
The Exocyst Is Required for CD36 Fatty Acid Translocase Trafficking and Free Fatty Acid Uptake in Skeletal Muscle Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Immunofluorescent Staining
2.3. Proximity Ligation Assay
2.4. In-Cell Western to Quantify Cell-Surface Delivery of CD36 in Cultured Cells
2.5. Fatty Acid Uptake Assay
3. Results and Discussion
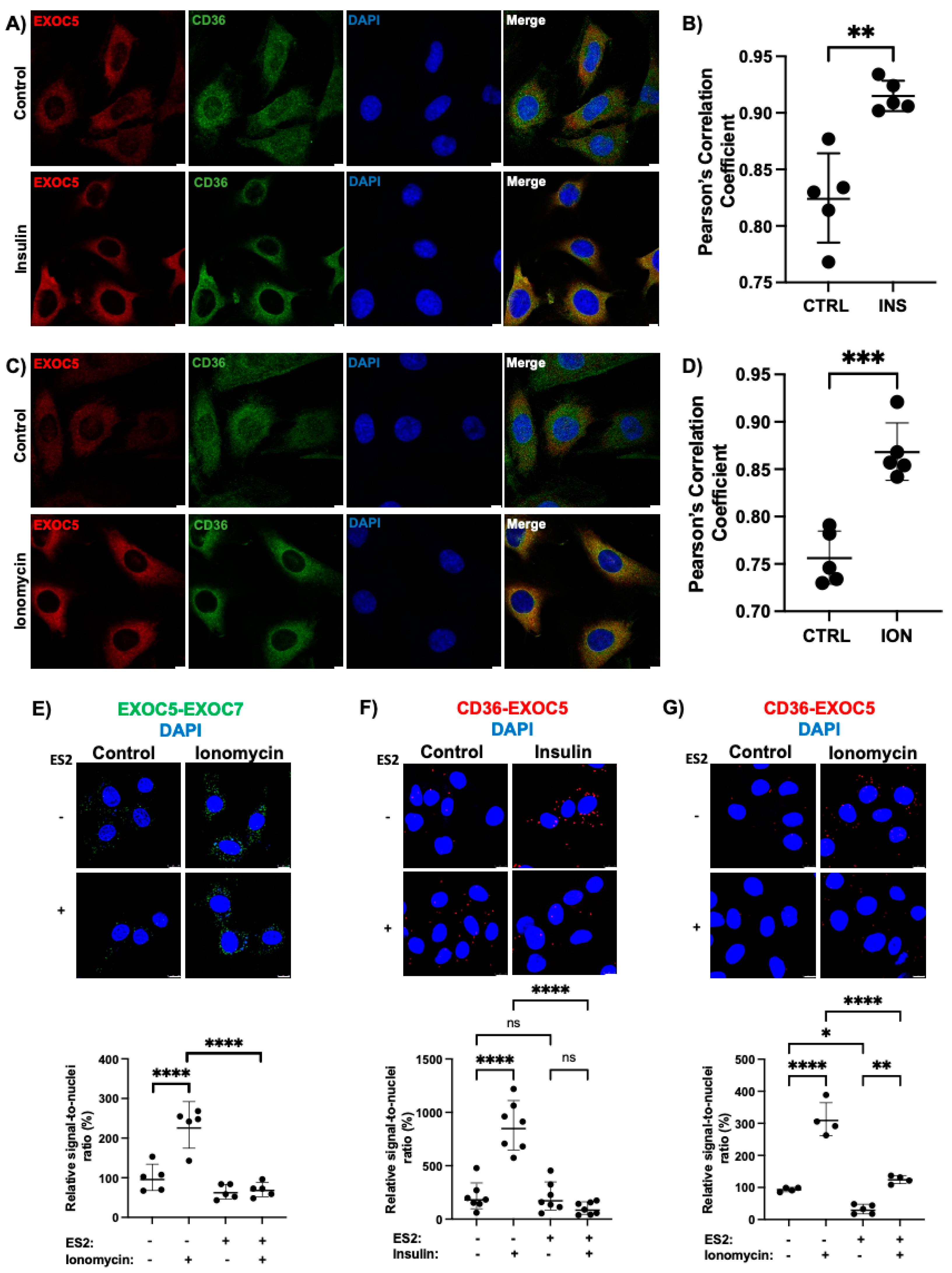
3.1. Insulin and Ionomycin Stimulate Exocyst-CD36 Colocalization
3.2. The Exocyst Is Recruited to CD36 Vesicles in Response to Stimuli
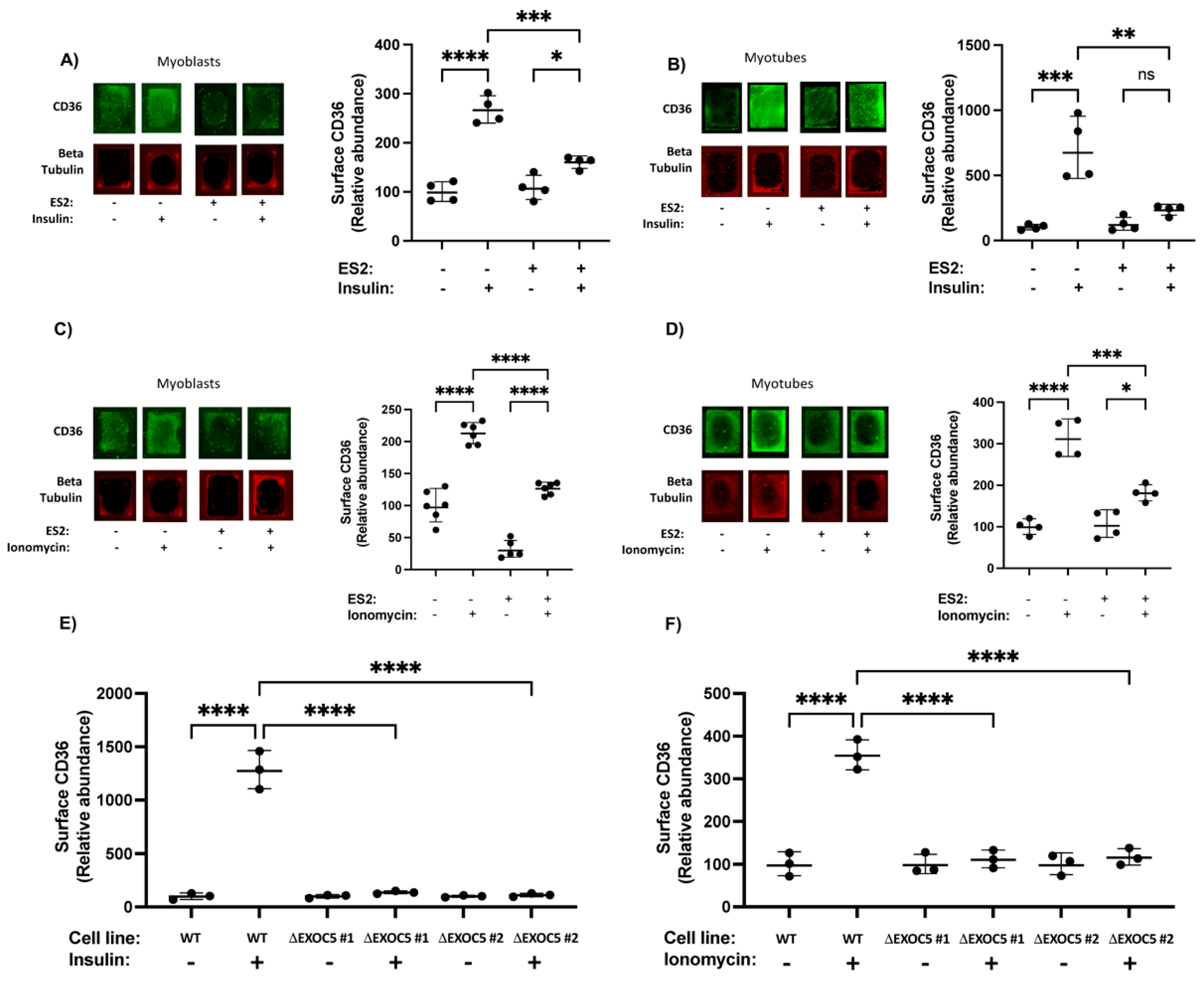
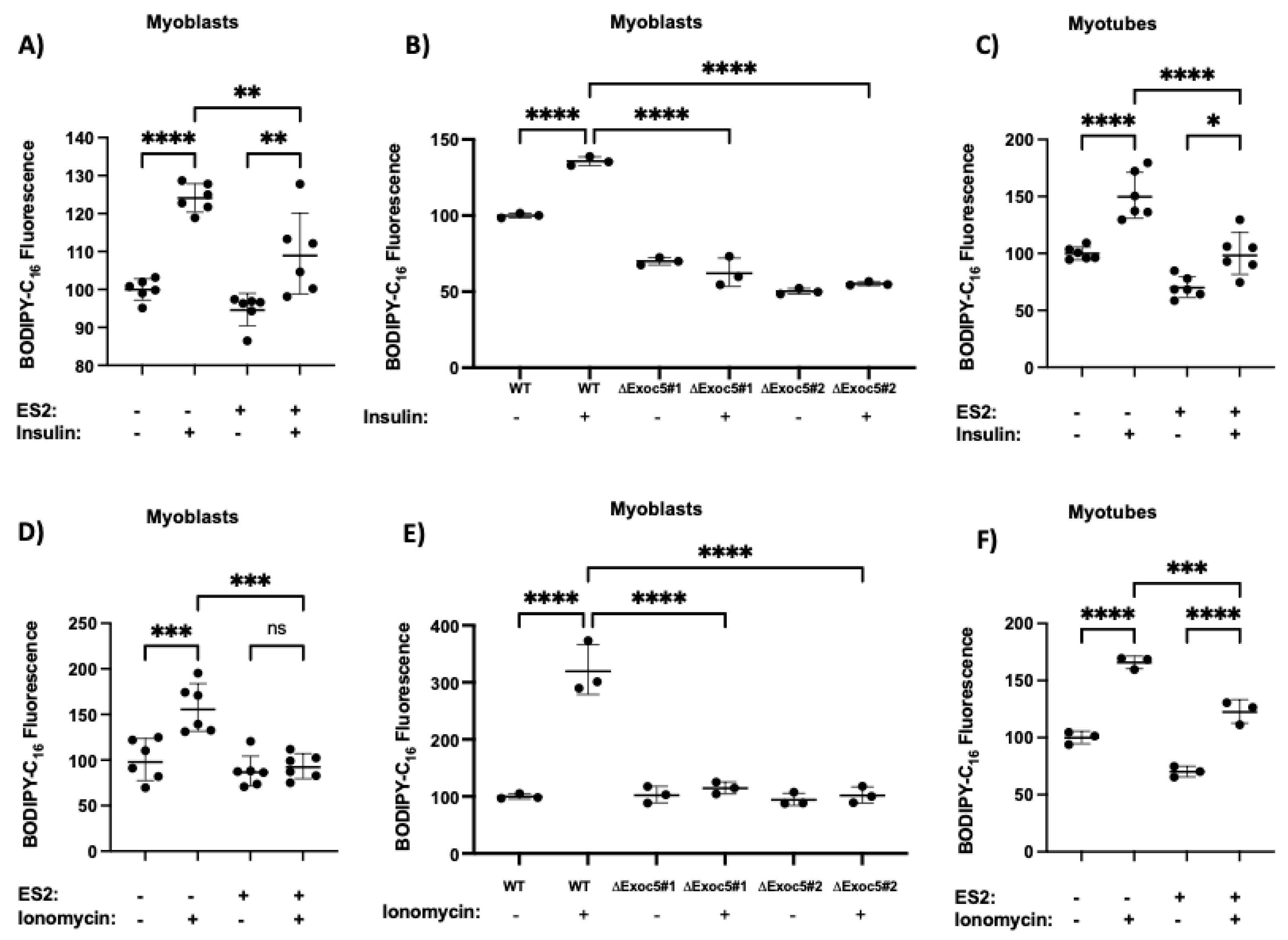
3.3. Disrupted Exocyst Function Impairs CD36 Membrane Delivery and FFA Uptake in Response to Stimuli
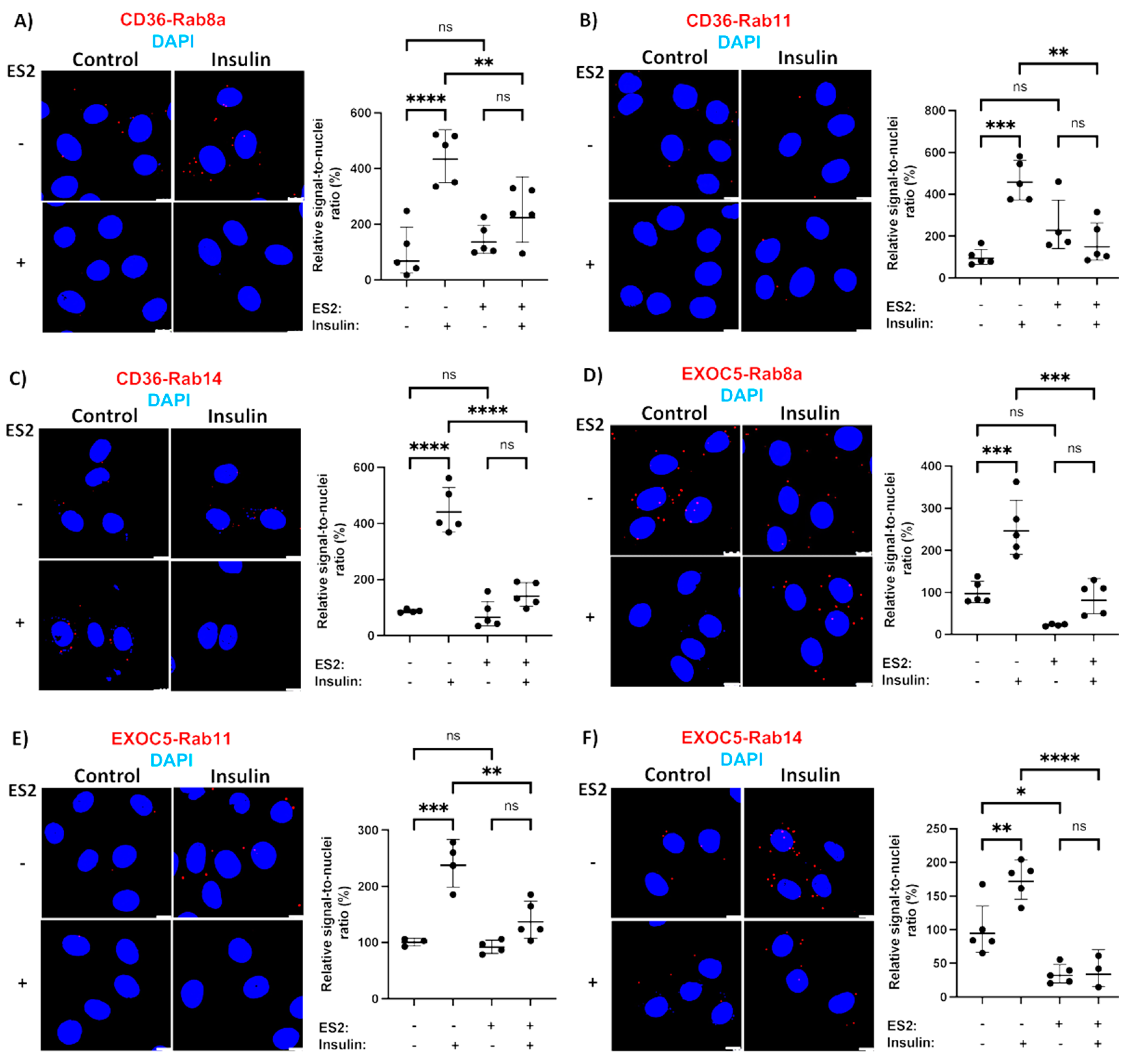
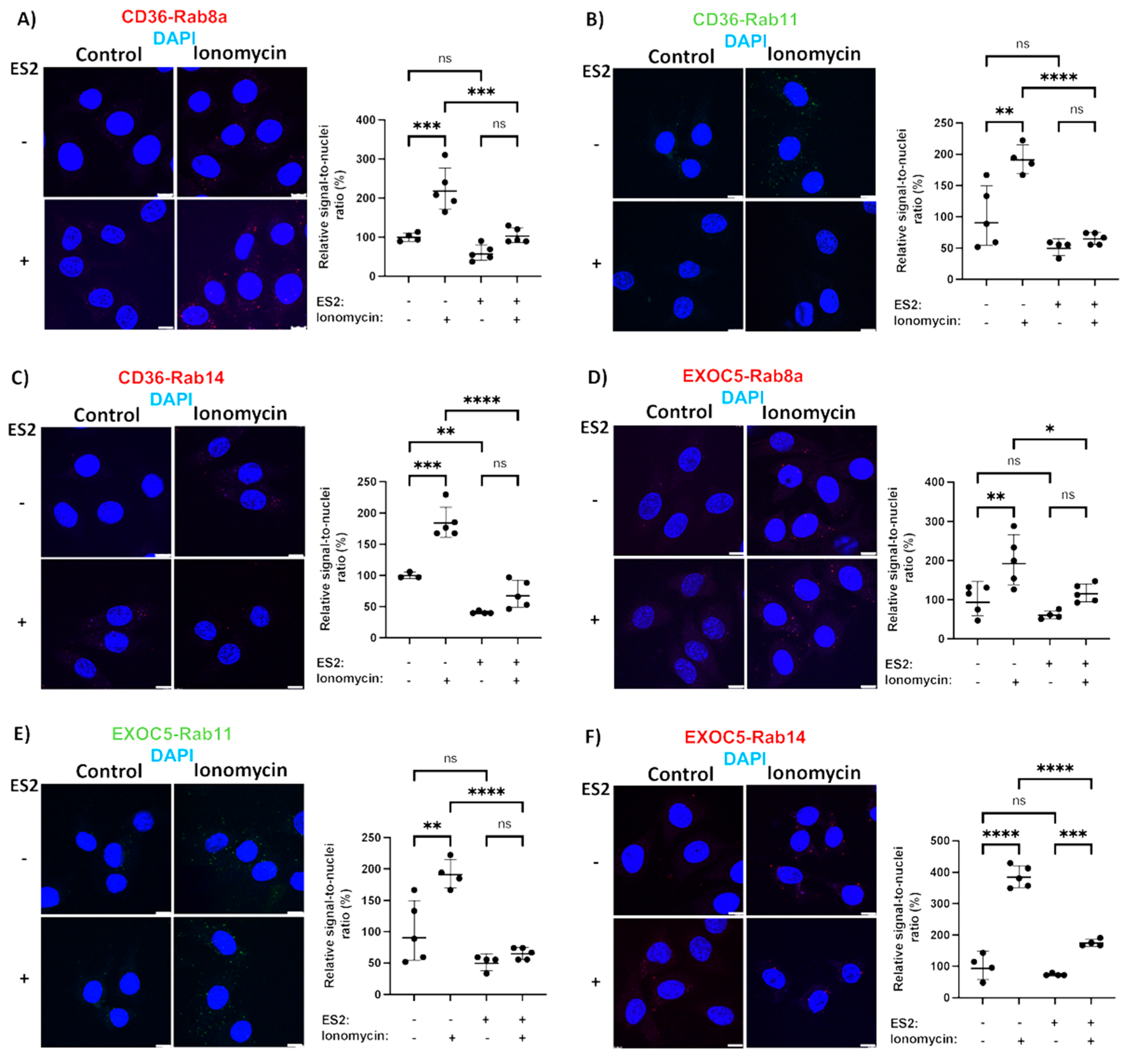
3.4. Rab GTPases Associate with the Exocyst and CD36 in Response to Stimuli
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- CDC. National Diabetes Statistics Report; US Department of Health and Human Services: Atlanta, GA, USA, 2017.
- Belfort, R.; Mandarino, L.; Kashyap, S.; Wirfel, K.; Pratipanawatr, T.; Berria, R.; Defronzo, R.A.; Cusi, K. Dose-response effect of elevated plasma free fatty acid on insulin signaling. Diabetes 2005, 54, 1640–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickersgill, L.; Litherland, G.J.; Greenberg, A.S.; Walker, M.; Yeaman, S.J. Key role for ceramides in mediating insulin resistance in human muscle cells. J. Biol. Chem. 2007, 282, 12583–12589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dresner, A.; Laurent, D.; Marcucci, M.; Griffin, M.E.; Dufour, S.; Cline, G.W.; Slezak, L.A.; Andersen, D.K.; Hundal, R.S.; Rothman, D.L.; et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J. Clin. Investig. 1999, 103, 253–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boden, G.; Chen, X.; Ruiz, J.; White, J.V.; Rossetti, L. Mechanisms of fatty acid-induced inhibition of glucose uptake. J. Clin. Investig. 1994, 93, 2438–2446. [Google Scholar] [CrossRef] [PubMed]
- McFarlan, J.T.; Yoshida, Y.; Jain, S.S.; Han, X.X.; Snook, L.A.; Lally, J.; Smith, B.K.; Glatz, J.F.; Luiken, J.J.; Sayer, R.A.; et al. In vivo, fatty acid translocase (CD36) critically regulates skeletal muscle fuel selection, exercise performance, and training-induced adaptation of fatty acid oxidation. J. Biol. Chem. 2012, 287, 23502–23516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samovski, D.; Sun, J.; Pietka, T.; Gross, R.W.; Eckel, R.H.; Su, X.; Stahl, P.D.; Abumrad, N.A. Regulation of AMPK activation by CD36 links fatty acid uptake to beta-oxidation. Diabetes 2015, 64, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Luiken, J.J.; Dyck, D.J.; Han, X.X.; Tandon, N.N.; Arumugam, Y.; Glatz, J.F.; Bonen, A. Insulin induces the translocation of the fatty acid transporter FAT/CD36 to the plasma membrane. Am. J. Physiol.-Endocrinol. Metab. 2002, 282, E491–E495. [Google Scholar] [CrossRef] [Green Version]
- Luiken, J.J.; Coort, S.L.; Willems, J.; Coumans, W.A.; Bonen, A.; van der Vusse, G.J.; Glatz, J.F. Contraction-induced fatty acid translocase/CD36 translocation in rat cardiac myocytes is mediated through AMP-activated protein kinase signaling. Diabetes 2003, 52, 1627–1634. [Google Scholar] [CrossRef] [Green Version]
- Rachek, L.I. Free fatty acids and skeletal muscle insulin resistance. Prog. Mol. Biol. Transl. Sci. 2014, 121, 267–292. [Google Scholar] [CrossRef]
- Zhu, B.; Li, M.Y.; Lin, Q.; Liang, Z.; Xin, Q.; Wang, M.; He, Z.; Wang, X.; Wu, X.; Chen, G.G.; et al. Lipid oversupply induces CD36 sarcolemmal translocation via dual modulation of PKCzeta and TBC1D1: An early event prior to insulin resistance. Theranostics 2020, 10, 1332–1354. [Google Scholar] [CrossRef]
- Holloway, G.P.; Schwenk, R.W.; Luiken, J.J.; Glatz, J.F.; Bone, A. Fatty acid transport in skeletal muscle: Role in energy provision and insulin resistance. Clin. Lipidol. 2010, 5, 731–745. [Google Scholar] [CrossRef]
- Goudriaan, J.R.; Dahlmans, V.E.; Teusink, B.; Ouwens, D.M.; Febbraio, M.; Maassen, J.A.; Romijn, J.A.; Havekes, L.M.; Voshol, P.J. CD36 deficiency increases insulin sensitivity in muscle, but induces insulin resistance in the liver in mice. J. Lipid Res. 2003, 44, 2270–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glatz, J.F.; Luiken, J.J.; Bonen, A. Membrane fatty acid transporters as regulators of lipid metabolism: Implications for metabolic disease. Physiol. Rev. 2010, 90, 367–417. [Google Scholar] [CrossRef] [Green Version]
- Steinbusch, L.K.; Wijnen, W.; Schwenk, R.W.; Coumans, W.A.; Hoebers, N.T.; Ouwens, D.M.; Diamant, M.; Bonen, A.; Glatz, J.F.; Luiken, J.J. Differential regulation of cardiac glucose and fatty acid uptake by endosomal pH and actin filaments. Am. J. Physiol.-Cell Physiol. 2010, 298, C1549–C1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Oort, M.M.; van Doorn, J.M.; Bonen, A.; Glatz, J.F.; van der Horst, D.J.; Rodenburg, K.W.; Luiken, J.J. Insulin-induced translocation of CD36 to the plasma membrane is reversible and shows similarity to that of GLUT4. Biochim. Biophys. Acta 2008, 1781, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Ouwens, D.M.; Diamant, M.; Fodor, M.; Habets, D.D.J.; Pelsers, M.; El Hasnaoui, M.; Dang, Z.C.; van den Brom, C.E.; Vlasblom, R.; Rietdijk, A.; et al. Cardiac contractile dysfunction in insulin-resistant rats fed a high-fat diet is associated with elevated CD36-mediated fatty acid uptake and esterification. Diabetologia 2007, 50, 1938–1948. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Akama, T.; Jiang, Y.; Chun, T.H. The Exocyst Complex Regulates Free Fatty Acid Uptake by Adipocytes. PLoS ONE 2015, 10, e0120289. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Rong, P.; Xu, D.; Zhu, S.; Chen, L.; Xie, B.; Du, Q.; Quan, C.; Sheng, Y.; Zhao, T.J.; et al. Rab8a Deficiency in Skeletal Muscle Causes Hyperlipidemia and Hepatosteatosis by Impairing Muscle Lipid Uptake and Storage. Diabetes 2017, 66, 2387–2399. [Google Scholar] [CrossRef] [Green Version]
- Samovski, D.; Su, X.; Xu, Y.; Abumrad, N.A.; Stahl, P.D. Insulin and AMPK regulate FA translocase/CD36 plasma membrane recruitment in cardiomyocytes via Rab GAP AS160 and Rab8a Rab GTPase. J. Lipid Res. 2012, 53, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Chang, L.; Hwang, J.; Chiang, S.H.; Saltiel, A.R. The exocyst complex is required for targeting of Glut4 to the plasma membrane by insulin. Nature 2003, 422, 629–633. [Google Scholar] [CrossRef]
- Fujimoto, B.; Young, M.; Carter, L.; Pang, A.P.S.; Corley, M.J.; Fogelgren, B.; Polgar, N. The exocyst complex regulates insulin-stimulated glucose uptake of skeletal muscle cells. Am. J. Physiol.-Endocrinol. Metab. 2019, 317, E957–E972. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, R.W.; Angin, Y.; Steinbusch, L.K.; Dirkx, E.; Hoebers, N.; Coumans, W.A.; Bonen, A.; Broers, J.L.; van Eys, G.J.; Glatz, J.F.; et al. Overexpression of vesicle-associated membrane protein (VAMP) 3, but not VAMP2, protects glucose transporter (GLUT) 4 protein translocation in an in vitro model of cardiac insulin resistance. J. Biol. Chem. 2012, 287, 37530–37539. [Google Scholar] [CrossRef] [Green Version]
- Steinbusch, L.K.; Schwenk, R.W.; Ouwens, D.M.; Diamant, M.; Glatz, J.F.; Luiken, J.J. Subcellular trafficking of the substrate transporters GLUT4 and CD36 in cardiomyocytes. Cell Mol. Life Sci. 2011, 68, 2525–2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Palanivel, R.; Sweeney, G. Regulation of fatty acid uptake and metabolism in L6 skeletal muscle cells by resistin. FEBS Lett 2005, 579, 5049–5054. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Lopez, J.A.; James, D.E.; Hunziker, W. Snapin interacts with the Exo70 subunit of the exocyst and modulates GLUT4 trafficking. J. Biol. Chem. 2008, 283, 324–331. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Rong, P.; Zhu, S.; Yang, X.; Ouyang, Q.; Wang, H.Y.; Chen, S. Targeting RalGAPalpha1 in skeletal muscle to simultaneously improve postprandial glucose and lipid control. Sci. Adv. 2019, 5, eaav4116. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.S.; Folsch, H. An old dog learns new tricks: Novel functions of the exocyst complex in polarized epithelia in animals. F1000 Biol. Rep. 2009, 1, 83. [Google Scholar] [CrossRef]
- Yamashita, M.; Kurokawa, K.; Sato, Y.; Yamagata, A.; Mimura, H.; Yoshikawa, A.; Sato, K.; Nakano, A.; Fukai, S. Structural basis for the Rho- and phosphoinositide-dependent localization of the exocyst subunit Sec3. Nat. Struct. Mol. Biol. 2010, 17, 180–186. [Google Scholar] [CrossRef]
- Boyd, C.; Hughes, T.; Pypaert, M.; Novick, P. Vesicles carry most exocyst subunits to exocytic sites marked by the remaining two subunits, Sec3p and Exo70p. J. Cell Biol. 2004, 167, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Fredriksson, S.; Gullberg, M.; Jarvius, J.; Olsson, C.; Pietras, K.; Gustafsdottir, S.M.; Ostman, A.; Landegren, U. Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 2002, 20, 473–477. [Google Scholar] [CrossRef]
- Kioumourtzoglou, D.; Gould, G.W.; Bryant, N.J. Insulin stimulates syntaxin4 SNARE complex assembly via a novel regulatory mechanism. Mol. Cell Biol. 2014, 34, 1271–1279. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Brown, M.Q.; van de Ven, W.; Zhang, Z.M.; Wu, B.; Young, M.C.; Synek, L.; Borchardt, D.; Harrison, R.; Pan, S.; et al. Endosidin2 targets conserved exocyst complex subunit EXO70 to inhibit exocytosis. Proc. Natl. Acad. Sci. USA 2016, 113, E41–E50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glatz, J.F.C.; Luiken, J. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J. Lipid Res. 2018, 59, 1084–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koonen, D.P.; Glatz, J.F.; Bonen, A.; Luiken, J.J. Long-chain fatty acid uptake and FAT/CD36 translocation in heart and skeletal muscle. Biochim. Biophys. Acta 2005, 1736, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.M.; Nishida-Fukuda, H.; Li, Y.; McDonald, W.H.; Gradinaru, C.C.; Macara, I.G. Exocyst dynamics during vesicle tethering and fusion. Nat. Commun. 2018, 9, 5140. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.J.; Polgar, N.; Napoli, J.A.; Lui, V.H.; Tamashiro, K.K.; Fujimoto, B.A.; Thompson, K.S.; Fogelgren, B. Fibroproliferative response to urothelial failure obliterates the ureter lumen in a mouse model of prenatal congenital obstructive nephropathy. Sci. Rep. 2016, 6, 31137. [Google Scholar] [CrossRef] [Green Version]
- Zuo, X.; Guo, W.; Lipschutz, J.H. The exocyst protein Sec10 is necessary for primary ciliogenesis and cystogenesis in vitro. Mol. Biol. Cell 2009, 20, 2522–2529. [Google Scholar] [CrossRef] [Green Version]
- Fogelgren, B.; Lin, S.Y.; Zuo, X.; Jaffe, K.M.; Park, K.M.; Reichert, R.J.; Bell, P.D.; Burdine, R.D.; Lipschutz, J.H. The exocyst protein Sec10 interacts with Polycystin-2 and knockdown causes PKD-phenotypes. PLoS Genet. 2011, 7, e1001361. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Khayat, Z.; Kishi, K.; Ebina, Y.; Klip, A. GLUT4 translocation by insulin in intact muscle cells: Detection by a fast and quantitative assay. FEBS Lett. 1998, 427, 193–197. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Sportsman, R.; Harris, J.; Stahl, A. Real-time quantification of fatty acid uptake using a novel fluorescence assay. J. Lipid Res. 2005, 46, 597–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sin, J.; Andres, A.M.; Taylor, D.J.; Weston, T.; Hiraumi, Y.; Stotland, A.; Kim, B.J.; Huang, C.; Doran, K.S.; Gottlieb, R.A. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy 2016, 12, 369–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Roth, D.; Gatti, E.; De Camilli, P.; Novick, P. Identification and characterization of homologues of the Exocyst component Sec10p. FEBS Lett. 1997, 404, 135–139. [Google Scholar] [CrossRef] [Green Version]
- Ting, A.E.; Hazuka, C.D.; Hsu, S.C.; Kirk, M.D.; Bean, A.J.; Scheller, R.H. rSec6 and rSec8, mammalian homologs of yeast proteins essential for secretion. Proc. Natl. Acad. Sci. USA 1995, 92, 9613–9617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essid, M.; Gopaldass, N.; Yoshida, K.; Merrifield, C.; Soldati, T. Rab8a regulates the exocyst-mediated kiss-and-run discharge of the Dictyostelium contractile vacuole. Mol. Biol. Cell 2012, 23, 1267–1282. [Google Scholar] [CrossRef]
- Sano, H.; Peck, G.R.; Blachon, S.; Lienhard, G.E. A potential link between insulin signaling and GLUT4 translocation: Association of Rab10-GTP with the exocyst subunit Exoc6/6b. Biochem. Biophys. Res. Commun. 2015, 465, 601–605. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; Bist, P.; Wu, J.; Zhao, Q.; Li, Q.J.; Wan, Y.; Abraham, S.N. Collaboration between Distinct Rab Small GTPase Trafficking Circuits Mediates Bacterial Clearance from the Bladder Epithelium. Cell Host Microbe 2017, 22, 330–342. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Bilan, P.J.; Liu, Z.; Klip, A. Rab8A and Rab13 are activated by insulin and regulate GLUT4 translocation in muscle cells. Proc. Natl. Acad. Sci. USA 2010, 107, 19909–19914. [Google Scholar] [CrossRef] [Green Version]
- Ishikura, S.; Bilan, P.J.; Klip, A. Rabs 8A and 14 are targets of the insulin-regulated Rab-GAP AS160 regulating GLUT4 traffic in muscle cells. Biochem. Biophys. Res. Commun. 2007, 353, 1074–1079. [Google Scholar] [CrossRef]
- Schwenk, R.W.; Luiken, J.J.; Eckel, J. FIP2 and Rip11 specify Rab11a-mediated cellular distribution of GLUT4 and FAT/CD36 in H9c2-hIR cells. Biochem. Biophys. Res. Commun. 2007, 363, 119–125. [Google Scholar] [CrossRef]
- Müller, H.; Deckers, K.; Eckel, J. The fatty acid translocase (FAT)/CD36 and the glucose transporter GLUT4 are localized in different cellular compartments in rat cardiac muscle. Biochem. Biophys. Res. Commun. 2002, 293, 665–669. [Google Scholar] [CrossRef]
- Bonen, A.; Luiken, J.J.; Arumugam, Y.; Glatz, J.F.; Tandon, N.N. Acute regulation of fatty acid uptake involves the cellular redistribution of fatty acid translocase. J. Biol. Chem. 2000, 275, 14501–14508. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Kubo, K.; Waguri, S.; Yabashi, A.; Shin, H.W.; Katoh, Y.; Nakayama, K. Rab11 regulates exocytosis of recycling vesicles at the plasma membrane. J. Cell Sci. 2012, 125, 4049–4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jose, M.; Tollis, S.; Nair, D.; Mitteau, R.; Velours, C.; Massoni-Laporte, A.; Royou, A.; Sibarita, J.B.; McCusker, D. A quantitative imaging-based screen reveals the exocyst as a network hub connecting endocytosis and exocytosis. Mol. Biol. Cell 2015, 26, 2519–2534. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura, N.K.; Tokunaga, D.S.; Ha, H.Y.; Polgar, N. The Exocyst Is Required for CD36 Fatty Acid Translocase Trafficking and Free Fatty Acid Uptake in Skeletal Muscle Cells. Cells 2022, 11, 2440. https://doi.org/10.3390/cells11152440
Nakamura NK, Tokunaga DS, Ha HY, Polgar N. The Exocyst Is Required for CD36 Fatty Acid Translocase Trafficking and Free Fatty Acid Uptake in Skeletal Muscle Cells. Cells. 2022; 11(15):2440. https://doi.org/10.3390/cells11152440
Chicago/Turabian StyleNakamura, Nicole K., Darcy S. Tokunaga, Herena Y. Ha, and Noemi Polgar. 2022. "The Exocyst Is Required for CD36 Fatty Acid Translocase Trafficking and Free Fatty Acid Uptake in Skeletal Muscle Cells" Cells 11, no. 15: 2440. https://doi.org/10.3390/cells11152440