J. Exp. Clin. Cancer Res. 2017, 36, 130. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.A.; Jamaludin, S.Y.N.; Yapa, K.; Chalmers, S.; Wiegmans, A.P.; Lim, H.F.; Milevskiy, M.J.G.; Azimi, I.; Davis, F.M.; Northwood, K.S.; et al. Oncosis and apoptosis induction by activation of an overexpressed ion channel in breast cancer cells. Oncogene 2017, 36, 6490–6500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, V.R.; Perez-Neut, M.; Kaja, S.; Gentile, S. Voltage-gated ion channels in cancer cell proliferation. Cancers 2015, 7, 849–875. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Li, H.; Liang, K.; Du, Y.; Guo, D.; Huang, S. Imatinib has the potential to exert its antileukemia effects by down-regulating hERG1 K+ channels in chronic myelogenous leukemia. Med. Oncol. 2012, 29, 2127–2135. [Google Scholar] [CrossRef]
- Zheng, F.; Li, H.; Du, W.; Huang, S. Role of hERG1 K(+) channels in leukemia cells as a positive regulator in SDF-1a-induced proliferation. Hematology 2011, 16, 177–184. [Google Scholar] [CrossRef]
- Breuer, E.K.; Fukushiro-Lopes, D.; Dalheim, A.; Burnette, M.; Zartman, J.; Kaja, S.; Wells, C.; Campo, L.; Curtis, K.J.; Romero-Moreno, R.; et al. Potassium channel activity controls breast cancer metastasis by affecting beta-catenin signaling. Cell Death Dis. 2019, 10, 180. [Google Scholar] [CrossRef]
- Gentile, S. hERG1 potassium channel in cancer cells: A tool to reprogram immortality. Eur. Biophys. J. 2016, 45, 649–655. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, Y.; Cao, L.; Han, H.; Wang, J.; Yang, B.; Nattel, S.; Wang, Z. HERG K+ channel, a regulator of tumor cell apoptosis and proliferation. Cancer Res. 2002, 62, 4843–4848. [Google Scholar]
- Senyuk, V.; Eskandari, N.; Jiang, Y.; Garcia-Varela, R.; Sundstrom, R.; Leanza, L.; Peruzzo, R.; Burkard, M.; Minshall, R.D.; Gentile, S. Compensatory expression of NRF2-dependent antioxidant genes is required to overcome the lethal effects of Kv11.1 activation in breast cancer cells and PDOs. Redox Biol. 2021, 45, 102030. [Google Scholar]
- Eskandari, N.; Senyuk, V.; Moore, J.; Kalik, Z.; Luan, Q.; Papautsky, I.; Moshiri, A.; Bocchetta, M.; Salami, S.A.; Oryan, S.; et al. Molecular Activation of the Kv11.1 Channel Reprograms EMT in Colon Cancer by Inhibiting TGFbeta Signaling via Activation of Calcineurin. Cancers 2021, 13, 6025. [Google Scholar] [CrossRef]
- Liu, P.; Rudick, M.; Anderson, R.G. Multiple functions of caveolin-1. J. Biol. Chem. 2002, 277, 41295–41298. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, R.; Diaz, J.; Diaz-Valdivia, N.; Martinez, S.; Simon, L.; Contreras, P.; Lobos-Gonzalez, L.; Guerrero, S.; Leyton, L.; Quest, A.F.G. Src-family kinase inhibitors block early steps of caveolin-1-enhanced lung metastasis by melanoma cells. Biochem. Pharmacol. 2020, 177, 113941. [Google Scholar] [CrossRef]
- Martinez-Meza, S.; Diaz, J.; Sandoval-Borquez, A.; Valenzuela-Valderrama, M.; Diaz-Valdivia, N.; Rojas-Celis, V.; Contreras, P.; Huilcaman, R.; Ocaranza, M.P.; Chiong, M.; et al. AT2 Receptor Mediated Activation of the Tyrosine Phosphatase PTP1B Blocks Caveolin-1 Enhanced Migration, Invasion and Metastasis of Cancer Cells. Cancers 2019, 11, 1299. [Google Scholar] [CrossRef] [Green Version]
- Hau, A.M.; Gupta, S.; Leivo, M.Z.; Nakashima, K.; Macias, J.; Zhou, W.; Hodge, A.; Wulfkuhle, J.; Conkright, B.; Bhuvaneshwar, K.; et al. Dynamic Regulation of Caveolin-1 Phosphorylation and Caveolae Formation by Mammalian Target of Rapamycin Complex 2 in Bladder Cancer Cells. Am. J. Pathol. 2019, 189, 1846–1862. [Google Scholar] [CrossRef]
- Yang, J.; Zhu, T.; Zhao, R.; Gao, D.; Cui, Y.; Wang, K.; Guo, Y. Caveolin-1 Inhibits Proliferation, Migration, and Invasion of Human Colorectal Cancer Cells by Suppressing Phosphorylation of Epidermal Growth Factor Receptor. Med. Sci. Monit. 2018, 24, 332–341. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Park, J.W.; Lee, G.W.; Li, L.; Chun, Y.S. Inhibition of neddylation facilitates cell migration through enhanced phosphorylation of caveolin-1 in PC3 and U373MG cells. BMC Cancer 2018, 18, 30. [Google Scholar] [CrossRef] [Green Version]
- Piegeler, T.; Schlapfer, M.; Dull, R.O.; Schwartz, D.E.; Borgeat, A.; Minshall, R.D.; Beck-Schimmer, B. Clinically relevant concentrations of lidocaine and ropivacaine inhibit TNFalpha-induced invasion of lung adenocarcinoma cells in vitro by blocking the activation of Akt and focal adhesion kinase. Br. J. Anaesth 2015, 115, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Oliveira, S.D.; Zimnicka, A.M.; Jiang, Y.; Sharma, T.; Chen, S.; Lazarov, O.; Bonini, M.G.; Haus, J.M.; Minshall, R.D. Reciprocal regulation of eNOS and caveolin-1 functions in endothelial cells. Mol. Biol. Cell 2018, 29, 1190–1202. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Sverdlov, M.S.; Toth, P.T.; Huang, L.S.; Du, G.; Liu, Y.; Natarajan, V.; Minshall, R.D. Phosphatidic Acid Produced by RalA-activated PLD2 Stimulates Caveolae-mediated Endocytosis and Trafficking in Endothelial Cells. J. Biol. Chem. 2016, 291, 20729–20738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, T.H.; Dickson, F.H.; Timmins, L.R.; Nabi, I.R. Tyrosine phosphorylation of tumor cell caveolin-1: Impact on cancer progression. Cancer Metastasis Rev. 2020, 39, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Joshi, B.; Strugnell, S.S.; Goetz, J.G.; Kojic, L.D.; Cox, M.E.; Griffith, O.L.; Chan, S.K.; Jones, S.J.; Leung, S.P.; Masoudi, H.; et al. Phosphorylated caveolin-1 regulates Rho/ROCK-dependent focal adhesion dynamics and tumor cell migration and invasion. Cancer Res. 2008, 68, 8210–8220. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Wen, Z.; Wang, Y.; Liu, Y.J.; Shi, K.; Jiu, Y. Feedback-Driven Mechanisms Between Phosphorylated Caveolin-1 and Contractile Actin Assemblies Instruct Persistent Cell Migration. Front. Cell Dev. Biol. 2021, 9, 665919. [Google Scholar] [CrossRef]
- Rodriguez, D.A.; Tapia, J.C.; Fernandez, J.G.; Torres, V.A.; Munoz, N.; Galleguillos, D.; Leyton, L.; Quest, A.F. Caveolin-1-mediated suppression of cyclooxygenase-2 via a beta-catenin-Tcf/Lef-dependent transcriptional mechanism reduced prostaglandin E2 production and survivin expression. Mol. Biol. Cell 2009, 20, 2297–2310. [Google Scholar] [CrossRef] [Green Version]
- Torres, V.A.; Tapia, J.C.; Rodriguez, D.A.; Lladser, A.; Arredondo, C.; Leyton, L.; Quest, A.F. E-cadherin is required for caveolin-1-mediated down-regulation of the inhibitor of apoptosis protein survivin via reduced beta-catenin-Tcf/Lef-dependent transcription. Mol. Cell. Biol. 2007, 27, 7703–7717. [Google Scholar] [CrossRef] [Green Version]
- Torres, V.A.; Tapia, J.C.; Rodriguez, D.A.; Parraga, M.; Lisboa, P.; Montoya, M.; Leyton, L.; Quest, A.F. Caveolin-1 controls cell proliferation and cell death by suppressing expression of the inhibitor of apoptosis protein survivin. J. Cell Sci. 2006, 119, 1812–1823. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wang, N.; Li, W.; Liu, P.; Chen, Q.; Situ, H.; Zhong, S.; Guo, L.; Lin, Y.; Shen, J.; et al. Caveolin-1 mediates chemoresistance in breast cancer stem cells via beta-catenin/ABCG2 signaling pathway. Carcinogenesis 2014, 35, 2346–2356. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Krantz, S.; Qin, X.; Li, S.; Gunasekara, H.; Kim, Y.M.; Zimnicka, A.; Bae, M.; Ma, K.; Toth, P.T.; et al. Caveolin-1 controls mitochondrial damage and ROS production by regulating fission—Fusion dynamics and mitophagy. Redox Biol. 2022, 52, 102304. [Google Scholar] [CrossRef]
- Zimnicka, A.M.; Husain, Y.S.; Shajahan, A.N.; Sverdlov, M.; Chaga, O.; Chen, Z.; Toth, P.T.; Klomp, J.; Karginov, A.V.; Tiruppathi, C.; et al. Src-dependent phosphorylation of caveolin-1 Tyr-14 promotes swelling and release of caveolae. Mol. Biol. Cell 2016, 27, 2090–2106. [Google Scholar] [CrossRef]
- Grande-Garcia, A.; del Pozo, M.A. Caveolin-1 in cell polarization and directional migration. Eur. J. Cell Biol. 2008, 87, 641–647. [Google Scholar] [CrossRef]
- Glenney, J.R., Jr.; Zokas, L. Novel tyrosine kinase substrates from Rous sarcoma virus-transformed cells are present in the membrane skeleton. J. Cell Biol. 1989, 108, 2401–2408. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure and regulation. Biochem. Biophys. Res. Commun. 2004, 324, 1155–1164. [Google Scholar] [CrossRef]
- Perez-Neut, M.; Shum, A.; Cuevas, B.D.; Miller, R.; Gentile, S. Stimulation of hERG1 channel activity promotes a calcium-dependent degradation of cyclin E2, but not cyclin E1, in breast cancer cells. Oncotarget 2015, 6, 1631–1639. [Google Scholar] [CrossRef] [Green Version]
- Troyanovsky, S. Adherens junction assembly. Subcell. Biochem. 2012, 60, 89–108. [Google Scholar]
- Kowalczyk, A.P.; Nanes, B.A. Adherens junction turnover: Regulating adhesion through cadherin endocytosis, degradation, and recycling. Subcell. Biochem. 2012, 60, 197–222. [Google Scholar]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 2008, 1778, 660–669. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.T.; Stewart, D.B.; Nelson, W.J. Coupling assembly of the E-cadherin/beta-catenin complex to efficient endoplasmic reticulum exit and basal-lateral membrane targeting of E-cadherin in polarized MDCK cells. J. Cell Biol. 1999, 144, 687–699. [Google Scholar] [CrossRef]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef] [Green Version]
- Orford, K.; Crockett, C.; Jensen, J.P.; Weissman, A.M.; Byers, S.W. Serine phosphorylation-regulated ubiquitination and degradation of beta-catenin. J. Biol. Chem. 1997, 272, 24735–24738. [Google Scholar] [CrossRef] [Green Version]
- Vandenberg, J.I.; Perry, M.D.; Perrin, M.J.; Mann, S.A.; Ke, Y.; Hill, A.P. hERG K(+) channels: Structure, function, and clinical significance. Physiol. Rev. 2012, 92, 1393–1478. [Google Scholar] [CrossRef] [Green Version]
- Sanguinetti, M.C.; Tristani-Firouzi, M. hERG potassium channels and cardiac arrhythmia. Nature 2006, 440, 463–469. [Google Scholar] [CrossRef]
- Bentzen, B.H.; Bahrke, S.; Wu, K.; Larsen, A.P.; Odening, K.E.; Franke, G.; Storm vans Gravesande, K.; Biermann, J.; Peng, X.; Koren, G.; et al. Pharmacological activation of Kv11.1 in transgenic long QT-1 rabbits. J. Cardiovasc. Pharmacol. 2011, 57, 223–230. [Google Scholar] [CrossRef]
- Fukushiro-Lopes, D.F.; Hegel, A.D.; Rao, V.; Wyatt, D.; Baker, A.; Breuer, E.K.; Osipo, C.; Zartman, J.J.; Burnette, M.; Kaja, S.; et al. Preclinical study of a Kv11.1 potassium channel activator as antineoplastic approach for breast cancer. Oncotarget 2018, 9, 3321–3337. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.W.; Reimer, C.L.; Oh, P.; Campbell, D.B.; Schnitzer, J.E. Tumor cell growth inhibition by caveolin re-expression in human breast cancer cells. Oncogene 1998, 16, 1391–1397. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Xie, L.; Luo, Y.; Lee, S.Y.; Lawrence, D.S.; Wang, X.B.; Sotgia, F.; Lisanti, M.P.; Zhang, Z.Y. Identification of phosphocaveolin-1 as a novel protein tyrosine phosphatase 1B substrate. Biochemistry 2006, 45, 234–240. [Google Scholar] [CrossRef]
- Devreotes, P.; Horwitz, A.R. Signaling networks that regulate cell migration. Cold Spring Harb. Perspect. Biol. 2015, 7, a005959. [Google Scholar] [CrossRef] [Green Version]
- Farquhar, M.G.; Palade, G.E. Junctional complexes in various epithelia. J. Cell Biol. 1963, 17, 375–412. [Google Scholar] [CrossRef] [Green Version]
- Niessen, C.M.; Gottardi, C.J. Molecular components of the adherens junction. Biochim. Biophys. Acta 2008, 1778, 562–571. [Google Scholar] [CrossRef] [Green Version]
- Eslami Amirabadi, H.; Tuerlings, M.; Hollestelle, A.; SahebAli, S.; Luttge, R.; van Donkelaar, C.C.; Martens, J.W.M.; den Toonder, J.M.J. Characterizing the invasion of different breast cancer cell lines with distinct E-cadherin status in 3D using a microfluidic system. Biomed. Microdevices 2019, 21, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarrio, D.; Palacios, J.; Hergueta-Redondo, M.; Gomez-Lopez, G.; Cano, A.; Moreno-Bueno, G. Functional characterization of E- and P-cadherin in invasive breast cancer cells. BMC Cancer 2009, 9, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieman, M.T.; Prudoff, R.S.; Johnson, K.R.; Wheelock, M.J. N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J. Cell Biol. 1999, 147, 631–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Luo, Q.C.; Chen, J.B.; Lin, L.E.; Luo, M.X.; Ren, H.Y.; Chen, P.Q.; Shi, L.G. Efficient isolation and proteomic analysis of cell plasma membrane proteins in gastric cancer reveal a novel differentiation and progression related cell surface marker, R-cadherin. Tumour Biol. 2016, 37, 11775–11787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, L.; Hazan, R.; Roy, P. Profilin-1 overexpression restores adherens junctions in MDA-MB-231 breast cancer cells in R-cadherin-dependent manner. Cell Motil. Cytoskeleton. 2009, 66, 1048–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miotto, E.; Sabbioni, S.; Veronese, A.; Calin, G.A.; Gullini, S.; Liboni, A.; Gramantieri, L.; Bolondi, L.; Ferrazzi, E.; Gafa, R.; et al. Frequent aberrant methylation of the CDH4 gene promoter in human colorectal and gastric cancer. Cancer Res. 2004, 64, 8156–8159. [Google Scholar] [CrossRef] [Green Version]
- Slater, S.C.; Koutsouki, E.; Jackson, C.L.; Bush, R.C.; Angelini, G.D.; Newby, A.C.; George, S.J. R-cadherin:beta-catenin complex and its association with vascular smooth muscle cell proliferation. Arterioscler Thromb. Vasc. Biol. 2004, 24, 1204–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlichenko, L.; Weller, S.G.; Cao, H.; Krueger, E.W.; Awoniyi, M.; Beznoussenko, G.; Buccione, R.; McNiven, M.A. Caveolae mediate growth factor-induced disassembly of adherens junctions to support tumor cell dissociation. Mol. Biol. Cell 2009, 20, 4140–4152. [Google Scholar] [CrossRef] [Green Version]
- Nomura, R.; Fujimoto, T. Tyrosine-phosphorylated caveolin-1: Immunolocalization and molecular characterization. Mol. Biol. Cell 1999, 10, 975–986. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/beta-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target Ther. 2022, 7, 3. [Google Scholar] [CrossRef]
- Nagano, M.; Hoshino, D.; Koshikawa, N.; Akizawa, T.; Seiki, M. Turnover of focal adhesions and cancer cell migration. Int. J. Cell Biol. 2012, 2012, 310616. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Saxena, S.; Liu, Y.; Joshi, B.; Wong, T.H.; Shankar, J.; Foster, L.J.; Bernatchez, P.; Nabi, I.R. The phospho-caveolin-1 scaffolding domain dampens force fluctuations in focal adhesions and promotes cancer cell migration. Mol. Biol. Cell 2017, 28, 2190–2201. [Google Scholar] [CrossRef]
- Borradori, L.; Sonnenberg, A. Structure and function of hemidesmosomes: More than simple adhesion complexes. J. Investig. Dermatol. 1999, 112, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Brennan, D.; Peltonen, S.; Dowling, A.; Medhat, W.; Green, K.J.; Wahl, J.K., 3rd; Del Galdo, F.; Mahoney, M.G. A role for caveolin-1 in desmoglein binding and desmosome dynamics. Oncogene 2012, 31, 1636–1648. [Google Scholar] [CrossRef] [Green Version]
- Resnik, N.; Sepcic, K.; Plemenitas, A.; Windoffer, R.; Leube, R.; Veranic, P. Desmosome assembly and cell-cell adhesion are membrane raft-dependent processes. J. Biol. Chem. 2011, 286, 1499–1507. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Y.; Senyuk, V.; Ma, K.; Chen, H.; Qin, X.; Li, S.; Liu, Y.; Gentile, S.; Minshall, R.D. Pharmacological Activation of Potassium Channel Kv11.1 with NS1643 Attenuates Triple Negative Breast Cancer Cell Migration by Promoting the Dephosphorylation of Caveolin-1. Cells 2022, 11, 2461. https://doi.org/10.3390/cells11152461
Jiang Y, Senyuk V, Ma K, Chen H, Qin X, Li S, Liu Y, Gentile S, Minshall RD. Pharmacological Activation of Potassium Channel Kv11.1 with NS1643 Attenuates Triple Negative Breast Cancer Cell Migration by Promoting the Dephosphorylation of Caveolin-1. Cells. 2022; 11(15):2461. https://doi.org/10.3390/cells11152461
Chicago/Turabian StyleJiang, Ying, Vitalyi Senyuk, Ke Ma, Hui Chen, Xiang Qin, Shun Li, Yiyao Liu, Saverio Gentile, and Richard D. Minshall. 2022. "Pharmacological Activation of Potassium Channel Kv11.1 with NS1643 Attenuates Triple Negative Breast Cancer Cell Migration by Promoting the Dephosphorylation of Caveolin-1" Cells 11, no. 15: 2461. https://doi.org/10.3390/cells11152461