
Immunoregulation via Cell Density and Quorum Sensing-like Mechanisms: An Underexplored Emerging Field with Potential Translational Implications


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Quorum Sensing-like Mechanisms of T Cells and Regulation of the Adaptive Immune System
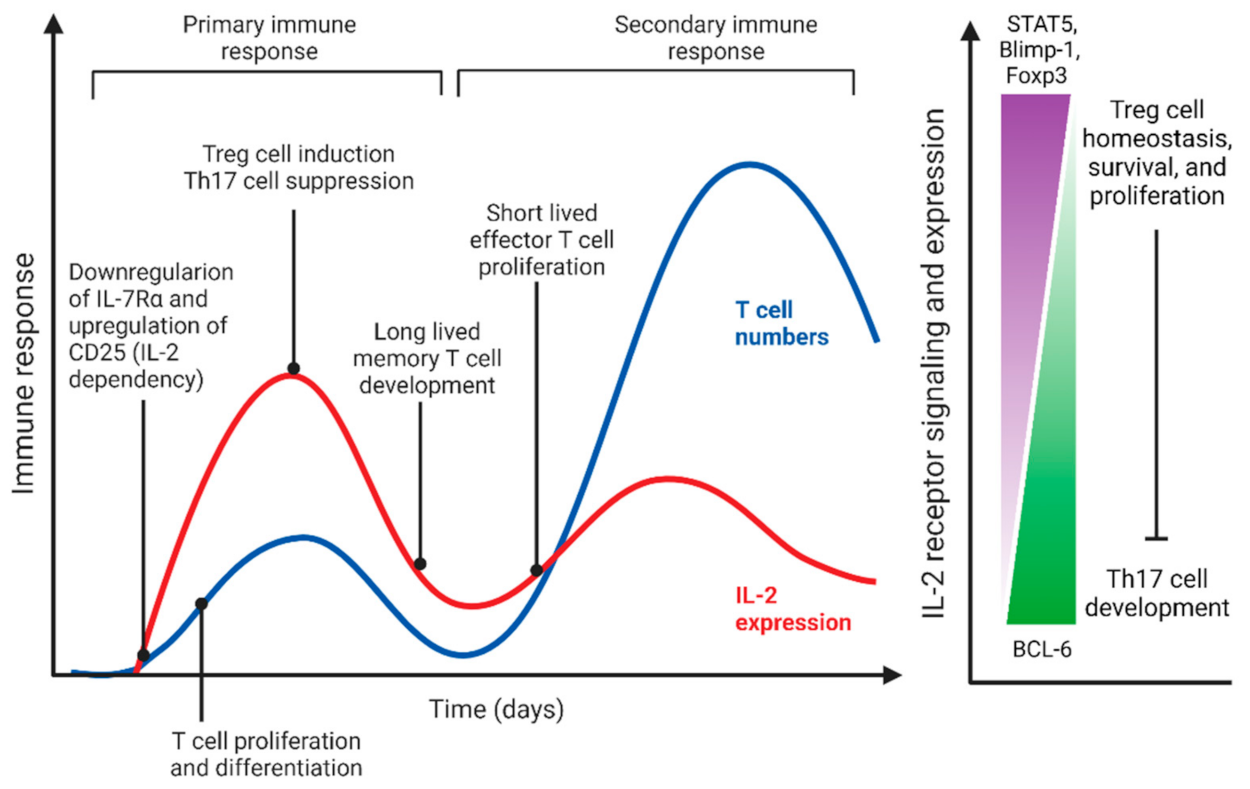
2.1. Common-Gamma Chain (γc) Receptor and Cytokines as QS-like Mechanism for Coordinating T Lymphocyte Responses and Homeostasis
2.1.1. IL-2 Mediated QS-like Behavior of Effector and Regulatory T Cells
2.1.2. QS-like Behavior Mediated by IL-7 and IL-15 in Homeostatic Regulation of Naïve and Memory T Cells
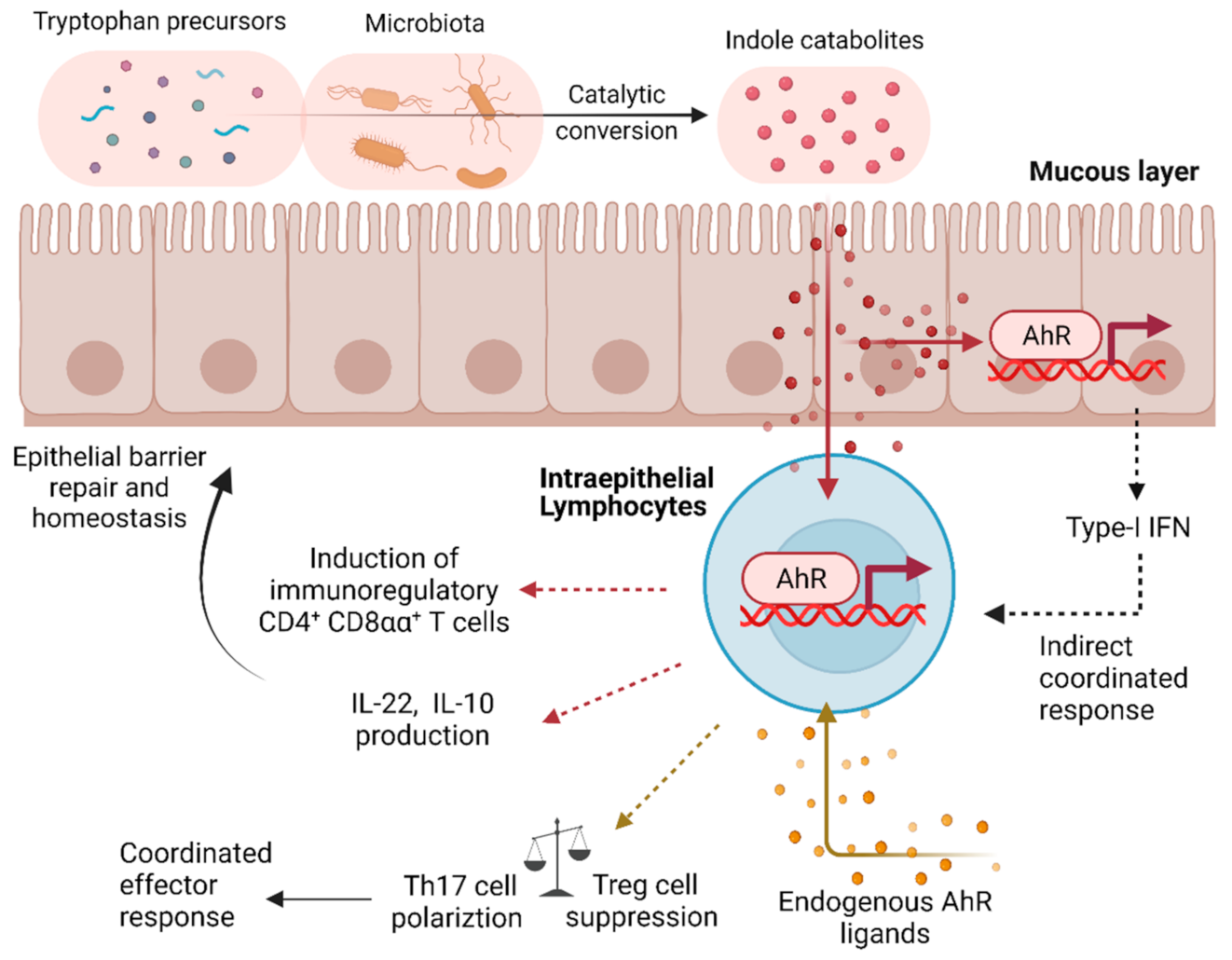
2.2. The Aryl Hydrocarbon Receptor (AhR) as a QS-like Regulator in Immune Cells
2.3. Bystander Activation of T Cells: QS-like Acute Phase Response of Specific T Cell Subsets
3. QS-like Regulation of Myeloid Cells and the Innate Immune System
3.1. QS as a Myeloid Lineage Modulator: Population Density of Tissue-Resident Macrophages Contributes to Spatiotemporal Regulation
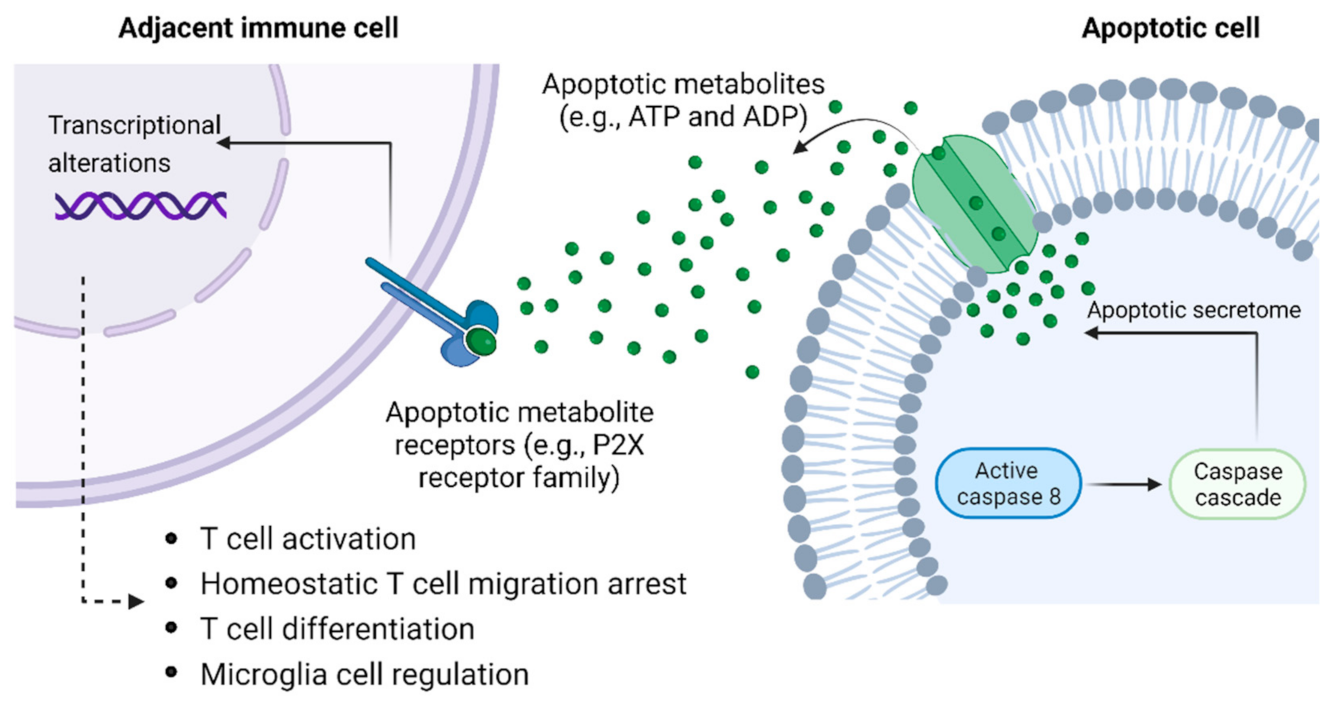
3.2. Apoptotic Metabolite Release as a Putative QS-like Mechanism Governing Macrophage Density
3.3. QS-like Density-Dependent Polarization of Macrophages
3.4. Intersection of Microbial QS with Regulation of Myeloid Cells and Tissue Macrophages
4. Clinical Implications
4.1. Exploring QS-like Regulation of T Cells for Inflammatory Diseases and GvHD
4.2. QS-like Modulation of T Cells for Cancer Immunotherapy
4.3. Implications of QS for Treatment of Infectious or Inflammatory Diseases via Regulation of Macrophage and Myeloid Cells
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ng, W.L.; Bassler, B.L. Bacterial quorum-sensing network architectures. Annu. Rev. Genet. 2009, 43, 197–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, H.H.; Gurich, N.; Gonzalez, J.E. Regulation of motility by the ExpR/Sin quorum-sensing system in Sinorhizobium meliloti. J. Bacteriol. 2008, 190, 861–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutherford, S.T.; Bassler, B.L. Bacterial quorum sensing: Its role in virulence and possibilities for its control. Cold Spring Harb Perspect. Med. 2012, 2, a012427. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Bassler, B.L. Bacterial quorum sensing in complex and dynamically changing environments. Nat. Rev. Microbiol. 2019, 17, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Padder, S.A.; Prasad, R.; Shah, A.H. Quorum sensing: A less known mode of communication among fungi. Microbiol. Res. 2018, 210, 51–58. [Google Scholar] [CrossRef]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Guilliams, M.; Hasko, G. Quorum sensing in the immune system. Nat. Rev. Immunol. 2018, 18, 537–538. [Google Scholar] [CrossRef]
- Brodin, P.; Davis, M.M. Human immune system variation. Nat. Rev. Immunol. 2017, 17, 21–29. [Google Scholar] [CrossRef]
- Parkin, J.; Cohen, B. An overview of the immune system. Lancet 2001, 357, 1777–1789. [Google Scholar] [CrossRef]
- Netea, M.G.; Dominguez-Andres, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef]
- Boehm, T. Design principles of adaptive immune systems. Nat. Rev. Immunol. 2011, 11, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Liston, A.; Humblet-Baron, S.; Duffy, D.; Goris, A. Human immune diversity: From evolution to modernity. Nat. Immunol. 2021, 22, 1479–1489. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, G.; Rudensky, A.Y. Interactions between innate and adaptive lymphocytes. Nat. Rev. Immunol. 2014, 14, 631–639. [Google Scholar] [CrossRef]
- Vivier, E.; Malissen, B. Innate and adaptive immunity: Specificities and signaling hierarchies revisited. Nat. Immunol. 2005, 6, 17–21. [Google Scholar] [CrossRef]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Guilliams, M.; Hasko, G. Rethinking Communication in the Immune System: The Quorum Sensing Concept. Trends Immunol. 2019, 40, 88–97. [Google Scholar] [CrossRef]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Raphael, I.; Joern, R.R.; Forsthuber, T.G. Memory CD4(+) T Cells in Immunity and Autoimmune Diseases. Cells 2020, 9, 531. [Google Scholar] [CrossRef] [Green Version]
- Negron, A.; Robinson, R.R.; Stuve, O.; Forsthuber, T.G. The role of B cells in multiple sclerosis: Current and future therapies. Cell Immunol. 2019, 339, 10–23. [Google Scholar] [CrossRef] [PubMed]
- van Bladel, D.A.G.; van den Brand, M.; Rijntjes, J.; Pamidimarri Naga, S.; Haacke, D.; Luijks, J.; Hebeda, K.M.; van Krieken, J.; Groenen, P.; Scheijen, B. Clonality assessment and detection of clonal diversity in classic Hodgkin lymphoma by next-generation sequencing of immunoglobulin gene rearrangements. Mod Pathol. 2021, 35, 757–766. [Google Scholar] [CrossRef]
- Gascoigne, N.R.; Rybakin, V.; Acuto, O.; Brzostek, J. TCR Signal Strength and T Cell Development. Annu. Rev. Cell Dev. Biol. 2016, 32, 327–348. [Google Scholar] [CrossRef] [Green Version]
- Niiro, H.; Clark, E.A. Regulation of B-cell fate by antigen-receptor signals. Nat. Rev. Immunol. 2002, 2, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Adams, N.M.; Grassmann, S.; Sun, J.C. Clonal expansion of innate and adaptive lymphocytes. Nat. Rev. Immunol. 2020, 20, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Hesse, M.D.; Karulin, A.Y.; Boehm, B.O.; Lehmann, P.V.; Tary-Lehmann, M. A T cell clone’s avidity is a function of its activation state. J. Immunol. 2001, 167, 1353–1361. [Google Scholar] [CrossRef] [Green Version]
- Chiffelle, J.; Genolet, R.; Perez, M.A.; Coukos, G.; Zoete, V.; Harari, A. T-cell repertoire analysis and metrics of diversity and clonality. Curr. Opin. Biotechnol. 2020, 65, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Szabo, P.A.; Levitin, H.M.; Miron, M.; Snyder, M.E.; Senda, T.; Yuan, J.; Cheng, Y.L.; Bush, E.C.; Dogra, P.; Thapa, P.; et al. Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat. Commun. 2019, 10, 4706. [Google Scholar] [CrossRef]
- Obar, J.J.; Khanna, K.M.; Lefrancois, L. Endogenous naive CD8+ T cell precursor frequency regulates primary and memory responses to infection. Immunity 2008, 28, 859–869. [Google Scholar] [CrossRef] [Green Version]
- Badovinac, V.P.; Haring, J.S.; Harty, J.T. Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8(+) T cell response to infection. Immunity 2007, 26, 827–841. [Google Scholar] [CrossRef] [Green Version]
- Marzo, A.L.; Klonowski, K.D.; Le Bon, A.; Borrow, P.; Tough, D.F.; Lefrancois, L. Initial T cell frequency dictates memory CD8+ T cell lineage commitment. Nat. Immunol. 2005, 6, 793–799. [Google Scholar] [CrossRef]
- Polonsky, M.; Rimer, J.; Kern-Perets, A.; Zaretsky, I.; Miller, S.; Bornstein, C.; David, E.; Kopelman, N.M.; Stelzer, G.; Porat, Z.; et al. Induction of CD4 T cell memory by local cellular collectivity. Science 2018, 360, eaaj1853. [Google Scholar] [CrossRef] [Green Version]
- Altan-Bonnet, G.; Mukherjee, R. Cytokine-mediated communication: A quantitative appraisal of immune complexity. Nat. Rev. Immunol. 2019, 19, 205–217. [Google Scholar] [CrossRef]
- Schluns, K.S.; Lefrancois, L. Cytokine control of memory T-cell development and survival. Nat. Rev. Immunol. 2003, 3, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Mackall, C.L.; Hakim, F.T.; Gress, R.E. Restoration of T-cell homeostasis after T-cell depletion. Semin. Immunol. 1997, 9, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.B.; Sparshott, S.M. The peripheral T-cell pool: Regulation by non-antigen induced proliferation? Semin. Immunol. 1997, 9, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Sprent, J.; Tough, D.F. Lymphocyte life-span and memory. Science 1994, 265, 1395–1400. [Google Scholar] [CrossRef]
- Surh, C.D.; Sprent, J. Homeostasis of naive and memory T cells. Immunity 2008, 29, 848–862. [Google Scholar] [CrossRef] [Green Version]
- Jameson, S.C. Maintaining the norm: T-cell homeostasis. Nat. Rev. Immunol. 2002, 2, 547–556. [Google Scholar] [CrossRef]
- Masse, G.X.; Corcuff, E.; Decaluwe, H.; Bommhardt, U.; Lantz, O.; Buer, J.; Di Santo, J.P. gamma(c) cytokines provide multiple homeostatic signals to naive CD4(+) T cells. Eur. J. Immunol. 2007, 37, 2606–2616. [Google Scholar] [CrossRef]
- Kinter, A.L.; Godbout, E.J.; McNally, J.P.; Sereti, I.; Roby, G.A.; O’Shea, M.A.; Fauci, A.S. The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J. Immunol. 2008, 181, 6738–6746. [Google Scholar] [CrossRef]
- Overwijk, W.W.; Schluns, K.S. Functions of gammaC cytokines in immune homeostasis: Current and potential clinical applications. Clin. Immunol. 2009, 132, 153–165. [Google Scholar] [CrossRef] [Green Version]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar] [CrossRef]
- Ross, S.H.; Cantrell, D.A. Signaling and Function of Interleukin-2 in T Lymphocytes. Annu. Rev. Immunol. 2018, 36, 411–433. [Google Scholar] [CrossRef] [PubMed]
- Rochman, Y.; Spolski, R.; Leonard, W.J. New insights into the regulation of T cells by gamma(c) family cytokines. Nat. Rev. Immunol. 2009, 9, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Di Santo, J.P.; Aifantis, I.; Rosmaraki, E.; Garcia, C.; Feinberg, J.; Fehling, H.J.; Fischer, A.; von Boehmer, H.; Rocha, B. The common cytokine receptor gamma chain and the pre-T cell receptor provide independent but critically overlapping signals in early alpha/beta T cell development. J. Exp. Med. 1999, 189, 563–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Santo, J.P.; Kuhn, R.; Muller, W. Common cytokine receptor gamma chain (gamma c)-dependent cytokines: Understanding in vivo functions by gene targeting. Immunol. Rev. 1995, 148, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Yamane, H.; Paul, W.E. Cytokines of the gamma(c) family control CD4+ T cell differentiation and function. Nat. Immunol. 2012, 13, 1037–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, H.H.; Kovanen, P.E.; Pise-Masison, C.A.; Berg, M.; Radovich, M.F.; Brady, J.N.; Leonard, W.J. IL-2 negatively regulates IL-7 receptor alpha chain expression in activated T lymphocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 13759–13764. [Google Scholar] [CrossRef] [Green Version]
- Kovanen, P.E.; Leonard, W.J. Cytokines and immunodeficiency diseases: Critical roles of the gamma(c)-dependent cytokines interleukins 2, 4, 7, 9, 15, and 21, and their signaling pathways. Immunol. Rev. 2004, 202, 67–83. [Google Scholar] [CrossRef]
- Lucey, D.R.; Clerici, M.; Shearer, G.M. Type 1 and type 2 cytokine dysregulation in human infectious, neoplastic, and inflammatory diseases. Clin. Microbiol. Rev. 1996, 9, 532–562. [Google Scholar] [CrossRef]
- Benczik, M.; Gaffen, S.L. The interleukin (IL)-2 family cytokines: Survival and proliferation signaling pathways in T lymphocytes. Immunol. Investig. 2004, 33, 109–142. [Google Scholar] [CrossRef]
- Ye, C.; Brand, D.; Zheng, S.G. Targeting IL-2: An unexpected effect in treating immunological diseases. Signal Transduct. Target 2018, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Piccirillo, C.A.; Thornton, A.M. Cornerstone of peripheral tolerance: Naturally occurring CD4+CD25+ regulatory T cells. Trends Immunol. 2004, 25, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Shimojima, Y.; Shirai, T.; Li, Y.; Ju, J.; Yang, Z.; Tian, L.; Goronzy, J.J.; Weyand, C.M. NADPH oxidase deficiency underlies dysfunction of aged CD8+ Tregs. J. Clin. Investig. 2016, 126, 1953–1967. [Google Scholar] [CrossRef] [PubMed]
- Setoguchi, R.; Hori, S.; Takahashi, T.; Sakaguchi, S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J. Exp. Med. 2005, 201, 723–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, A.L.; Yu, A.; Adeegbe, D.; Malek, T.R. Essential role for interleukin-2 for CD4(+)CD25(+) T regulatory cell development during the neonatal period. J. Exp. Med. 2005, 201, 769–777. [Google Scholar] [CrossRef]
- Fontenot, J.D.; Rudensky, A.Y. A well adapted regulatory contrivance: Regulatory T cell development and the forkhead family transcription factor Foxp3. Nat. Immunol. 2005, 6, 331–337. [Google Scholar] [CrossRef]
- Malek, T.R. The biology of interleukin-2. Annu. Rev. Immunol. 2008, 26, 453–479. [Google Scholar] [CrossRef]
- Almeida, A.R.; Zaragoza, B.; Freitas, A.A. Indexation as a novel mechanism of lymphocyte homeostasis: The number of CD4+CD25+ regulatory T cells is indexed to the number of IL-2-producing cells. J. Immunol. 2006, 177, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Busse, D.; de la Rosa, M.; Hobiger, K.; Thurley, K.; Flossdorf, M.; Scheffold, A.; Hofer, T. Competing feedback loops shape IL-2 signaling between helper and regulatory T lymphocytes in cellular microenvironments. Proc. Natl. Acad. Sci. USA 2010, 107, 3058–3063. [Google Scholar] [CrossRef] [Green Version]
- Sadlack, B.; Merz, H.; Schorle, H.; Schimpl, A.; Feller, A.C.; Horak, I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell 1993, 75, 253–261. [Google Scholar] [CrossRef]
- Sadlack, B.; Löhler, J.; Schorle, H.; Klebb, G.; Haber, H.; Sickel, E.; Noelle, R.J.; Horak, I.; Horak, I. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur. J. Immunol. 1995, 25, 3053–3059. [Google Scholar] [CrossRef]
- Suzuki, H.; Ündig, T.; Furlonger, C.; Wakeham, A.; Timms, E.; Matsuyama, T.; Schmits, R.; Simard, J.J.; Ohashi, P.S.; Griesser, H.; et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science 1995, 268, 1472–1476. [Google Scholar] [CrossRef] [PubMed]
- Willerford, D.M.; Chen, J.; Ferry, J.A.; Davidson, L.; Ma, A.; Alt, F.W. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity 1995, 3, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Caudy, A.A.; Reddy, S.T.; Chatila, T.; Atkinson, J.P.; Verbsky, J.W. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J. Allergy Clin. Immunol. 2007, 119, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef] [PubMed]
- Josefowicz, S.Z.; Niec, R.E.; Kim, H.Y.; Treuting, P.; Chinen, T.; Zheng, Y.; Umetsu, D.T.; Rudensky, A.Y. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 2012, 482, 395–399. [Google Scholar] [CrossRef]
- Haribhai, D.; Williams, J.B.; Jia, S.; Nickerson, D.; Schmitt, E.G.; Edwards, B.; Ziegelbauer, J.; Yassai, M.; Li, S.H.; Relland, L.M.; et al. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity 2011, 35, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Haribhai, D.; Lin, W.; Edwards, B.; Ziegelbauer, J.; Salzman, N.H.; Carlson, M.R.; Li, S.H.; Simpson, P.M.; Chatila, T.A.; Williams, C.B. A central role for induced regulatory T cells in tolerance induction in experimental colitis. J. Immunol. 2009, 182, 3461–3468. [Google Scholar] [CrossRef] [Green Version]
- Butler, T.C.; Kardar, M.; Chakraborty, A.K. Quorum sensing allows T cells to discriminate between self and nonself. Proc. Natl. Acad. Sci. USA 2013, 110, 11833–11838. [Google Scholar] [CrossRef] [Green Version]
- Feinerman, O.; Jentsch, G.; Tkach, K.E.; Coward, J.W.; Hathorn, M.M.; Sneddon, M.W.; Emonet, T.; Smith, K.A.; Altan-Bonnet, G. Single-cell quantification of IL-2 response by effector and regulatory T cells reveals critical plasticity in immune response. Mol. Syst. Biol. 2010, 6, 437. [Google Scholar] [CrossRef]
- Heufler, C.; Topar, G.; Grasseger, A.; Stanzl, U.; Koch, F.; Romani, N.; Namen, A.E.; Schuler, G. Interleukin 7 is produced by murine and human keratinocytes. J. Exp. Med. 1993, 178, 1109–1114. [Google Scholar] [CrossRef]
- Fry, T.J.; Mackall, C.L. The many faces of IL-7: From lymphopoiesis to peripheral T cell maintenance. J. Immunol. 2005, 174, 6571–6576. [Google Scholar] [CrossRef] [PubMed]
- Link, A.; Vogt, T.K.; Favre, S.; Britschgi, M.R.; Acha-Orbea, H.; Hinz, B.; Cyster, J.G.; Luther, S.A. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat. Immunol. 2007, 8, 1255–1265. [Google Scholar] [CrossRef]
- Hofmeister, R.; Khaled, A.R.; Benbernou, N.; Rajnavolgyi, E.; Muegge, K.; Durum, S.K. Interleukin-7: Physiological roles and mechanisms of action. Cytokine Growth Factor Rev. 1999, 10, 41–60. [Google Scholar] [CrossRef]
- Hataye, J.; Moon, J.; Khoruts, A.; Reilly, C.; Jenkins, M.K. Naive and memory CD4+ T cell survival controlled by clonal abundance. Science 2006, 312, 114–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, A.; Koka, R.; Burkett, P. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu. Rev. Immunol. 2006, 24, 657–679. [Google Scholar] [CrossRef] [PubMed]
- Schluns, K.S.; Stoklasek, T.; Lefrancois, L. The roles of interleukin-15 receptor alpha: Trans-presentation, receptor component, or both? Int. J. Biochem. Cell. Biol. 2005, 37, 1567–1571. [Google Scholar] [CrossRef]
- Anderson, D.M.; Kumaki, S.; Ahdieh, M.; Bertles, J.; Tometsko, M.; Loomis, A.; Giri, J.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; et al. Functional characterization of the human interleukin-15 receptor alpha chain and close linkage of IL15RA and IL2RA genes. J. Biol. Chem. 1995, 270, 29862–29869. [Google Scholar] [CrossRef] [Green Version]
- Colpitts, S.L.; Stonier, S.W.; Stoklasek, T.A.; Root, S.H.; Aguila, H.L.; Schluns, K.S.; Lefrancois, L. Transcriptional regulation of IL-15 expression during hematopoiesis. J. Immunol. 2013, 191, 3017–3024. [Google Scholar] [CrossRef] [Green Version]
- Seddon, B.; Tomlinson, P.; Zamoyska, R. Interleukin 7 and T cell receptor signals regulate homeostasis of CD4 memory cells. Nat. Immunol. 2003, 4, 680–686. [Google Scholar] [CrossRef]
- Kondrack, R.M.; Harbertson, J.; Tan, J.T.; McBreen, M.E.; Surh, C.D.; Bradley, L.M. Interleukin 7 regulates the survival and generation of memory CD4 cells. J. Exp. Med. 2003, 198, 1797–1806. [Google Scholar] [CrossRef]
- Purton, J.F.; Tan, J.T.; Rubinstein, M.P.; Kim, D.M.; Sprent, J.; Surh, C.D. Antiviral CD4+ memory T cells are IL-15 dependent. J. Exp. Med. 2007, 204, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.M.; MacLeod, M.; Marsden, V.S.; Kappler, J.W.; Marrack, P. Not all CD4+ memory T cells are long lived. Immunol. Rev. 2006, 211, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Lenz, D.C.; Kurz, S.K.; Lemmens, E.; Schoenberger, S.P.; Sprent, J.; Oldstone, M.B.; Homann, D. IL-7 regulates basal homeostatic proliferation of antiviral CD4+T cell memory. Proc. Natl. Acad. Sci. USA 2004, 101, 9357–9362. [Google Scholar] [CrossRef] [Green Version]
- Swain, S.L.; Hu, H.; Huston, G. Class II-independent generation of CD4 memory T cells from effectors. Science 1999, 286, 1381–1383. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.L. CD4 T-cell memory can persist in the absence of class II. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2000, 355, 407–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murali-Krishna, K.; Lau, L.L.; Sambhara, S.; Lemonnier, F.; Altman, J.; Ahmed, R. Persistence of memory CD8 T cells in MHC class I-deficient mice. Science 1999, 286, 1377–1381. [Google Scholar] [CrossRef]
- Li, J.; Huston, G.; Swain, S.L. IL-7 promotes the transition of CD4 effectors to persistent memory cells. J. Exp. Med. 2003, 198, 1807–1815. [Google Scholar] [CrossRef]
- Hahn, M.E.; Karchner, S.; Shapiro, M.A.; Perera, S.A. Molecular evolution of two vertebrate aryl hydrocarbon (dioxin) receptors (AHR1 and AHR2) and the PAS family. J. Biochem. 2003. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, Y.; Ohsawa, S.; Mimura, J.; Ema, M.; Takasaki, C.; Sogawa, K.; Fujii-Kuriyama, Y. Heterodimers of bHLH-PAS protein fragments derived from AhR, AhRR, and Arnt prepared by co-expression in Escherichia coli: Characterization of their DNA binding activity and preparation of a DNA complex. J. Biochem. 2003, 134, 83–90. [Google Scholar] [CrossRef]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Entschladen, F.; Lindquist, J.; Serfling, E.; Thiel, G.; Kieser, A.; Giehl, K.; Ehrhardt, C.; Feller, S.M.; Ullrich, O.; Schaper, F.; et al. Signal transduction-receptors, mediators, and genes. Cell Commun. Signal 2009, 2, mr3. [Google Scholar]
- Shinde, R.; McGaha, T.L. The Aryl Hydrocarbon Receptor: Connecting Immunity to the Microenvironment. Trends Immunol. 2018, 39, 1005–1020. [Google Scholar] [CrossRef] [PubMed]
- Minton, K. Intercepting bacterial communications. Nat. Rev. Immunol. 2020, 20, 138–139. [Google Scholar] [CrossRef]
- Moura-Alves, P.; Puyskens, A.; Stinn, A.; Klemm, M.; Guhlich-Bornhof, U.; Dorhoi, A.; Furkert, J.; Kreuchwig, A.; Protze, J.; Lozza, L.; et al. Host monitoring of quorum sensing during Pseudomonas aeruginosa infection. Science 2019, 366. [Google Scholar] [CrossRef]
- Perdew, G.H.; Babbs, C.F. Production of Ah receptor ligands in rat fecal suspensions containing tryptophan or indole-3-carbinol. Nutr. Cancer 1991, 16, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Wattenberg Lw Fau - Loub, W.D.; Loub, W.D. Inhibition of polycyclic aromatic hydrocarbon-induced neoplasia by naturally occurring indoles. Cancer Res. 1978, 38, 1410–1413. [Google Scholar]
- Cheng, Y.; Jin, U.H.; Allred, C.D.; Jayaraman, A.; Chapkin, R.S.; Safe, S. Aryl Hydrocarbon Receptor Activity of Tryptophan Metabolites in Young Adult Mouse Colonocytes. Drug Metab. Dispos. 2015, 43, 1536–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, T.D.; Murray, I.A.; Bisson, W.H.; Lahoti, T.S.; Gowda, K.; Amin, S.G.; Patterson, A.D.; Perdew, G.H. Adaptation of the human aryl hydrocarbon receptor to sense microbiota-derived indoles. Sci. Rep. 2015, 5, 12689. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Barragan, L.; Chai, J.N.; Tianero, M.D.; Di Luccia, B.; Ahern, P.P.; Merriman, J.; Cortez, V.S.; Caparon, M.G.; Donia, M.S.; Gilfillan, S.; et al. Lactobacillus reuteri induces gut intraepithelial CD4(+)CD8alphaalpha(+) T cells. Science 2017, 357, 806–810. [Google Scholar] [CrossRef] [Green Version]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef] [Green Version]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swimm, A.; Giver, C.R.; DeFilipp, Z.; Rangaraju, S.; Sharma, A.A.-O.; Ulezko Antonova, A.; Sonowal, R.; Capaldo, C.; Powell, D.; Qayed, M.; et al. Indoles derived from intestinal microbiota act via type I interferon signaling to limit graft-versus-host disease. Blood 2018, 132, 2506–2519. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Guo, X.; Chen, Z.M.; He, L.; Sonnenberg, G.F.; Artis, D.; Fu, Y.X.; Zhou, L. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity 2013, 39, 386–399. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Heller, J.J.; Guo, X.; Chen, Z.M.; Fish, K.; Fu, Y.X.; Zhou, L. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 2012, 36, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Lindemans, C.A.; Calafiore, M.; Mertelsmann, A.M.; O’Connor, M.H.; Dudakov, J.A.; Jenq, R.R.; Velardi, E.; Young, L.F.; Smith, O.M.; Lawrence, G.; et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 2015, 528, 560–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.P.; Michel, M.L.; Da Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef]
- Lee, G.R. The Balance of Th17 versus Treg Cells in Autoimmunity. Int. J. Mol. Sci. 2018, 19, 730. [Google Scholar] [CrossRef] [Green Version]
- Zi, C.; He, L.; Yao, H.; Ren, Y.; He, T.; Gao, Y. Changes of Th17 cells, regulatory T cells, Treg/Th17, IL-17 and IL-10 in patients with type 2 diabetes mellitus: A systematic review and meta-analysis. Endocrine 2022, 76, 263–272. [Google Scholar] [CrossRef]
- Lan, Y.T.; Wang, Z.L.; Tian, P.; Gong, X.N.; Fan, Y.C.; Wang, K. Treg/Th17 imbalance and its clinical significance in patients with hepatitis B-associated liver cirrhosis. Diagn Pathol. 2019, 14, 114. [Google Scholar] [CrossRef] [Green Version]
- Kleinewietfeld, M.; Hafler, D.A. The plasticity of human Treg and Th17 cells and its role in autoimmunity. Semin. Immunol. 2013, 25, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.B.; Luo, M.M.; Chen, Z.Y.; He, B.H. The Function and Role of the Th17/Treg Cell Balance in Inflammatory Bowel Disease. J. Immunol. Res. 2020, 2020, 8813558. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, Z.; Si, Z.; Yang, Y.; Li, S.; Xue, Y. Dapagliflozin reverses the imbalance of T helper 17 and T regulatory cells by inhibiting SGK1 in a mouse model of diabetic kidney disease. FEBS Open Bio 2021, 11, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Elson, C.O.; Cong, Y.; Weaver, C.T.; Schoeb, T.R.; McClanahan, T.K.; Fick, R.B.; Kastelein, R.A. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology 2007, 132, 2359–2370. [Google Scholar] [CrossRef]
- Tao, L.; Liu, H.; Gong, Y. Role and mechanism of the Th17/Treg cell balance in the development and progression of insulin resistance. Mol. Cell. Biochem. 2019, 459, 183–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleczynska, W.; Jakiela, B.; Plutecka, H.; Milewski, M.; Sanak, M.; Musial, J. Imbalance between Th17 and regulatory T-cells in systemic lupus erythematosus. Folia Histochem. Cytobiol. 2011, 49, 646–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef]
- Veldhoen, M.; Hirota, K.; Westendorf, A.M.; Buer, J.; Dumoutier, L.; Renauld, J.C.; Stockinger, B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 2008, 453, 106–109. [Google Scholar] [CrossRef]
- Xie, J.; Wang, Z.; Wang, W. Semaphorin 4D Induces an Imbalance of Th17/Treg Cells by Activating the Aryl Hydrocarbon Receptor in Ankylosing Spondylitis. Front. Immunol. 2020, 11, 2151. [Google Scholar] [CrossRef]
- McAleer, J.P.; Fan, J.; Roar, B.; Primerano, D.A.; Denvir, J. Cytokine Regulation in Human CD4 T Cells by the Aryl Hydrocarbon Receptor and Gq-Coupled Receptors. Sci. Rep. 2018, 8, 10954. [Google Scholar] [CrossRef] [Green Version]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef]
- Talbot, J.; Peres, R.S.; Pinto, L.G.; Oliveira, R.D.R.; Lima, K.A.; Donate, P.B.; Silva, J.R.; Ryffel, B.; Cunha, T.M.; Alves-Filho, J.C.; et al. Smoking-induced aggravation of experimental arthritis is dependent of aryl hydrocarbon receptor activation in Th17 cells. Arthritis Res. Ther. 2018, 20, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, A.; Naka, T.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc. Natl. Acad. Sci. USA 2008, 105, 9721–9726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, F.; Martínez, A.C.; Camonis, J.; Rebollo, A. Aiolos transcription factor controls cell death in T cells by regulating Bcl-2 expression and its cellular localization. EMBO J 1999, 18, 3419–3430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintana, F.J.; Jin, H.; Burns, E.J.; Nadeau, M.; Yeste, A.; Kumar, D.; Rangachari, M.; Zhu, C.; Xiao, S.; Seavitt, J.; et al. Aiolos promotes TH17 differentiation by directly silencing Il2 expression. Nat. Immunol. 2012, 13, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, S.K.; Snook, J.P.; Williams, M.A.; Weis, J.J. Bystander T Cells: A Balancing Act of Friends and Foes. Trends Immunol. 2018, 39, 1021–1035. [Google Scholar] [CrossRef]
- Pacheco, Y.; Acosta-Ampudia, Y.; Monsalve, D.M.; Chang, C.; Gershwin, M.E.; Anaya, J.M. Bystander activation and autoimmunity. J. Autoimmun. 2019, 103, 102301. [Google Scholar] [CrossRef]
- Maurice, N.J.; Taber, A.K.; Prlic, M. The Ugly Duckling Turned to Swan: A Change in Perception of Bystander-Activated Memory CD8 T Cells. J. Immunol. 2021, 206, 455–462. [Google Scholar] [CrossRef]
- Tough, D.F.; Borrow, P.; Sprent, J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science 1996, 272, 1947–1950. [Google Scholar] [CrossRef]
- Zarozinski, C.C.; Welsh, R.M. Minimal bystander activation of CD8 T cells during the virus-induced polyclonal T cell response. J. Exp. Med. 1997, 185, 1629–1639. [Google Scholar] [CrossRef]
- Murali-Krishna, K.; Altman, J.D.; Suresh, M.; Sourdive, D.J.; Zajac, A.J.; Miller, J.D.; Slansky, J.; Ahmed, R. Counting antigen-specific CD8 T cells: A reevaluation of bystander activation during viral infection. Immunity 1998, 8, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Montoya, D.; Cruz, D.; Teles, R.M.; Lee, D.J.; Ochoa, M.T.; Krutzik, S.R.; Chun, R.; Schenk, M.; Zhang, X.; Ferguson, B.G.; et al. Divergence of macrophage phagocytic and antimicrobial programs in leprosy. Cell Host. Microbe 2009, 6, 343–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussell, T.; Bell, T.J. Alveolar macrophages: Plasticity in a tissue-specific context. Nat. Rev. Immunol. 2014, 14, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; De Kleer, I.; Henri, S.; Post, S.; Vanhoutte, L.; De Prijck, S.; Deswarte, K.; Malissen, B.; Hammad, H.; Lambrecht, B.N. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 2013, 210, 1977–1992. [Google Scholar] [CrossRef] [Green Version]
- Hoeffel, G.; Wang, Y.; Greter, M.; See, P.; Teo, P.; Malleret, B.; Leboeuf, M.; Low, D.; Oller, G.; Almeida, F.; et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J. Exp. Med. 2012, 209, 1167–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christian Schulz, E.G.P.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Eirik, S.; Jacobsen, W.; Frampton, J.; et al. A Lineage of Myeloid Cells Independent of Myb and Hematopoietic Stem Cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Benoit, M.; Desnues, B.; Mege, J.L. Macrophage polarization in bacterial infections. J. Immunol. 2008, 181, 3733–3739. [Google Scholar] [CrossRef] [Green Version]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Scott, C.L.; Zheng, F.; De Baetselier, P.; Martens, L.; Saeys, Y.; De Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun. 2016, 7, 10321. [Google Scholar] [CrossRef]
- Zigmond, E.; Samia-Grinberg, S.; Pasmanik-Chor, M.; Brazowski, E.; Shibolet, O.; Halpern, Z.; Varol, C. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J. Immunol. 2014, 193, 344–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, C.B.; Mehrotra, P.; Arandjelovic, S.; Perry, J.S.A.; Guo, Y.; Morioka, S.; Barron, B.; Walk, S.F.; Ghesquiere, B.; Krupnick, A.S.; et al. Metabolites released from apoptotic cells act as tissue messengers. Nature 2020, 580, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Baxter, A.A.; Poon, I.K. Apoptotic cells secrete metabolites to regulate immune homeostasis. Immunol. Cell Biol. 2020, 98, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Dunkle, A.; He, Y.W. Apoptosis and autophagy in the regulation of T lymphocyte function. Immunol. Res 2011, 49, 70–86. [Google Scholar] [CrossRef] [Green Version]
- Birk, A.V.; Broekman, M.J.; Gladek, E.M.; Robertson, H.D.; Drosopoulos, J.H.; Marcus, A.J.; Szeto, H.H. Role of extracellular ATP metabolism in regulation of platelet reactivity. J. Lab. Clin. Med. 2002, 140, 166–175. [Google Scholar] [CrossRef]
- Yip, L.; Woehrle, T.; Corriden, R.; Hirsh, M.; Chen, Y.; Inoue, Y.; Ferrari, V.; Insel, P.A.; Junger, W.G. Autocrine regulation of T-cell activation by ATP release and P2 × 7 receptors. FASEB J. 2009, 23, 1685–1693. [Google Scholar] [CrossRef] [Green Version]
- Winoto, A. Cell death in the regulation of immune responses. Curr. Opin. Immunol. 1997, 9, 365–370. [Google Scholar] [CrossRef]
- Ganeshan, K.; Chawla, A. Metabolic regulation of immune responses. Annu. Rev. Immunol. 2014, 32, 609–634. [Google Scholar] [CrossRef] [Green Version]
- van Rossum, D.; Hanisch, U.K. Microglia. Metab. Brain Dis. 2004, 19, 393–411. [Google Scholar] [CrossRef]
- Amici, S.A.; Dong, J.; Guerau-de-Arellano, M. Molecular Mechanisms Modulating the Phenotype of Macrophages and Microglia. Front. Immunol. 2017, 8, 1520. [Google Scholar] [CrossRef] [Green Version]
- DePaula-Silva, A.B.; Gorbea, C.; Doty, D.J.; Libbey, J.E.; Sanchez, J.M.S.; Hanak, T.J.; Cazalla, D.; Fujinami, R.S. Differential transcriptional profiles identify microglial- and macrophage-specific gene markers expressed during virus-induced neuroinflammation. J. Neuroinflamm. 2019, 16, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichhoff, G.; Brawek, B.; Garaschuk, O. Microglial calcium signal acts as a rapid sensor of single neuron damage in vivo. Biochim. Biophys. Acta 2011, 1813, 1014–1024. [Google Scholar] [CrossRef] [Green Version]
- Zarruk, J.G.; Greenhalgh, A.D.; David, S. Microglia and macrophages differ in their inflammatory profile after permanent brain ischemia. Exp. Neurol. 2018, 301, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Valin, K.L.; Dixon, M.L.; Leavenworth, J.W. The Role of Microglia and Macrophages in CNS Homeostasis, Autoimmunity, and Cancer. J. Immunol. Res. 2017, 2017, 5150678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tay, T.L.; Mai, D.; Dautzenberg, J.; Fernandez-Klett, F.; Lin, G.; Sagar; Datta, M.; Drougard, A.; Stempfl, T.; Ardura-Fabregat, A.; et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci. 2017, 20, 793–803. [Google Scholar] [CrossRef]
- Boechat, N.; Bouchonnet, F.; Bonay, M.; Grodet, A.; Pelicic, V.; Gicquel, B.; Hance, A.J. Culture at high density improves the ability of human macrophages to control mycobacterial growth. J. Immunol. 2001, 166, 6203–6211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.C.; Wang, L.; Plikus, M.V.; Jiang, T.X.; Murray, P.J.; Ramos, R.; Guerrero-Juarez, C.F.; Hughes, M.W.; Lee, O.K.; Shi, S.; et al. Organ-level quorum sensing directs regeneration in hair stem cell populations. Cell 2015, 161, 277–290. [Google Scholar] [CrossRef] [Green Version]
- Muldoon, J.J.; Chuang, Y.; Bagheri, N.; Leonard, J.N. Macrophages employ quorum licensing to regulate collective activation. Nat. Commun. 2020, 11, 878. [Google Scholar] [CrossRef] [Green Version]
- Postat, J.; Bousso, P. Quorum Sensing by Monocyte-Derived Populations. Front. Immunol. 2019, 10, 2140. [Google Scholar] [CrossRef]
- Xu, D.; Andrew, R.G.R.; Hapel, J.; Melissa, G.; Dominguez, R.; Russell, G.; Sara Kapp, V.S.; Stanley, E.R. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 2002, 99, 111–120. [Google Scholar]
- Groh, J.; Weis, J.; Zieger, H.; Stanley, E.R.; Heuer, H.; Martini, R. Colony-stimulating factor-1 mediates macrophage-related neural damage in a model for Charcot-Marie-Tooth disease type 1X. Brain 2012, 135, 88–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenzo, J.C.; Turner, A.L.; Cook, A.D.; Vlahos, R.; Anderson, G.P.; Reynolds, E.C.; Hamilton, J.A. Control of macrophage lineage populations by CSF-1 receptor and GM-CSF in homeostasis and inflammation. Immunol. Cell Biol. 2012, 90, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Nandi, S.; Akhter, M.P.; Seifert, M.F.; Dai, X.M.; Stanley, E.R. Developmental and functional significance of the CSF-1 proteoglycan chondroitin sulfate chain. Blood 2006, 107, 786–795. [Google Scholar] [CrossRef] [Green Version]
- Groh, J.; Basu, R.; Stanley, E.R.; Martini, R. Cell-Surface and Secreted Isoforms of CSF-1 Exert Opposing Roles in Macrophage-Mediated Neural Damage in Cx32-Deficient Mice. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 1890–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, F.P.; Jacqueline, C.; Poschmann, J.; Roquilly, A. Alveolar Macrophages: Adaptation to Their Anatomic Niche during and after Inflammation. Cells 2021, 10, 2720. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Rani, A.; Alam, A.; Zarin, S.; Pandey, S.; Singh, H.; Hasnain, S.E.; Ehtesham, N.Z. Macrophage: A Cell With Many Faces and Functions in Tuberculosis. Front. Immunol. 2022, 13, 747799. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.T.; Pestana, N.T. The in vivo extracellular life of facultative intracellular bacterial parasites: Role in pathogenesis. Immunobiology 2013, 218, 325–337. [Google Scholar] [CrossRef]
- Srinivasan, L.; Ahlbrand, S.; Briken, V. Interaction of Mycobacterium tuberculosis with host cell death pathways. Cold Spring Harb. Perspect. Med. 2014, 4, a022459. [Google Scholar] [CrossRef] [Green Version]
- Behar, S.M.; Martin, C.; Booty, M.; Nishimura, T.; Zhao, X.; Gan, H.X.; Divangahi, M.; Remold, H.G.; Remold, H.G. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol. 2011, 4, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Keane, J.; Remold, H.; Kornfeld, H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J. Immunol. 2000, 164, 2016–2020. [Google Scholar] [CrossRef] [Green Version]
- Danelishvili, L.; McGarvey, J.; Li, Y.; Bermudez, L.E. Mycobacterium tuberculosis infection causes different levels of apoptosis and necrosis in human macrophages and alveolar epithelial cells. Cell Microbiol. 2003, 5, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Afriyie-Asante, A.; Dabla, A.; Dagenais, A.; Berton, S.; Smyth, R.; Sun, J. Mycobacterium tuberculosis Exploits Focal Adhesion Kinase to Induce Necrotic Cell Death and Inhibit Reactive Oxygen Species Production. Front. Immunol. 2021, 12, 742370. [Google Scholar] [CrossRef] [PubMed]
- Lerner, T.R.; Borel, S.A.-O.X.; Greenwood, D.A.-O.X.; Repnik, U.; Russell, M.A.-O.; Herbst, S.; Jones, M.A.-O.; Collinson, L.M.; Griffiths, G.; Gutierrez, M.A.-O. Mycobacterium tuberculosis replicates within necrotic human macrophages. J. Cell Biol. 2017, 216, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.H.; Chen, H.Y.; Huang, C.; Yan, J.M.; Yin, Z.; Zhang, X.L.; Pan, Q. Accumulation of EBI3 induced by virulent Mycobacterium tuberculosis inhibits apoptosis in murine macrophages. Pathog Dis. 2019, 77, ftz007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longhi, M.P.; Trumpfheller, C.; Idoyaga, J.; Caskey, M.; Matos, I.; Kluger, C.; Salazar, A.M.; Colonna, M.; Steinman, R.M. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J. Exp. Med. 2009, 206, 1589–1602. [Google Scholar] [CrossRef]
- Bardou, M.; Postat, J.; Loaec, C.; Lemaitre, F.; Ronteix, G.; Garcia, Z.; Bousso, P. Quorum sensing governs collective dendritic cell activation in vivo. EMBO J. 2021, 40, e107176. [Google Scholar] [CrossRef]
- Celli, S.; Day, M.; Müller, A.J.; Molina-Paris, C.; Lythe, G.; Bousso, P. How many dendritic cells are required to initiate a T-cell response? Blood 2012, 120, 3945–3948. [Google Scholar] [CrossRef] [Green Version]
- Xue, Q.; Lu, Y.; Eisele, M.R.; Sulistijo, E.S.; Khan, N.; Fan, R.; Miller-Jensen, K. Analysis of single-cell cytokine secretion reveals a role for paracrine signaling in coordinating macrophage responses to TLR4 stimulation. Sci. Signal 2015, 8, ra59. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.B.; Bell, C.J.M.; Howlett, S.K.; Pekalski, M.L.; Brady, K.; Hinton, H.; Sauter, D.; Todd, J.A.; Umana, P.; Ast, O.; et al. A long-lived IL-2 mutein that selectively activates and expands regulatory T cells as a therapy for autoimmune disease. J. Autoimmun. 2018, 95, 1–14. [Google Scholar] [CrossRef]
- Mullard, A. Restoring IL-2 to its cancer immunotherapy glory. Nat. Rev. Drug Discov. 2021, 20, 163–165. [Google Scholar] [CrossRef]
- Tahvildari, M.; Dana, R. Low-Dose IL-2 Therapy in Transplantation, Autoimmunity, and Inflammatory Diseases. J. Immunol. 2019, 203, 2749–2755. [Google Scholar] [CrossRef] [PubMed]
- Churlaud, G.; Abbara, C.; Vinot, P.A.; Fourcade, G.; Ritvo, P.G.; Lorenzon, R.; Rosenzwajg, M.; Diquet, B.; Klatzmann, D. Pharmacodynamics of regulatory T cells in mice and humans treated with low-dose IL-2. J. Allergy Clin. Immunol. 2018, 142, 1344–1346.e3. [Google Scholar] [CrossRef]
- Yu, A.; Snowhite, I.; Vendrame, F.; Rosenzwajg, M.; Klatzmann, D.; Pugliese, A.; Malek, T.R. Selective IL-2 responsiveness of regulatory T cells through multiple intrinsic mechanisms supports the use of low-dose IL-2 therapy in type 1 diabetes. Diabetes 2015, 64, 2172–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klatzmann, D.; Abbas, A.K. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat. Rev. Immunol. 2015, 15, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, D.; Rosenzwajg, M.; Joly, F.; Six, A.; Carrat, F.; Thibault, V.; Sene, D.; Cacoub, P.; Klatzmann, D. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N. Engl. J. Med. 2011, 365, 2067–2077. [Google Scholar] [CrossRef]
- Baeyens, A.; Perol, L.; Fourcade, G.; Cagnard, N.; Carpentier, W.; Woytschak, J.; Boyman, O.; Hartemann, A.; Piaggio, E. Limitations of IL-2 and rapamycin in immunotherapy of type 1 diabetes. Diabetes 2013, 62, 3120–3131. [Google Scholar] [CrossRef] [Green Version]
- Rosenzwajg, M.; Churlaud, G.; Mallone, R.; Six, A.; Derian, N.; Chaara, W.; Lorenzon, R.; Long, S.A.; Buckner, J.H.; Afonso, G.; et al. Low-dose interleukin-2 fosters a dose-dependent regulatory T cell tuned milieu in T1D patients. J. Autoimmun. 2015, 58, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Humrich, J.Y.; von Spee-Mayer, C.; Siegert, E.; Alexander, T.; Hiepe, F.; Radbruch, A.; Burmester, G.R.; Riemekasten, G. Rapid induction of clinical remission by low-dose interleukin-2 in a patient with refractory SLE. Ann. Rheum. Dis. 2015, 74, 791–792. [Google Scholar] [CrossRef]
- He, J.; Zhang, X.; Wei, Y.; Sun, X.; Chen, Y.; Deng, J.; Jin, Y.; Gan, Y.; Hu, X.; Jia, R.; et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat. Med. 2016, 22, 991–993. [Google Scholar] [CrossRef]
- Castela, E.; Le Duff, F.; Butori, C.; Ticchioni, M.; Hofman, P.; Bahadoran, P.; Lacour, J.P.; Passeron, T. Effects of low-dose recombinant interleukin 2 to promote T-regulatory cells in alopecia areata. JAMA Derm. 2014, 150, 748–751. [Google Scholar] [CrossRef]
- Kennedy-Nasser, A.A.; Ku, S.; Castillo-Caro, P.; Hazrat, Y.; Wu, M.F.; Liu, H.; Melenhorst, J.; Barrett, A.J.; Ito, S.; Foster, A.; et al. Ultra low-dose IL-2 for GVHD prophylaxis after allogeneic hematopoietic stem cell transplantation mediates expansion of regulatory T cells without diminishing antiviral and antileukemic activity. Clin. Cancer Res. 2014, 20, 2215–2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koreth, J.; Matsuoka, K.; Kim, H.T.; McDonough, S.M.; Bindra, B.; Alyea, E.P., 3rd; Armand, P.; Cutler, C.; Ho, V.T.; Treister, N.S.; et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N. Engl. J. Med. 2011, 365, 2055–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.; Jeon, Y.W.; Nam, Y.S.; Lim, J.Y.; Im, K.I.; Lee, E.S.; Cho, S.G. Therapeutic potential of low-dose IL-2 in a chronic GVHD patient by in vivo expansion of regulatory T cells. Cytokine 2016, 78, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, K.; Koreth, J.; Kim, H.T.; Bascug, G.; McDonough, S.; Kawano, Y.; Murase, K.; Cutler, C.; Ho, V.T.; Alyea, E.P.; et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci. Transl. Med. 2013, 5, 179ra143. [Google Scholar] [CrossRef] [Green Version]
- Zorn, E.; Mohseni, M.; Kim, H.; Porcheray, F.; Lynch, A.; Bellucci, R.; Canning, C.; Alyea, E.P.; Soiffer, R.J.; Ritz, J. Combined CD4+ donor lymphocyte infusion and low-dose recombinant IL-2 expand FOXP3+ regulatory T cells following allogeneic hematopoietic stem cell transplantation. Biol. Blood Marrow Transpl. 2009, 15, 382–388. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, B.; Vigneron, J.; Levacher, B.; Vazquez, T.; Pitoiset, F.; Brimaud, F.; Churlaud, G.; Klatzmann, D.; Bellier, B. Low-Dose IL-2 Induces Regulatory T Cell-Mediated Control of Experimental Food Allergy. J. Immunol. 2016, 197, 188–198. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Horn, G.; Moulton, K.; Oza, A.; Byler, S.; Kokolus, S.; Longacre, M. Cancer development, progression, and therapy: An epigenetic overview. Int. J. Mol. Sci. 2013, 14, 21087–21113. [Google Scholar] [CrossRef]
- Kaye, S.B. New antimetabolites in cancer chemotherapy and their clinical impact. Br. J. Cancer 1998, 78 (Suppl. 3), 1–7. [Google Scholar] [CrossRef] [Green Version]
- Ahmadzadeh, M.; Rosenberg, S.A. IL-2 administration increases CD4+ CD25(hi) Foxp3+ regulatory T cells in cancer patients. Blood 2006, 107, 2409–2414. [Google Scholar] [CrossRef] [Green Version]
- Klapper, J.A.; Downey, S.G.; Smith, F.O.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Sherry, R.M.; Royal, R.E.; Steinberg, S.M.; Rosenberg, S. High-dose interleukin-2 for the treatment of metastatic renal cell carcinoma: A retrospective analysis of response and survival in patients treated in the surgery branch at the National Cancer Institute between 1986 and 2006. Cancer 2008, 113, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Buchbinder, E.I.; Dutcher, J.P.; Daniels, G.A.; Curti, B.D.; Patel, S.P.; Holtan, S.G.; Miletello, G.P.; Fishman, M.N.; Gonzalez, R.; Clark, J.I.; et al. Therapy with high-dose Interleukin-2 (HD IL-2) in metastatic melanoma and renal cell carcinoma following PD1 or PDL1 inhibition. J. Immunother. Cancer 2019, 7, 49. [Google Scholar] [CrossRef] [Green Version]
- Davar, D.; Ding, F.; Saul, M.; Sander, C.; Tarhini, A.A.; Kirkwood, J.M.; Tawbi, H.A. High-dose interleukin-2 (HD IL-2) for advanced melanoma: A single center experience from the University of Pittsburgh Cancer Institute. J. Immunother. Cancer 2017, 5, 74. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L.; Liu, K.D. Overview of interleukin-2 function, production and clinical applications. Cytokine 2004, 28, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Moon, B.I.; Kim, T.H.; Seoh, J.Y. Functional Modulation of Regulatory T Cells by IL-2. PLoS ONE 2015, 10, e0141864. [Google Scholar] [CrossRef]
- Dockery, J.D.; Keener, J.P. A mathematical model for quorum sensing in Pseudomonas aeruginosa. Bull. Math. Biol. 2001, 63, 95–116. [Google Scholar] [CrossRef]
- Vital-Lopez, F.G.; Reifman, J.; Wallqvist, A. Biofilm Formation Mechanisms of Pseudomonas aeruginosa Predicted via Genome-Scale Kinetic Models of Bacterial Metabolism. PLoS Comput. Biol. 2015, 11, e1004452. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.R.; Amado, I.F.; Reynolds, J.; Berges, J.; Lythe, G.; Molina-París, C.; Freitas, A.A. Quorum-Sensing in CD4(+) T Cell Homeostasis: A Hypothesis and a Model. Front. Immunol. 2012, 3, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, J.; Amado, I.F.; Freitas, A.A.; Lythe, G.; Molina-París, C. A mathematical perspective on CD4(+) T cell quorum-sensing. J. Biol. 2014, 347, 160–175. [Google Scholar] [CrossRef]
- Schrom, E.C., 2nd; Levin, S.A.; Graham, A.L. Quorum sensing via dynamic cytokine signaling comprehensively explains divergent patterns of effector choice among helper T cells. PLoS Comput. Biol. 2020, 16, e1008051. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naoun, A.A.; Raphael, I.; Forsthuber, T.G. Immunoregulation via Cell Density and Quorum Sensing-like Mechanisms: An Underexplored Emerging Field with Potential Translational Implications. Cells 2022, 11, 2442. https://doi.org/10.3390/cells11152442
Naoun AA, Raphael I, Forsthuber TG. Immunoregulation via Cell Density and Quorum Sensing-like Mechanisms: An Underexplored Emerging Field with Potential Translational Implications. Cells. 2022; 11(15):2442. https://doi.org/10.3390/cells11152442
Chicago/Turabian StyleNaoun, Adrian A., Itay Raphael, and Thomas G. Forsthuber. 2022. "Immunoregulation via Cell Density and Quorum Sensing-like Mechanisms: An Underexplored Emerging Field with Potential Translational Implications" Cells 11, no. 15: 2442. https://doi.org/10.3390/cells11152442