
TGF-β Superfamily Signaling in the Eye: Implications for Ocular Pathologies


Abstract
:1. Introduction
2. The TGF-β Superfamily
2.1. Soluble Factors of the TGF-β Superfamily
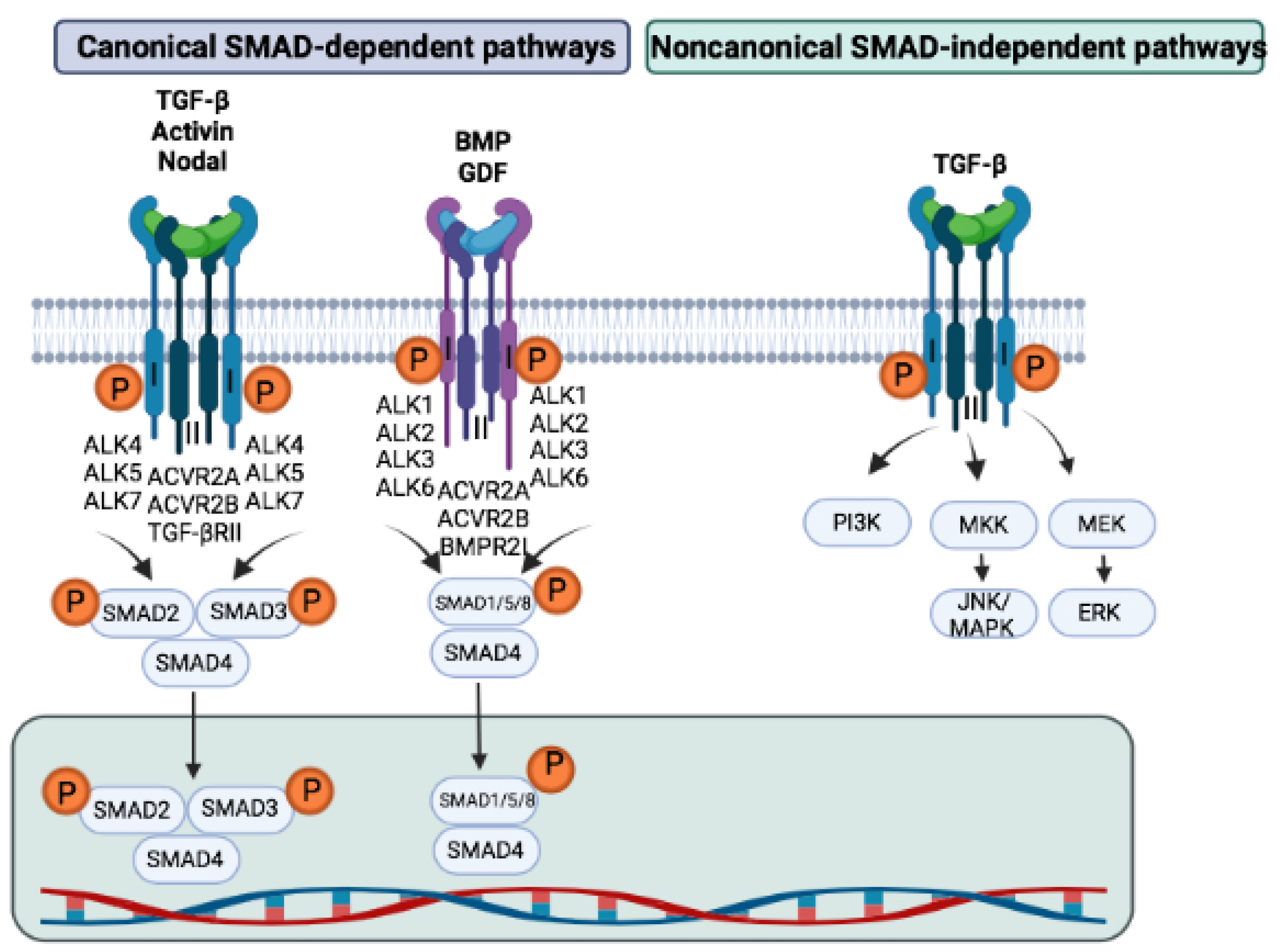
2.2. Receptors and Their Signaling
2.3. SMADs
2.4. The Regulation of TGF-β Signaling
3. Biological Roles of TGF-β
3.1. In Vascular Development
3.2. In Immune Cells
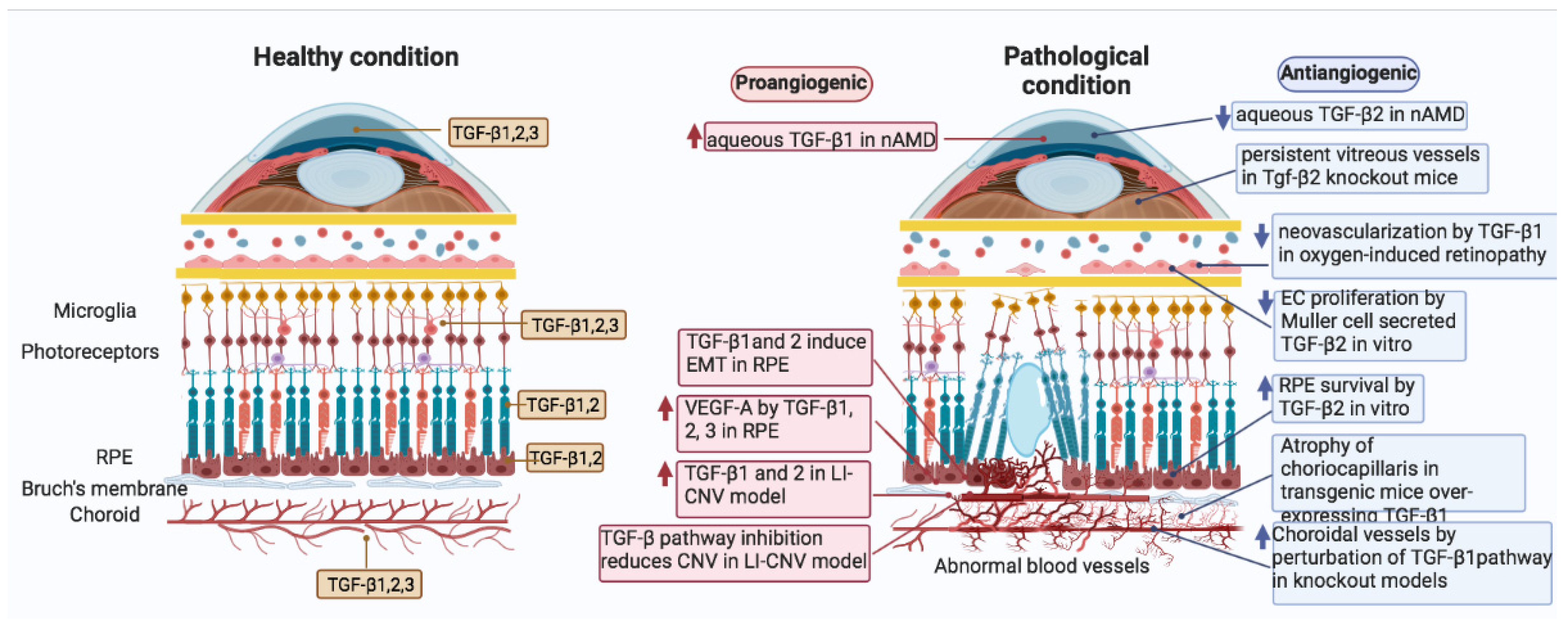
4. Expression and Distribution of TGF-β Family Members in the Eye

{kind=link}
{kind=link}
{kind=link}
| Gene | Human/Animal Model | Ocular Pathologies | References |
|---|---|---|---|
| TGF-β1↑ | In human plasma | Primary open-angle glaucoma | [137] |
| TGF-β1↑ | In human conjunctiva and minor salivary glands | Inflammatory ocular surface [138] | |
| TGF-β2↑ | In human aqueous humor | Proliferative vitreoretinopathy | [114] |
| TGF-β2↑ | In human vitreous | Diabetic retinopathy | [113] |
| TGF-β2↑ | In human aqueous humor | Open-angle glaucoma + increase of intraocular pressure in a glaucomatous eye | [139] |
| Activin A↑ | In human vitreous specimens obtained from eyes with retinal ischemia | Regulation of angiogenesis and tissue fibrosis | [136] |
| BMP4↑ | In adult retinal pigment epithelium-19 (ARPE-19) cells | Ocular angiogenesis associated with diabetic retinopathy via stimulation of VEGF by RPE cells | [131] |
| Loss of SMAD3 | Human RPE-cell | Attenuation of PVR development | [126] |
| TGF-β1↑ | In lens epithelium in mice | EMT-related fibrosis in lens epithelium | [126] |
| TGF-β1↑ | In transgenic mice | Cataracts in the lens epithelial cells in association with EMT and accumulation of fibrous/collagenous extracellular matrix | [140] |
| Over-expression of TGF-β1 | α-crystalline promoter in TGF-β2-null mice | Inhibition of abnormalities in ocular development (caused by the deletion of TGFβ2) | [141] |
| TGF-β2↓ | In mouse embryo lacking TGF-β2 | Loss of the corneal endothelium and anterior chamber, immature retina, and persistent vitreous vessels | [142] |
| Administration of anti-TGF-β2 neutralizing antibody | In mouse lens epithelium | Suppression of SMAD2/3 nuclear translocation following cataract surgery | [143] |
| BMP antagonist noggin | In chicken embryo lenses | Increase of cell death in lens epithelium | [144] |
| BMP4↓ | In embryos lacking BMP4 | Involvement in eye development | [145,146] |
| BMP4 antagonist ventroptin | In the chick eye | Alteration of several genes’ expression in the retina | [147] |
| Heterozygous deficiency of BMP4 | In mice | Elevated intraocular pressure and optic nerve abnormalities | [130] |
| BMP7↓ | In embryos lacking BMP7 | Involvement in eye development | [145] |
| Adenoviral gene transfer of BMP7 | In mouse lens epithelium | Suppression of injury induced EMT of lens epithelial cells and sealing of the capsular break | [148] |
| Targeted deletion of the BMPRIb gene | In mice | Significant elevation of apoptosis in the inner retina during postnatal development | [149] |
| BMPR1A↓ | In lenses lacking Alk3 | Abnormal lens development | [150] |
| TGF-βRI/RII↑ | In the lens fibers of transgenic mice | Nuclear cataracts | [150] |
| TGF-βRII↑ | In mice | Corneal opacification | [151] |
| Blockade of TGF-β using an adenovirus expressing an entire ectodomain of the human type II TGF-β receptor | In mice | Inhibition of the process of cornea opacification, edema and angiogenesis | [151] |
| SMAD3-null mice | In mouse lens epithelium with corneal exposure to alkali | Severe intraocular inflammation | [126] |
| Mice lacking SMAD3 | In mice | Acceleration of cutaneous wound healing | |
| Loss of SMAD3 | In mice | Blocking of morphological changes of lens epithelium and the expression of the EMT markers | [152] |
| Loss of SMAD3 | In mice | Suppression of macrophage infiltration and growth factor expression associated to tissue destruction of the healing cornea | [152] |
| SMAD3 gene ablation | In mice | Attenuation of injury induced EMT of lens epithelial cells | [126] |
| Adenoviral gene introduction of cDNAs for SMAD7 | In lens epithelium in mice | Attenuation of injury induced EMT of the lens epithelium | [153] |
| SMAD7 gene introduction | In mice | Attenuation of PVR development | [126] |
| Topical administration of SMAD7 gene introduction | In mouse cornea | Suppression of scarring and neovascularization | [126] |
5. TGF-β in Ocular Pathologies
5.1. Age-Related Macular Degeneration (AMD)
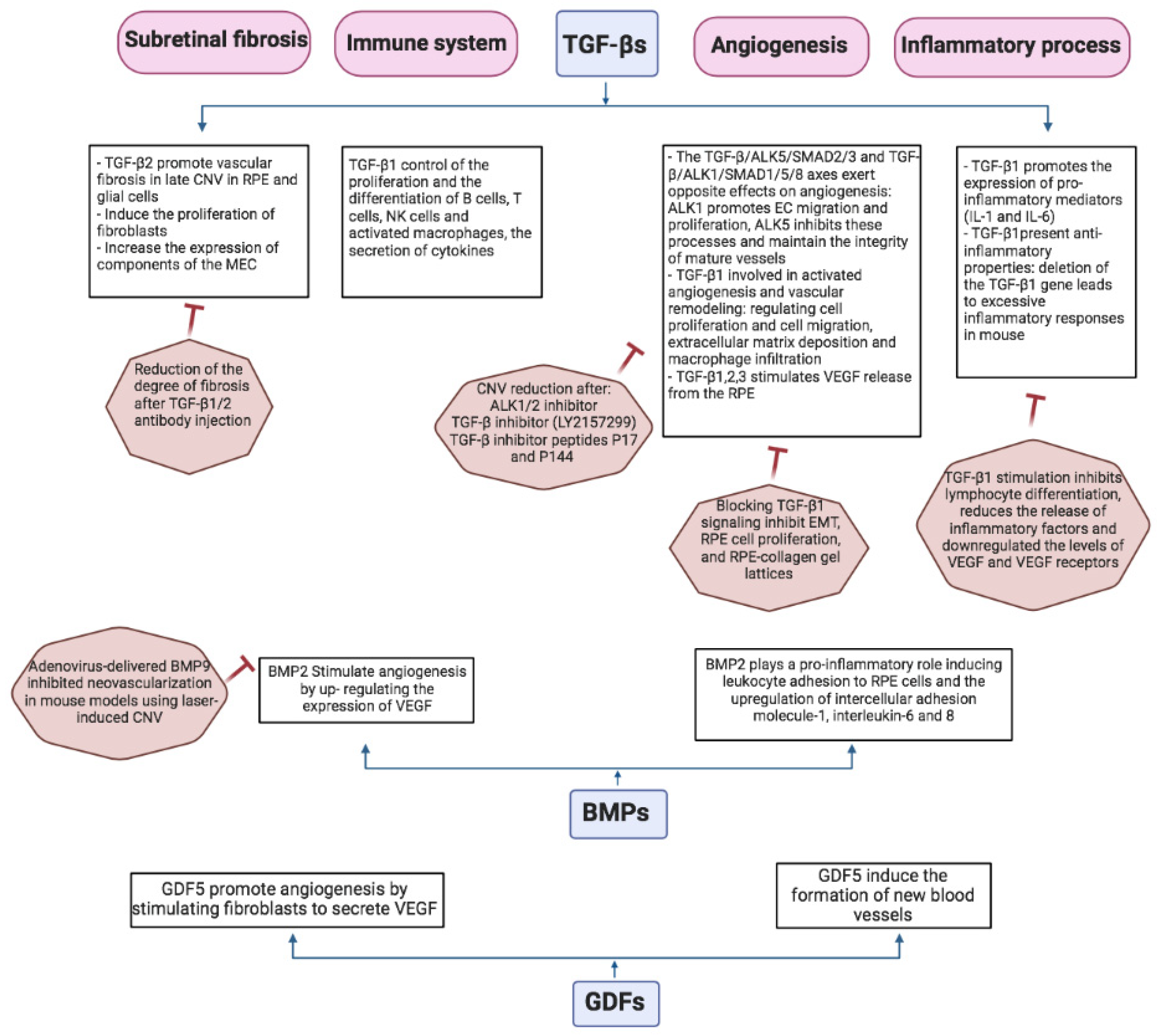
5.2. TGF-β Signaling in CNV Angiogenesis
5.3. TGF-β and Subretinal Fibrosis in Wet AMD
5.4. TGF-β Mediates Inflammatory Process Associated with Wet AMD
6. TGF-β in Other Ocular Pathologies
6.1. In Proliferative Vitreoretinopathy (PVR)
6.2. In Diabetic Retinopathy
6.3. In Corneal Injury
6.4. In Glaucoma
6.5. In Eye Tumors
7. Therapeutic Uses of TGF-β Signaling Inhibition
7.1. Targeting the TGF-β Pathway for Wet AMD
7.2. Targeting TGF-β Signaling in Glaucoma
7.3. Targeting TGF-β Signaling in Corneal Wound Healing
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Choudhary, M.; Malek, G. A Review of Pathogenic Drivers of Age-Related Macular Degeneration, Beyond Complement, with a Focus on Potential Endpoints for Testing Therapeutic Interventions in Preclinical Studies. Adv. Exp. Med. Biol. 2019, 1185, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [Green Version]
- El-Amir, A.N.; Sagoo, M.S.; da Cruz, L. Age-related macular degeneration. Br. J. Hosp. Med. 2005, 66, 677–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keane, P.A.; Sadda, S.R. Development of Anti-VEGF Therapies for Intraocular Use: A Guide for Clinicians. J. Ophthalmol. 2012, 2012, 483034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saint-Geniez, M.; Maharaj, A.S.; Walshe, T.E.; Tucker, B.A.; Sekiyama, E.; Kurihara, T.; Darland, D.C.; Young, M.J.; D’Amore, P.A. Endogenous VEGF is required for visual function: Evidence for a survival role on muller cells and photoreceptors. PLoS ONE 2008, 3, e3554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, A.W.; Bressler, S.B. Long-term follow-up of vascular endothelial growth factor inhibitor therapy for neovascular age-related macular degeneration. Curr. Opin. Ophthalmol. 2013, 24, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Nesmith, B.L.; Ihnen, M.; Schaal, S. Poor responders to bevacizumab pharmacotherapy in age-related macular degeneration and in diabetic macular edema demonstrate increased risk for obstructive sleep apnea. Retina 2014, 34, 2423–2430. [Google Scholar] [CrossRef] [PubMed]
- Santibanez, J.F.; Quintanilla, M.; Bernabeu, C. TGF-β/TGF-β receptor system and its role in physiological and pathological conditions. Clin. Sci. 2011, 121, 233–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, A.C.; Wrana, J.L. TGF-β Family Signaling in Embryonic and Somatic Stem-Cell Renewal and Differentiation. Cold Spring Harb. Perspect. Biol. 2017, 9, a022186. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Audiger, C.; Popovic, N.; Akla, N.; Lanthier, K.; Legault-Navarrete, I.; Melichar, H.; Costantino, S.; Lesage, S.; Larrivee, B. BMP9 signaling promotes the normalization of tumor blood vessels. Oncogene 2020, 39, 2996–3014. [Google Scholar] [CrossRef]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.Y.; Flavell, R.A. TGF-β and regulatory T cell in immunity and autoimmunity. J. Clin. Immunol. 2008, 28, 647–659. [Google Scholar] [CrossRef] [Green Version]
- Bressler, N.M. Antiangiogenic approaches to age-related macular degeneration today. Ophthalmology 2009, 116, S15–S23. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ma, W.; Han, S.; Meng, Z.; Zhao, L.; Yin, Y.; Wang, Y.; Li, J. TGF-β participates choroid neovascularization through Smad2/3-VEGF/TNF-alpha signaling in mice with Laser-induced wet age-related macular degeneration. Sci. Rep. 2017, 7, 9672. [Google Scholar] [CrossRef] [Green Version]
- Sporn, M.B. The early history of TGF-β, and a brief glimpse of its future. Cytokine Growth Factor Rev. 2006, 17, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Govinden, R.; Bhoola, K.D. Genealogy, expression, and cellular function of transforming growth factor-β. Pharmacol. Ther. 2003, 98, 257–265. [Google Scholar] [CrossRef]
- Schmierer, B.; Hill, C.S. TGFβ-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef]
- Matzuk, M.M.; Kumar, T.R.; Vassalli, A.; Bickenbach, J.R.; Roop, D.R.; Jaenisch, R.; Bradley, A. Functional analysis of activins during mammalian development. Nature 1995, 374, 354–356. [Google Scholar] [CrossRef] [PubMed]
- Munz, B.; Hubner, G.; Tretter, Y.; Alzheimer, C.; Werner, S. A novel role of activin in inflammation and repair. J. Endocrinol. 1999, 161, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Hinck, A.P.; Mueller, T.D.; Springer, T.A. Structural Biology and Evolution of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Schneyer, A.L. The biology of activin: Recent advances in structure, regulation and function. J. Endocrinol. 2009, 202, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellor, S.L.; Cranfield, M.; Ries, R.; Pedersen, J.; Cancilla, B.; de Kretser, D.; Groome, N.P.; Mason, A.J.; Risbridger, G.P. Localization of activin β(A)-, β(B)-, and β(C)-subunits in humanprostate and evidence for formation of new activin heterodimers of β(C)-subunit. J. Clin. Endocrinol. Metab. 2000, 85, 4851–4858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabicovsky, M.; Herkner, K.; Rossmanith, W. Overexpression of activin β(C) or activin β(E) in the mouse liver inhibits regenerative deoxyribonucleic acid synthesis of hepatic cells. Endocrinology 2003, 144, 3497–3504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Jiang, W.; Phillips, F.M.; Haydon, R.C.; Peng, Y.; Zhou, L.; Luu, H.H.; An, N.; Breyer, B.; Vanichakarn, P.; et al. Osteogenic activity of the fourteen types of human bone morphogenetic proteins (BMPs). J. Bone Jt. Surg. Am. 2003, 85, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Beederman, M.; Lamplot, J.D.; Nan, G.; Wang, J.; Liu, X.; Yin, L.; Li, R.; Shui, W.; Zhang, H.; Kim, S.H.; et al. BMP signaling in mesenchymal stem cell differentiation and bone formation. J. Biomed. Sci. Eng. 2013, 6, 32–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.S.; Akhtar, N.; Jamil, H.M.; Banik, R.S.; Asaduzzaman, S.M. TGF-β/BMP signaling and other molecular events: Regulation of osteoblastogenesis and bone formation. Bone Res. 2015, 3, 15005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Q.; Sun, M.H.; Cheng, H.; Peng, Y.; Montag, A.G.; Deyrup, A.T.; Jiang, W.; Luu, H.H.; Luo, J.; Szatkowski, J.P.; et al. Characterization of the distinct orthotopic bone-forming activity of 14 BMPs using recombinant adenovirus-mediated gene delivery. Gene. Ther. 2004, 11, 1312–1320. [Google Scholar] [CrossRef] [Green Version]
- Luther, G.; Wagner, E.R.; Zhu, G.; Kang, Q.; Luo, Q.; Lamplot, J.; Bi, Y.; Luo, X.; Luo, J.; Teven, C.; et al. BMP-9 induced osteogenic differentiation of mesenchymal stem cells: Molecular mechanism and therapeutic potential. Curr. Gene. Ther. 2011, 11, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Schier, A.F. Nodal morphogens. Cold Spring Harb. Perspect. Biol. 2009, 1, a003459. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, O.; Cunha, G.R.; Lawrence, W.D.; Robboy, S.J. Timing and irreversibility of Mullerian duct inhibition in the embryonic reproductive tract of the human male. Dev. Biol. 1984, 106, 394–398. [Google Scholar] [CrossRef]
- Tzavlaki, K.; Moustakas, A. TGF-β Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Haim, N.; Lu, C.; Guzman-Ayala, M.; Pescatore, L.; Mesnard, D.; Bischofberger, M.; Naef, F.; Robertson, E.J.; Constam, D.B. The nodal precursor acting via activin receptors induces mesoderm by maintaining a source of its convertases and BMP4. Dev. Cell 2006, 11, 313–323. [Google Scholar] [CrossRef]
- Ge, G.; Hopkins, D.R.; Ho, W.B.; Greenspan, D.S. GDF11 forms a bone morphogenetic protein 1-activated latent complex that can modulate nerve growth factor-induced differentiation of PC12 cells. Mol. Cell Biol. 2005, 25, 5846–5858. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Massague, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Massague, J.; Gomis, R.R. The logic of TGFβ signaling. FEBS Lett. 2006, 580, 2811–2820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Chen, Y.G.; Ozdamar, B.; Gyuricza, C.A.; Chong, P.A.; Wrana, J.L.; Massague, J.; Shi, Y. Structural basis of Smad2 recognition by the Smad anchor for receptor activation. Science 2000, 287, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Chen, Y.G. Controlling TGF-β signaling. Genes Dev. 2000, 14, 627–644. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.; Hill, C.S. How the Smads regulate transcription. Int. J. Biochem. Cell Biol. 2008, 40, 383–408. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Vanlandewijck, M.; Moustakas, A. Regulation of EMT by TGFβ in cancer. FEBS Lett. 2012, 586, 1959–1970. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Chen, Y.G. Regulation of TGF-β Signal Transduction. Cell Biosci. 2012, 2, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Gatza, C.E.; Oh, S.Y.; Blobe, G.C. Roles for the type III TGF-β receptor in human cancer. Cell Signal. 2010, 22, 1163–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andres, J.L.; Stanley, K.; Cheifetz, S.; Massague, J. Membrane-anchored and soluble forms of betaglycan, a polymorphic proteoglycan that binds transforming growth factor-beta. J. Cell Biol. 1989, 109, 3137–3145. [Google Scholar] [CrossRef]
- Blanco, F.J.; Santibanez, J.F.; Guerrero-Esteo, M.; Langa, C.; Vary, C.P.; Bernabeu, C. Interaction and functional interplay between endoglin and ALK-1, two components of the endothelial transforming growth factor-β receptor complex. J. Cell Physiol. 2005, 204, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Baeyens, N.; Larrivee, B.; Ola, R.; Hayward-Piatkowskyi, B.; Dubrac, A.; Huang, B.; Ross, T.D.; Coon, B.G.; Min, E.; Tsarfati, M.; et al. Defective fluid shear stress mechanotransduction mediates hereditary hemorrhagic telangiectasia. J. Cell Biol. 2016, 214, 807–816. [Google Scholar] [CrossRef] [Green Version]
- Sasai, Y.; De Robertis, E.M. Ectodermal patterning in vertebrate embryos. Dev. Biol. 1997, 182, 5–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammerschmidt, M.; Serbedzija, G.N.; McMahon, A.P. Genetic analysis of dorsoventral pattern formation in the zebrafish: Requirement of a BMP-like ventralizing activity and its dorsal repressor. Genes Dev. 1996, 10, 2452–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrighton, K.H.; Lin, X.; Feng, X.H. Critical regulation of TGFβ signaling by Hsp90. Proc. Natl. Acad. Sci. USA 2008, 105, 9244–9249. [Google Scholar] [CrossRef] [Green Version]
- Tosi, G.M.; Regoli, M.; Altera, A.; Galvagni, F.; Arcuri, C.; Bacci, T.; Elia, I.; Realini, G.; Orlandini, M.; Bertelli, E. Heat Shock Protein 90 Involvement in the Development of Idiopathic Epiretinal Membranes. Investig. Ophthalmol. Vis. Sci. 2020, 61, 34. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Kulkarni, A.B. Extracellular heat shock protein HSP90β secreted by MG63 osteosarcoma cells inhibits activation of latent TGF-β1. Biochem. Biophys. Res. Commun. 2010, 398, 525–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Mare, J.A.; Jurgens, T.; Edkins, A.L. Extracellular Hsp90 and TGFβ regulate adhesion, migration and anchorage independent growth in a paired colon cancer cell line model. BMC Cancer 2017, 17, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ten Dijke, P.; Goumans, M.J.; Itoh, F.; Itoh, S. Regulation of cell proliferation by Smad proteins. J. Cell Physiol. 2002, 191, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.P.; Seki, T.; Goss, K.A.; Imamura, T.; Yi, Y.; Donahoe, P.K.; Li, L.; Miyazono, K.; ten Dijke, P.; Kim, S.; et al. Activin receptor-like kinase 1 modulates transforming growth factor-β 1 signaling in the regulation of angiogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 2626–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goumans, M.J.; Liu, Z.; ten Dijke, P. TGF-β signaling in vascular biology and dysfunction. Cell Res. 2009, 19, 116–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardali, E.; Goumans, M.J.; ten Dijke, P. Signaling by members of the TGF-β family in vascular morphogenesis and disease. Trends Cell Biol. 2010, 20, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Walshe, T.E.; Leach, L.L.; D’Amore, P.A. TGF-β signaling is required for maintenance of retinal ganglion cell differentiation and survival. Neuroscience 2011, 189, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, C.M.; Darland, D.C.; Massingham, L.J.; D’Amore, P.A. Endothelial cell-astrocyte interactions and TGF β are required for induction of blood-neural barrier properties. Dev. Brain Res. 2004, 152, 25–38. [Google Scholar] [CrossRef]
- Khan, Z.A.; Chakrabarti, S. Growth factors in proliferative diabetic retinopathy. Exp. Diabesity Res. 2003, 4, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Nagineni, C.N.; Samuel, W.; Nagineni, S.; Pardhasaradhi, K.; Wiggert, B.; Detrick, B.; Hooks, J.J. Transforming growth factor-β induces expression of vascular endothelial growth factor in human retinal pigment epithelial cells: Involvement of mitogen-activated protein kinases. J. Cell Physiol. 2003, 197, 453–462. [Google Scholar] [CrossRef]
- Nagineni, C.N.; Cherukuri, K.S.; Kutty, V.; Detrick, B.; Hooks, J.J. Interferon-gamma differentially regulates TGF-β1 and TGF-β2 expression in human retinal pigment epithelial cells through JAK-STAT pathway. J. Cell Physiol. 2007, 210, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Mummery, C. Functional analysis of the TGFβ receptor/Smad pathway through gene ablation in mice. Int. J. Dev. Biol. 2000, 44, 253–265. [Google Scholar] [PubMed]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; ten Dijke, P. Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- McAllister, K.A.; Grogg, K.M.; Johnson, D.W.; Gallione, C.J.; Baldwin, M.A.; Jackson, C.E.; Helmbold, E.A.; Markel, D.S.; McKinnon, W.C.; Murrell, J.; et al. Endoglin, a TGF-β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 1994, 8, 345–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayrak-Toydemir, P.; McDonald, J.; Akarsu, N.; Toydemir, R.M.; Calderon, F.; Tuncali, T.; Tang, W.; Miller, F.; Mao, R. A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am. J. Med. Genet. A 2006, 140, 2155–2162. [Google Scholar] [CrossRef]
- Park, S.O.; Wankhede, M.; Lee, Y.J.; Choi, E.J.; Fliess, N.; Choe, S.W.; Oh, S.H.; Walter, G.; Raizada, M.K.; Sorg, B.S.; et al. Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J. Clin. Investig. 2009, 119, 3487–3496. [Google Scholar] [CrossRef]
- Jakobsson, L.; van Meeteren, L.A. Transforming growth factor β family members in regulation of vascular function: In the light of vascular conditional knockouts. Exp. Cell Res. 2013, 319, 1264–1270. [Google Scholar] [CrossRef]
- Watabe, T.; Nishihara, A.; Mishima, K.; Yamashita, J.; Shimizu, K.; Miyazawa, K.; Nishikawa, S.; Miyazono, K. TGF-β receptor kinase inhibitor enhances growth and integrity of embryonic stem cell-derived endothelial cells. J. Cell Biol. 2003, 163, 1303–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birukova, A.A.; Adyshev, D.; Gorshkov, B.; Birukov, K.G.; Verin, A.D. ALK5 and Smad4 are involved in TGF-β1-induced pulmonary endothelial permeability. FEBS Lett. 2005, 579, 4031–4037. [Google Scholar] [CrossRef] [Green Version]
- Seki, T.; Hong, K.H.; Oh, S.P. Nonoverlapping expression patterns of ALK1 and ALK5 reveal distinct roles of each receptor in vascular development. Lab. Investig. 2006, 86, 116–129. [Google Scholar] [CrossRef]
- Zarkada, G.; Howard, J.P.; Xiao, X.; Park, H.; Bizou, M.; Leclerc, S.; Kunzel, S.E.; Boisseau, B.; Li, J.; Cagnone, G.; et al. Specialized endothelial tip cells guide neuroretina vascularization and blood-retina-barrier formation. Dev. Cell 2021, 56, 2237–2251.e6. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Yang, X.; Friesel, R.E.; Vary, C.P.; Liaw, L. Mechanisms of TGF-β-induced differentiation in human vascular smooth muscle cells. J. Vasc. Res. 2011, 48, 485–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebrin, F.; Goumans, M.J.; Jonker, L.; Carvalho, R.L.; Valdimarsdottir, G.; Thorikay, M.; Mummery, C.; Arthur, H.M.; ten Dijke, P. Endoglin promotes endothelial cell proliferation and TGF-β/ALK1 signal transduction. EMBO J. 2004, 23, 4018–4028. [Google Scholar] [CrossRef] [Green Version]
- Fonsatti, E.; Altomonte, M.; Nicotra, M.R.; Natali, P.G.; Maio, M. Endoglin (CD105): A powerful therapeutic target on tumor-associated angiogenetic blood vessels. Oncogene 2003, 22, 6557–6563. [Google Scholar] [CrossRef] [Green Version]
- Larrivee, B.; Prahst, C.; Gordon, E.; del Toro, R.; Mathivet, T.; Duarte, A.; Simons, M.; Eichmann, A. ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev. Cell 2012, 22, 489–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Llorente, L.; Gallardo-Vara, E.; Rossi, E.; Smadja, D.M.; Botella, L.M.; Bernabeu, C. Endoglin and alk1 as therapeutic targets for hereditary hemorrhagic telangiectasia. Expert Opin. Ther. Targets 2017, 21, 933–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorai, H.; Vukicevic, S.; Sampath, T.K. Bone morphogenetic protein-7 (osteogenic protein-1) inhibits smooth muscle cell proliferation and stimulates the expression of markers that are characteristic of SMC phenotype in vitro. J. Cell Physiol. 2000, 184, 37–45. [Google Scholar] [CrossRef]
- Hansmann, G.; de Jesus Perez, V.A.; Alastalo, T.P.; Alvira, C.M.; Guignabert, C.; Bekker, J.M.; Schellong, S.; Urashima, T.; Wang, L.; Morrell, N.W.; et al. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J. Clin. Investig. 2008, 118, 1846–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y.; Montagne, K.; Nishihara, A.; Watabe, T.; Miyazono, K. BMPs promote proliferation and migration of endothelial cells via stimulation of VEGF-A/VEGFR2 and angiopoietin-1/Tie2 signalling. J. Biochem. 2008, 143, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Goldman, D.C.; Donley, N.; Christian, J.L. Genetic interaction between Bmp2 and Bmp4 reveals shared functions during multiple aspects of mouse organogenesis. Mech. Dev. 2009, 126, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdimarsdottir, G.; Goumans, M.J.; Rosendahl, A.; Brugman, M.; Itoh, S.; Lebrin, F.; Sideras, P.; ten Dijke, P. Stimulation of Id1 expression by bone morphogenetic protein is sufficient and necessary for bone morphogenetic protein-induced activation of endothelial cells. Circulation 2002, 106, 2263–2270. [Google Scholar] [CrossRef] [PubMed]
- David, L.; Feige, J.J.; Bailly, S. Emerging role of bone morphogenetic proteins in angiogenesis. Cytokine Growth Factor Rev. 2009, 20, 203–212. [Google Scholar] [CrossRef]
- David, L.; Mallet, C.; Mazerbourg, S.; Feige, J.J.; Bailly, S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 2007, 109, 1953–1961. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Brady Ridgway, J.; Sai, T.; Lai, J.; Warming, S.; Chen, H.; Roose-Girma, M.; Zhang, G.; Shou, W.; Yan, M. Context-dependent signaling defines roles of BMP9 and BMP10 in embryonic and postnatal development. Proc. Natl. Acad. Sci. USA 2013, 110, 11887–11892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goumans, M.J.; Zwijsen, A.; Ten Dijke, P.; Bailly, S. Bone Morphogenetic Proteins in Vascular Homeostasis and Disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a031989. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Lebrin, F.; Larsson, J.; Mummery, C.; Karlsson, S.; ten Dijke, P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFβ/ALK5 signaling. Mol. Cell 2003, 12, 817–828. [Google Scholar] [CrossRef]
- Itoh, F.; Itoh, S.; Adachi, T.; Ichikawa, K.; Matsumura, Y.; Takagi, T.; Festing, M.; Watanabe, T.; Weinstein, M.; Karlsson, S.; et al. Smad2/Smad3 in endothelium is indispensable for vascular stability via S1PR1 and N-cadherin expressions. Blood 2012, 119, 5320–5328. [Google Scholar] [CrossRef] [Green Version]
- Ricard, N.; Ciais, D.; Levet, S.; Subileau, M.; Mallet, C.; Zimmers, T.A.; Lee, S.J.; Bidart, M.; Feige, J.J.; Bailly, S. BMP9 and BMP10 are critical for postnatal retinal vascular remodeling. Blood 2012, 119, 6162–6171. [Google Scholar] [CrossRef] [Green Version]
- Cunha, S.I.; Pardali, E.; Thorikay, M.; Anderberg, C.; Hawinkels, L.; Goumans, M.J.; Seehra, J.; Heldin, C.H.; ten Dijke, P.; Pietras, K. Genetic and pharmacological targeting of activin receptor-like kinase 1 impairs tumor growth and angiogenesis. J. Exp. Med. 2010, 207, 85–100. [Google Scholar] [CrossRef] [Green Version]
- Garrido-Martin, E.M.; Blanco, F.J.; Roque, M.; Novensa, L.; Tarocchi, M.; Lang, U.E.; Suzuki, T.; Friedman, S.L.; Botella, L.M.; Bernabeu, C. Vascular injury triggers Kruppel-like factor 6 mobilization and cooperation with specificity protein 1 to promote endothelial activation through upregulation of the activin receptor-like kinase 1 gene. Circ. Res. 2013, 112, 113–127. [Google Scholar] [CrossRef]
- Tual-Chalot, S.; Garcia-Collado, M.; Redgrave, R.E.; Singh, E.; Davison, B.; Park, C.; Lin, H.; Luli, S.; Jin, Y.; Wang, Y.; et al. Loss of Endothelial Endoglin Promotes High-Output Heart Failure Through Peripheral Arteriovenous Shunting Driven by VEGF Signaling. Circ. Res. 2020, 126, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Kehrl, J.H.; Wakefield, L.M.; Roberts, A.B.; Jakowlew, S.; Alvarez-Mon, M.; Derynck, R.; Sporn, M.B.; Fauci, A.S. Production of transforming growth factor β by human T lymphocytes and its potential role in the regulation of T cell growth. J. Exp. Med. 1986, 163, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Sillett, H.K.; Cruickshank, S.M.; Southgate, J.; Trejdosiewicz, L.K. Transforming growth factor-β promotes ‘death by neglect’ in post-activated human T cells. Immunology 2001, 102, 310–316. [Google Scholar] [CrossRef]
- Littman, D.R.; Rudensky, A.Y. Th17 and regulatory T cells in mediating and restraining inflammation. Cell 2010, 140, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Travis, M.A.; Sheppard, D. TGF-β activation and function in immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 2004, 22, 531–562. [Google Scholar] [CrossRef] [PubMed]
- Pyzik, M.; Piccirillo, C.A. TGF-β1 modulates Foxp3 expression and regulatory activity in distinct CD4+ T cell subsets. J. Leukoc. Biol. 2007, 82, 335–346. [Google Scholar] [CrossRef]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limon, P. The polarization of immune cells in the tumour environment by TGFβ. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Nickel, J.; Ten Dijke, P.; Mueller, T.D. TGF-β family co-receptor function and signaling. Acta Biochim. Biophys. Sin. 2018, 50, 12–36. [Google Scholar] [CrossRef] [Green Version]
- Kehrl, J.H.; Roberts, A.B.; Wakefield, L.M.; Jakowlew, S.; Sporn, M.B.; Fauci, A.S. Transforming growth factor β is an important immunomodulatory protein for human B lymphocytes. J. Immunol. 1986, 137, 3855–3860. [Google Scholar] [PubMed]
- Stavnezer, J.; Kang, J. The surprising discovery that TGF β specifically induces the IgA class switch. J. Immunol. 2009, 182, 5–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strober, W. Regulation of IgA B-cell development in the mucosal immune system. J. Clin. Immunol. 1990, 10, 56S–61S, discussion 61S–63S. [Google Scholar] [CrossRef] [PubMed]
- Coffman, R.L.; Lebman, D.A.; Shrader, B. Transforming growth factor β specifically enhances IgA production by lipopolysaccharide-stimulated murine B lymphocytes. J. Exp. Med. 1989, 170, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Winnier, G.; Blessing, M.; Labosky, P.A.; Hogan, B.L. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 1995, 9, 2105–2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleul, C.C.; Boehm, T. BMP signaling is required for normal thymus development. J. Immunol. 2005, 175, 5213–5221. [Google Scholar] [CrossRef] [Green Version]
- Licona-Limon, P.; Soldevila, G. The role of TGF-β superfamily during T cell development: New insights. Immunol. Lett. 2007, 109, 1–12. [Google Scholar] [CrossRef]
- Martinez, V.G.; Sacedon, R.; Hidalgo, L.; Valencia, J.; Fernandez-Sevilla, L.M.; Hernandez-Lopez, C.; Vicente, A.; Varas, A. The BMP Pathway Participates in Human Naive CD4+ T Cell Activation and Homeostasis. PLoS ONE 2015, 10, e0131453. [Google Scholar] [CrossRef] [Green Version]
- Gu, A.D.; Wang, Y.; Lin, L.; Zhang, S.S.; Wan, Y.Y. Requirements of transcription factor Smad-dependent and -independent TGF-β signaling to control discrete T-cell functions. Proc. Natl. Acad. Sci. USA 2012, 109, 905–910. [Google Scholar] [CrossRef] [Green Version]
- Takimoto, T.; Wakabayashi, Y.; Sekiya, T.; Inoue, N.; Morita, R.; Ichiyama, K.; Takahashi, R.; Asakawa, M.; Muto, G.; Mori, T.; et al. Smad2 and Smad3 are redundantly essential for the TGF-β-mediated regulation of regulatory T plasticity and Th1 development. J. Immunol. 2010, 185, 842–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakao, A.; Miike, S.; Hatano, M.; Okumura, K.; Tokuhisa, T.; Ra, C.; Iwamoto, I. Blockade of transforming growth factor β/Smad signaling in T cells by overexpression of Smad7 enhances antigen-induced airway inflammation and airway reactivity. J. Exp. Med. 2000, 192, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darland, D.C.; Link, B.A.; Nishi, R. Activin A and follistatin expression in developing targets of ciliary ganglion neurons suggests a role in regulating neurotransmitter phenotype. Neuron 1995, 15, 857–866. [Google Scholar] [CrossRef] [Green Version]
- Hirase, K.; Ikeda, T.; Sotozono, C.; Nishida, K.; Sawa, H.; Kinoshita, S. Transforming growth factor β2 in the vitreous in proliferative diabetic retinopathy. Arch. Ophthalmol. 1998, 116, 738–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connor, T.B., Jr.; Roberts, A.B.; Sporn, M.B.; Danielpour, D.; Dart, L.L.; Michels, R.G.; de Bustros, S.; Enger, C.; Kato, H.; Lansing, M.; et al. Correlation of fibrosis and transforming growth factor-β type 2 levels in the eye. J. Clin. Investig. 1989, 83, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, B.A.; Flanders, K.C.; Guerin, C.J.; Danielpour, D.; Anderson, D.H. Transforming growth factor β 2 is the predominant isoform in the neural retina, retinal pigment epithelium-choroid and vitreous of the monkey eye. Exp. Eye Res. 1994, 59, 323–333. [Google Scholar] [CrossRef]
- Luo, G.; Hofmann, C.; Bronckers, A.L.; Sohocki, M.; Bradley, A.; Karsenty, G. BMP-7 is an inducer of nephrogenesis, and is also required for eye development and skeletal patterning. Genes Dev. 1995, 9, 2808–2820. [Google Scholar] [CrossRef] [Green Version]
- Nishida, K.; Kinoshita, S.; Yokoi, N.; Kaneda, M.; Hashimoto, K.; Yamamoto, S. Immunohistochemical localization of transforming growth factor-β 1, -β 2, and -β 3 latency-associated peptide in human cornea. Investig. Ophthalmol. Vis. Sci 1994, 35, 3289–3294. [Google Scholar]
- Nishida, K.; Sotozono, C.; Adachi, W.; Yamamoto, S.; Yokoi, N.; Kinoshita, S. Transforming growth factor-β 1, -β 2 and -β 3 mRNA expression in human cornea. Curr. Eye Res. 1995, 14, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Kokawa, N.; Sotozono, C.; Nishida, K.; Kinoshita, S. High total TGF-β 2 levels in normal human tears. Curr. Eye Res. 1996, 15, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Pasquale, L.R.; Dorman-Pease, M.E.; Lutty, G.A.; Quigley, H.A.; Jampel, H.D. Immunolocalization of TGF-β 1, TGF-β 2, and TGF-β 3 in the anterior segment of the human eye. Investig. Ophthalmol. Vis. Sci 1993, 34, 23–30. [Google Scholar]
- Lutty, G.A.; Merges, C.; Threlkeld, A.B.; Crone, S.; McLeod, D.S. Heterogeneity in localization of isoforms of TGF-β in human retina, vitreous, and choroid. Investig. Ophthalmol. Vis. Sci. 1993, 34, 477–487. [Google Scholar]
- Tosi, G.M.; Orlandini, M.; Galvagni, F. The Controversial Role of TGF-β in Neovascular Age-Related Macular Degeneration Pathogenesis. Int. J. Mol. Sci. 2018, 19, 3363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukhikh, G.T.; Panova, I.G.; Smirnova, Y.A.; Milyushina, L.A.; Firsova, N.V.; Markitantova, Y.V.; Poltavtseva, R.A.; Zinov’eva, R.D. Expression of transforming growth factor-β2in vitreous body and adjacent tissues during prenatal development of human eye. Bull. Exp. Biol. Med. 2010, 150, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Tanihara, H.; Yoshida, M.; Matsumoto, M.; Yoshimura, N. Identification of transforming growth factor-β expressed in cultured human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 1993, 34, 413–419. [Google Scholar]
- Hirsch, L.; Nazari, H.; Sreekumar, P.G.; Kannan, R.; Dustin, L.; Zhu, D.; Barron, E.; Hinton, D.R. TGF-β2 secretion from RPE decreases with polarization and becomes apically oriented. Cytokine 2015, 71, 394–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saika, S. TGFβ pathobiology in the eye. Lab. Investig. 2006, 86, 106–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jobling, A.I.; Nguyen, M.; Gentle, A.; McBrien, N.A. Isoform-specific changes in scleral transforming growth factor-β expression and the regulation of collagen synthesis during myopia progression. J. Biol. Chem. 2004, 279, 18121–18126. [Google Scholar] [CrossRef] [Green Version]
- You, L.; Kruse, F.E.; Pohl, J.; Volcker, H.E. Bone morphogenetic proteins and growth and differentiation factors in the human cornea. Investig. Ophthalmol. Vis. Sci. 1999, 40, 296–311. [Google Scholar]
- Ebendal, T.; Bengtsson, H.; Soderstrom, S. Bone morphogenetic proteins and their receptors: Potential functions in the brain. J. Neurosci. Res. 1998, 51, 139–146. [Google Scholar] [CrossRef]
- Chang, B.; Smith, R.S.; Peters, M.; Savinova, O.V.; Hawes, N.L.; Zabaleta, A.; Nusinowitz, S.; Martin, J.E.; Davisson, M.L.; Cepko, C.L.; et al. Haploinsufficient Bmp4 ocular phenotypes include anterior segment dysgenesis with elevated intraocular pressure. BMC Genet. 2001, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Vogt, R.R.; Unda, R.; Yeh, L.C.; Vidro, E.K.; Lee, J.C.; Tsin, A.T. Bone morphogenetic protein-4 enhances vascular endothelial growth factor secretion by human retinal pigment epithelial cells. J. Cell Biochem. 2006, 98, 1196–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathura, J.R., Jr.; Jafari, N.; Chang, J.T.; Hackett, S.F.; Wahlin, K.J.; Della, N.G.; Okamoto, N.; Zack, D.J.; Campochiaro, P.A. Bone morphogenetic proteins-2 and -4: Negative growth regulators in adult retinal pigmented epithelium. Investig. Ophthalmol. Vis. Sci. 2000, 41, 592–600. [Google Scholar]
- Shen, W.; Finnegan, S.; Lein, P.; Sullivan, S.; Slaughter, M.; Higgins, D. Bone morphogenetic proteins regulate ionotropic glutamate receptors in human retina. Eur. J. Neurosci. 2004, 20, 2031–2037. [Google Scholar] [CrossRef] [PubMed]
- Wordinger, R.J.; Agarwal, R.; Talati, M.; Fuller, J.; Lambert, W.; Clark, A.F. Expression of bone morphogenetic proteins (BMP), BMP receptors, and BMP associated proteins in human trabecular meshwork and optic nerve head cells and tissues. Mol. Vis. 2002, 8, 241–250. [Google Scholar] [PubMed]
- Belecky-Adams, T.L.; Scheurer, D.; Adler, R. Activin family members in the developing chick retina: Expression patterns, protein distribution, and in vitro effects. Dev. Biol. 1999, 210, 107–123. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Takeuchi, S.; Suzuki, K.; Yamashita, H. Expression and possible roles of activin A in proliferative vitreoretinal diseases. Jpn. J. Ophthalmol. 2000, 44, 221–226. [Google Scholar] [CrossRef]
- Kuchtey, J.; Kunkel, J.; Burgess, L.G.; Parks, M.B.; Brantley, M.A., Jr.; Kuchtey, R.W. Elevated transforming growth factor β1 in plasma of primary open-angle glaucoma patients. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5291–5297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Paiva, C.S.; Volpe, E.A.; Gandhi, N.B.; Zhang, X.; Zheng, X.; Pitcher, J.D., 3rd; Farley, W.J.; Stern, M.E.; Niederkorn, J.Y.; Li, D.Q.; et al. Disruption of TGF-β signaling improves ocular surface epithelial disease in experimental autoimmune keratoconjunctivitis sicca. PLoS ONE 2011, 6, e29017. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Daher, A.M.; Agarwal, R. Aqueous humor TGF-β2 levels in patients with open-angle glaucoma: A meta-analysis. Mol. Vis. 2015, 21, 612–620. [Google Scholar]
- Srinivasan, Y.; Lovicu, F.J.; Overbeek, P.A. Lens-specific expression of transforming growth factor β1 in transgenic mice causes anterior subcapsular cataracts. J. Clin. Investig. 1998, 101, 625–634. [Google Scholar] [CrossRef]
- Zhao, S.; Overbeek, P.A. Elevated TGFβ signaling inhibits ocular vascular development. Dev. Biol. 2001, 237, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFβ2 knockout mice have multiple developmental defects that are non-overlapping with other TGFβ knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [CrossRef] [PubMed]
- Saika, S.; Okada, Y.; Miyamoto, T.; Ohnishi, Y.; Ooshima, A.; McAvoy, J.W. Smad translocation and growth suppression in lens epithelial cells by endogenous TGFβ2 during wound repair. Exp. Eye Res. 2001, 72, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Wordinger, R.J.; Clark, A.F. Bone morphogenetic proteins and their receptors in the eye. Exp. Biol. Med. 2007, 232, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Dudley, A.T.; Lyons, K.M.; Robertson, E.J. A requirement for bone morphogenetic protein-7 during development of the mammalian kidney and eye. Genes Dev. 1995, 9, 2795–2807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, Y.; Hogan, B.L. BMP4 is essential for lens induction in the mouse embryo. Genes Dev. 1998, 12, 3764–3775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakuta, H.; Suzuki, R.; Takahashi, H.; Kato, A.; Shintani, T.; Iemura, S.; Yamamoto, T.S.; Ueno, N.; Noda, M. Ventroptin: A BMP-4 antagonist expressed in a double-gradient pattern in the retina. Science 2001, 293, 111–115. [Google Scholar] [CrossRef]
- Saika, S.; Ikeda, K.; Yamanaka, O.; Flanders, K.C.; Ohnishi, Y.; Nakajima, Y.; Muragaki, Y.; Ooshima, A. Adenoviral gene transfer of BMP-7, Id2, or Id3 suppresses injury-induced epithelial-to-mesenchymal transition of lens epithelium in mice. Am. J. Physiol.-Cell Physiol. 2006, 290, C282–C289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Wilson, S.; Reh, T. BMP receptor 1b is required for axon guidance and cell survival in the developing retina. Dev. Biol. 2003, 256, 34–48. [Google Scholar] [CrossRef] [Green Version]
- Beebe, D.; Garcia, C.; Wang, X.; Rajagopal, R.; Feldmeier, M.; Kim, J.Y.; Chytil, A.; Moses, H.; Ashery-Padan, R.; Rauchman, M. Contributions by members of the TGFβ superfamily to lens development. Int. J. Dev. Biol. 2004, 48, 845–856. [Google Scholar] [CrossRef]
- Sakamoto, T.; Ueno, H.; Sonoda, K.; Hisatomi, T.; Shimizu, K.; Ohashi, H.; Inomata, H. Blockade of TGF-β by in vivo gene transfer of a soluble TGF-β type II receptor in the muscle inhibits corneal opacification, edema and angiogenesis. Gene Ther. 2000, 7, 1915–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saika, S.; Ikeda, K.; Yamanaka, O.; Miyamoto, T.; Ohnishi, Y.; Sato, M.; Muragaki, Y.; Ooshima, A.; Nakajima, Y.; Kao, W.W.; et al. Expression of Smad7 in mouse eyes accelerates healing of corneal tissue after exposure to alkali. Am. J. Pathol. 2005, 166, 1405–1418. [Google Scholar] [CrossRef] [Green Version]
- Saika, S.; Ikeda, K.; Yamanaka, O.; Sato, M.; Muragaki, Y.; Ohnishi, Y.; Ooshima, A.; Nakajima, Y.; Namikawa, K.; Kiyama, H.; et al. Transient adenoviral gene transfer of Smad7 prevents injury-induced epithelial-mesenchymal transition of lens epithelium in mice. Lab. Investig. 2004, 84, 1259–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraccaro, P.; Nicolo, M.; Bonetto, M.; Giacomini, M.; Weller, P.; Traverso, C.E.; Prosperi, M.; OSullivan, D. Combining macula clinical signs and patient characteristics for age-related macular degeneration diagnosis: A machine learning approach. BMC Ophthalmol. 2015, 15, 10. [Google Scholar] [CrossRef] [Green Version]
- Dastgheib, K.; Green, W.R. Granulomatous reaction to Bruch’s membrane in age-related macular degeneration. Arch. Ophthalmol. 1994, 112, 813–818. [Google Scholar] [CrossRef]
- Zarbin, M.A. Current concepts in the pathogenesis of age-related macular degeneration. Arch. Ophthalmol. 2004, 122, 598–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lommatzsch, A.; Hermans, P.; Weber, B.; Pauleikhoff, D. Complement factor H variant Y402H and basal laminar deposits in exudative age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 2007, 245, 1713–1716. [Google Scholar] [CrossRef]
- Liu, R.T.; Gao, J.; Cao, S.; Sandhu, N.; Cui, J.Z.; Chou, C.L.; Fang, E.; Matsubara, J.A. Inflammatory mediators induced by amyloid-β in the retina and RPE in vivo: Implications for inflammasome activation in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2225–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullins, R.F.; Russell, S.R.; Anderson, D.H.; Hageman, G.S. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000, 14, 835–846. [Google Scholar] [CrossRef]
- Mammadzada, P.; Corredoira, P.M.; Andre, H. The role of hypoxia-inducible factors in neovascular age-related macular degeneration: A gene therapy perspective. Cell Mol. Life Sci. 2019, 77, 819–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semkova, I.; Kociok, N.; Karagiannis, D.; Nischt, R.; Smyth, N.; Paulsson, M.; Strauss, O.; Joussen, A.M. Anti-angiogenic effect of the basement membrane protein nidogen-1 in a mouse model of choroidal neovascularization. Exp. Eye Res. 2014, 118, 80–88. [Google Scholar] [CrossRef]
- Wang, K.; Li, H.; Sun, R.; Liu, C.; Luo, Y.; Fu, S.; Ying, Y. Emerging roles of transforming growth factor β signaling in wet age-related macular degeneration. Acta Biochim Biophys. Sin. 2019, 51, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkman, J. Seminars in Medicine of the Beth Israel Hospital, Boston. Clinical applications of research on angiogenesis. N. Engl. J. Med. 1995, 333, 1757–1763. [Google Scholar] [CrossRef] [PubMed]
- Witmer, A.N.; Vrensen, G.F.; Van Noorden, C.J.; Schlingemann, R.O. Vascular endothelial growth factors and angiogenesis in eye disease. Prog. Retin. Eye Res. 2003, 22, 1–29. [Google Scholar] [CrossRef]
- McLeod, D.S.; Taomoto, M.; Otsuji, T.; Green, W.R.; Sunness, J.S.; Lutty, G.A. Quantifying changes in RPE and choroidal vasculature in eyes with age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1986–1993. [Google Scholar]
- Keiner, C.M.; Zhou, H.; Zhang, Q.; Wang, R.K.; Rinella, N.T.; Oldenburg, C.E.; Duncan, J.L.; Schwartz, D.M. Quantifying choriocapillaris hypoperfusion in patients with choroidal neovascularization using swept-source OCT angiography. Clin. Ophthalmol. 2019, 13, 1613–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, N.J.Y.; Chan, E.J.J.; Cheung, C. Choroidal Neovascularization: Mechanisms of Endothelial Dysfunction. Front. Pharmacol. 2019, 10, 1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Abraham, S.; McKenzie, J.A.G.; Jeffs, N.; Swire, M.; Tripathi, V.B.; Luhmann, U.F.O.; Lange, C.A.K.; Zhai, Z.; Arthur, H.M.; et al. LRG1 promotes angiogenesis by modulating endothelial TGF-β signalling. Nature 2013, 499, 306–311. [Google Scholar] [CrossRef] [Green Version]
- Iwanishi, H.; Fujita, N.; Tomoyose, K.; Okada, Y.; Yamanaka, O.; Flanders, K.C.; Saika, S. Inhibition of development of laser-induced choroidal neovascularization with suppression of infiltration of macrophages in Smad3-null mice. Lab. Investig. 2016, 96, 641–651. [Google Scholar] [CrossRef] [Green Version]
- Nunes, I.; Munger, J.; Harpel, J.G.; Nagano, Y.; Shapiro, R.; Gleizes, P.E.; Rifkin, D.B. Structure and activation of the large latent transforming growth factor-Β complex. J. Am. Optom. Assoc. 1998, 69, 643–648. [Google Scholar] [PubMed]
- Bai, Y.; Liang, S.; Yu, W.; Zhao, M.; Huang, L.; Zhao, M.; Li, X. Semaphorin 3A blocks the formation of pathologic choroidal neovascularization induced by transforming growth factor β. Mol. Vis. 2014, 20, 1258–1270. [Google Scholar]
- Tosi, G.M.; Caldi, E.; Neri, G.; Nuti, E.; Marigliani, D.; Baiocchi, S.; Traversi, C.; Cevenini, G.; Tarantello, A.; Fusco, F.; et al. HTRA1 and TGF-β1 Concentrations in the Aqueous Humor of Patients With Neovascular Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2017, 58, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef]
- Schlecht, A.; Leimbeck, S.V.; Jagle, H.; Feuchtinger, A.; Tamm, E.R.; Braunger, B.M. Deletion of Endothelial Transforming Growth Factor-β Signaling Leads to Choroidal Neovascularization. Am. J. Pathol. 2017, 187, 2570–2589. [Google Scholar] [CrossRef] [Green Version]
- Walshe, T.E.; Saint-Geniez, M.; Maharaj, A.S.; Sekiyama, E.; Maldonado, A.E.; D’Amore, P.A. TGF-β is required for vascular barrier function, endothelial survival and homeostasis of the adult microvasculature. PLoS ONE 2009, 4, e5149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, S.C.; Ju, M.; Liu, N.; Mo, J.R.; Ney, J.J.; Smith, L.E. Transforming growth factor β1 induction of vascular endothelial growth factor receptor 1: Mechanism of pericyte-induced vascular survival in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 15859–15864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radeke, M.J.; Radeke, C.M.; Shih, Y.H.; Hu, J.; Bok, D.; Johnson, L.V.; Coffey, P.J. Restoration of mesenchymal retinal pigmented epithelial cells by TGFβ pathway inhibitors: Implications for age-related macular degeneration. Genome Med. 2015, 7, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, N.; Yamamoto, C.; Miyashiro, M.; Yamada, H.; Matsushima, M.; Uyama, M. Expression of transforming growth factor-β mRNA in experimental choroidal neovascularization. Curr Eye Res. 1997, 16, 9–18. [Google Scholar] [CrossRef]
- Xu, J.; Zhu, D.; Sonoda, S.; He, S.; Spee, C.; Ryan, S.J.; Hinton, D.R. Over-expression of BMP4 inhibits experimental choroidal neovascularization by modulating VEGF and MMP-9. Angiogenesis 2012, 15, 213–227. [Google Scholar] [CrossRef]
- Dyer, L.A.; Pi, X.; Patterson, C. The role of BMPs in endothelial cell function and dysfunction. Trends Endocrinol. Metab. 2014, 25, 472–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ntumba, K.; Akla, N.; Oh, S.P.; Eichmann, A.; Larrivee, B. BMP9/ALK1 inhibits neovascularization in mouse models of age-related macular degeneration. Oncotarget 2016, 7, 55957–55969. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liu, M.; Luan, Y.; Liu, Y.; Zhang, Z.; Ma, B.; Liu, X.; Liu, Y. BMP6 protects retinal pigment epithelial cells from oxidative stressinduced injury by inhibiting the MAPK signaling pathways. Int. J. Mol. Med. 2018, 42, 1096–1105. [Google Scholar] [CrossRef]
- Zhu, D.; Wu, J.; Spee, C.; Ryan, S.J.; Hinton, D.R. BMP4 mediates oxidative stress-induced retinal pigment epithelial cell senescence and is overexpressed in age-related macular degeneration. J. Biol. Chem. 2009, 284, 9529–9539. [Google Scholar] [CrossRef] [Green Version]
- Scharpfenecker, M.; van Dinther, M.; Liu, Z.; van Bezooijen, R.L.; Zhao, Q.; Pukac, L.; Lowik, C.W.; ten Dijke, P. BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J. Cell Sci. 2007, 120, 964–972. [Google Scholar] [CrossRef] [Green Version]
- Hussein, K.A.; Choksi, K.; Akeel, S.; Ahmad, S.; Megyerdi, S.; El-Sherbiny, M.; Nawaz, M.; Abu El-Asrar, A.; Al-Shabrawey, M. Bone morphogenetic protein 2: A potential new player in the pathogenesis of diabetic retinopathy. Exp. Eye Res. 2014, 125, 79–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulaki, V.; Mitsiades, N.; Kruse, F.E.; Radetzky, S.; Iliaki, E.; Kirchhof, B.; Joussen, A.M. Activin a in the regulation of corneal neovascularization and vascular endothelial growth factor expression. Am. J. Pathol. 2004, 164, 1293–1302. [Google Scholar] [CrossRef] [Green Version]
- White, C.E.; Kwok, B.; Olabisi, R.M. Activin A improves retinal pigment epithelial cell survival on stiff but not soft substrates. J. Biomed. Mater. Res. A 2018, 106, 2871–2880. [Google Scholar] [CrossRef]
- Skeie, J.M.; Zeng, S.; Faidley, E.A.; Mullins, R.F. Angiogenin in age-related macular degeneration. Mol. Vis. 2011, 17, 576–582. [Google Scholar]
- Tosi, G.M.; Neri, G.; Caldi, E.; Fusco, F.; Bacci, T.; Tarantello, A.; Nuti, E.; Marigliani, D.; Baiocchi, S.; Traversi, C.; et al. TGF-β concentrations and activity are down-regulated in the aqueous humor of patients with neovascular age-related macular degeneration. Sci. Rep. 2018, 8, 8053. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Loboda, A.; Sobczak, M.; Jozkowicz, A.; Dulak, J. TGF-β1/Smads and miR-21 in Renal Fibrosis and Inflammation. Mediat. Inflamm. 2016, 2016, 8319283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Yang, T.; Lu, D.W.; Zhao, H.; Feng, Y.L.; Chen, H.; Chen, D.Q.; Vaziri, N.D.; Zhao, Y.Y. Central role of dysregulation of TGF-β/Smad in CKD progression and potential targets of its treatment. Biomed. Pharmacother. 2018, 101, 670–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Liu, Z.L. Transforming growth factor-β neutralizing antibodies inhibit subretinal fibrosis in a mouse model. Int. J. Ophthalmol. 2012, 5, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.; Lee, H.G.; Baek, W.K.; Lee, Y.; Kim, K.S.; Jun, J.H.; Kim, J.Y.; Joo, C.K. Bortezomib inhibits proliferation, migration, and TGF-β1-induced epithelial-mesenchymal transition of RPE cells. Mol. Vis. 2017, 23, 1029–1038. [Google Scholar]
- Hu, H.H.; Chen, D.Q.; Wang, Y.N.; Feng, Y.L.; Cao, G.; Vaziri, N.D.; Zhao, Y.Y. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem. Biol. Interact. 2018, 292, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Varga, J. Scleroderma and Smads: Dysfunctional Smad family dynamics culminating in fibrosis. Arthritis Rheum. 2002, 46, 1703–1713. [Google Scholar] [CrossRef]
- Fontana, A.; Constam, D.B.; Frei, K.; Malipiero, U.; Pfister, H.W. Modulation of the immune response by transforming growth factor β. Int. Arch. Allergy Immunol. 1992, 99, 1–7. [Google Scholar] [CrossRef]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted disruption of the mouse transforming growth factor-β 1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming growth factor β 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, A.; Wakabayashi, Y.; Mori, T. Cellular and molecular basis for the regulation of inflammation by TGF-beta. J. Biochem. 2010, 147, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Ahmad, M.; Smith, K.E.; Labinskyy, N.; Gao, Q.; Kaley, G.; Edwards, J.G.; Wolin, M.S.; Ungvari, Z. Bone morphogenetic protein-2 induces proinflammatory endothelial phenotype. Am. J. Pathol. 2006, 168, 629–638. [Google Scholar] [CrossRef] [Green Version]
- Pastor, J.C.; de la Rua, E.R.; Martin, F. Proliferative vitreoretinopathy: Risk factors and pathobiology. Prog. Retin. Eye Res. 2002, 21, 127–144. [Google Scholar] [CrossRef]
- Leiderman, Y.I.; Miller, J.W. Proliferative vitreoretinopathy: Pathobiology and therapeutic targets. Semin. Ophthalmol. 2009, 24, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Bochaton-Piallat, M.L.; Kapetanios, A.D.; Donati, G.; Redard, M.; Gabbiani, G.; Pournaras, C.J. TGF-β1, TGF-β receptor II and ED-A fibronectin expression in myofibroblast of vitreoretinopathy. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2336–2342. [Google Scholar]
- Kita, T.; Hata, Y.; Kano, K.; Miura, M.; Nakao, S.; Noda, Y.; Shimokawa, H.; Ishibashi, T. Transforming growth factor-β2 and connective tissue growth factor in proliferative vitreoretinal diseases: Possible involvement of hyalocytes and therapeutic potential of Rho kinase inhibitor. Diabetes 2007, 56, 231–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kita, T.; Hata, Y.; Arita, R.; Kawahara, S.; Miura, M.; Nakao, S.; Mochizuki, Y.; Enaida, H.; Goto, Y.; Shimokawa, H.; et al. Role of TGF-β in proliferative vitreoretinal diseases and ROCK as a therapeutic target. Proc. Natl. Acad Sci. USA 2008, 105, 17504–17509. [Google Scholar] [CrossRef] [Green Version]
- Hardwick, C.; Feist, R.; Morris, R.; White, M.; Witherspoon, D.; Angus, R.; Guidry, C. Tractional force generation by porcine Muller cells: Stimulation by growth factors in human vitreous. Investig. Ophthalmol. Vis. Sci. 1997, 38, 2053–2063. [Google Scholar]
- Saika, S.; Yamanaka, O.; Sumioka, T.; Miyamoto, T.; Miyazaki, K.; Okada, Y.; Kitano, A.; Shirai, K.; Tanaka, S.; Ikeda, K. Fibrotic disorders in the eye: Targets of gene therapy. Prog. Retin. Eye Res. 2008, 27, 177–196. [Google Scholar] [CrossRef]
- Saika, S.; Kono-Saika, S.; Tanaka, T.; Yamanaka, O.; Ohnishi, Y.; Sato, M.; Muragaki, Y.; Ooshima, A.; Yoo, J.; Flanders, K.C.; et al. Smad3 is required for dedifferentiation of retinal pigment epithelium following retinal detachment in mice. Lab. Investig. 2004, 84, 1245–1258. [Google Scholar] [CrossRef]
- Grotendorst, G.R.; Smale, G.; Pencev, D. Production of transforming growth factor β by human peripheral blood monocytes and neutrophils. J. Cell Physiol. 1989, 140, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Yadav, H.; Quijano, C.; Kamaraju, A.K.; Gavrilova, O.; Malek, R.; Chen, W.; Zerfas, P.; Zhigang, D.; Wright, E.C.; Stuelten, C.; et al. Protection from obesity and diabetes by blockade of TGF-β/Smad3 signaling. Cell Metab. 2011, 14, 67–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akla, N.; Viallard, C.; Popovic, N.; Lora Gil, C.; Sapieha, P.; Larrivee, B. BMP9 (Bone Morphogenetic Protein-9)/Alk1 (Activin-Like Kinase Receptor Type I) Signaling Prevents Hyperglycemia-Induced Vascular Permeability. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1821–1836. [Google Scholar] [CrossRef] [Green Version]
- Pauk, M.; Bordukalo-Niksic, T.; Brkljacic, J.; Paralkar, V.M.; Brault, A.L.; Dumic-Cule, I.; Borovecki, F.; Grgurevic, L.; Vukicevic, S. A novel role of bone morphogenetic protein 6 (BMP6) in glucose homeostasis. Acta Diabetol. 2019, 56, 365–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Li, L.; Xu, X.; Wu, T.; Yang, M.; Zhang, C.; Mou, H.; Zhou, T.; Jia, Y.; Cai, C.; et al. Decreased circulating BMP-9 levels in patients with Type 2 diabetes is a signature of insulin resistance. Clin. Sci. 2017, 131, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.E.; Lee, N.Y. Emerging Roles of Transforming Growth Factor β Signaling in Diabetic Retinopathy. J. Cell Physiol. 2017, 232, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; O’Meara, S.J.; O’Brien, C.; Kane, R. The role of gremlin, a BMP antagonist, and epithelial-to-mesenchymal transition in proliferative vitreoretinopathy. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4291–4299. [Google Scholar] [CrossRef] [PubMed]
- Kane, R.; Stevenson, L.; Godson, C.; Stitt, A.W.; O’Brien, C. Gremlin gene expression in bovine retinal pericytes exposed to elevated glucose. Br. J. Ophthalmol. 2005, 89, 1638–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tandon, A.; Tovey, J.C.; Sharma, A.; Gupta, R.; Mohan, R.R. Role of transforming growth factor Β in corneal function, biology and pathology. Curr. Mol. Med. 2010, 10, 565–578. [Google Scholar] [CrossRef]
- Huh, M.I.; Chang, Y.; Jung, J.C. Temporal and spatial distribution of TGF-β isoforms and signaling intermediates in corneal regenerative wound repair. Histol. Histopathol. 2009, 24, 1405–1416. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, J.; Kamiyama, K.; Iguchi, I.; Kita, M.; Sotozono, C.; Kinoshita, S. Growth factors: Importance in wound healing and maintenance of transparency of the cornea. Prog. Retin. Eye Res. 2000, 19, 113–129. [Google Scholar] [CrossRef]
- Zieske, J.D.; Hutcheon, A.E.; Guo, X.; Chung, E.H.; Joyce, N.C. TGF-β receptor types I and II are differentially expressed during corneal epithelial wound repair. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1465–1471. [Google Scholar]
- Lim, M.; Chuong, C.M.; Roy-Burman, P. PI3K, Erk signaling in BMP7-induced epithelial-mesenchymal transition (EMT) of PC-3 prostate cancer cells in 2- and 3-dimensional cultures. Horm. Cancer 2011, 2, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Saika, S.; Ikeda, K.; Yamanaka, O.; Flanders, K.C.; Nakajima, Y.; Miyamoto, T.; Ohnishi, Y.; Kao, W.W.; Muragaki, Y.; Ooshima, A. Therapeutic effects of adenoviral gene transfer of bone morphogenic protein-7 on a corneal alkali injury model in mice. Lab. Investig. 2005, 85, 474–486. [Google Scholar] [CrossRef] [Green Version]
- Tandon, A.; Sharma, A.; Rodier, J.T.; Klibanov, A.M.; Rieger, F.G.; Mohan, R.R. BMP7 gene transfer via gold nanoparticles into stroma inhibits corneal fibrosis in vivo. PLoS ONE 2013, 8, e66434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, R.; Agarwal, P. Future target molecules in antiglaucoma therapy: Tgf-Β may have a role to play. Ophthalmic. Res. 2010, 43, 1–10. [Google Scholar] [CrossRef]
- Prendes, M.A.; Harris, A.; Wirostko, B.M.; Gerber, A.L.; Siesky, B. The role of transforming growth factor β in glaucoma and the therapeutic implications. Br. J. Ophthalmol. 2013, 97, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.K.; Gabelt, B.T.; Ruiz, J.; Picciani, R.; Kaufman, P.L. Cochlin expression in anterior segment organ culture models after TGFβ2 treatment. Investig. Ophthalmol. Vis. Sci. 2009, 50, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Shepard, A.R.; Millar, J.C.; Pang, I.H.; Jacobson, N.; Wang, W.H.; Clark, A.F. Adenoviral gene transfer of active human transforming growth factor-β2 elevates intraocular pressure and reduces outflow facility in rodent eyes. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2067–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, J.V.; Golesic, E.; Gauldie, J.; West-Mays, J.A. Ocular gene transfer of active TGF-β induces changes in anterior segment morphology and elevated IOP in rats. Investig. Ophthalmol. Vis. Sci. 2010, 51, 308–318. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, R.C.; Li, J.; Chan, W.F.; Tripathi, B.J. Aqueous humor in glaucomatous eyes contains an increased level of TGF-β 2. Exp. Eye Res. 1994, 59, 723–727. [Google Scholar] [CrossRef]
- Ochiai, Y.; Ochiai, H. Higher concentration of transforming growth factor-β in aqueous humor of glaucomatous eyes and diabetic eyes. Jpn. J. Ophthalmol. 2002, 46, 249–253. [Google Scholar] [CrossRef]
- Inatani, M.; Tanihara, H.; Katsuta, H.; Honjo, M.; Kido, N.; Honda, Y. Transforming growth factor-β 2 levels in aqueous humor of glaucomatous eyes. Graefes Arch. Clin. Exp. Ophthalmol. 2001, 239, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Gottanka, J.; Chan, D.; Eichhorn, M.; Lutjen-Drecoll, E.; Ethier, C.R. Effects of TGF-β2 in perfused human eyes. Investig. Ophthalmol. Vis. Sci. 2004, 45, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Fukuchi, T.; Ueda, J.; Hanyu, T.; Abe, H.; Sawaguchi, S. Distribution and expression of transforming growth factor-β and platelet-derived growth factor in the normal and glaucomatous monkey optic nerve heads. Jpn. J. Ophthalmol. 2001, 45, 592–599. [Google Scholar] [CrossRef]
- Kirwan, R.P.; Crean, J.K.; Fenerty, C.H.; Clark, A.F.; O’Brien, C.J. Effect of cyclical mechanical stretch and exogenous transforming growth factor-β1 on matrix metalloproteinase-2 activity in lamina cribrosa cells from the human optic nerve head. J. Glaucoma. 2004, 13, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Wordinger, R.J.; Fleenor, D.L.; Hellberg, P.E.; Pang, I.H.; Tovar, T.O.; Zode, G.S.; Fuller, J.A.; Clark, A.F. Effects of TGF-β2, BMP-4, and gremlin in the trabecular meshwork: Implications for glaucoma. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1191–1200. [Google Scholar] [CrossRef]
- Fuchshofer, R.; Stephan, D.A.; Russell, P.; Tamm, E.R. Gene expression profiling of TGFβ2- and/or BMP7-treated trabecular meshwork cells: Identification of Smad7 as a critical inhibitor of TGF-β2 signaling. Exp. Eye Res. 2009, 88, 1020–1032. [Google Scholar] [CrossRef] [Green Version]
- Wordinger, R.J.; Clark, A.F.; Agarwal, R.; Lambert, W.; McNatt, L.; Wilson, S.E.; Qu, Z.; Fung, B.K. Cultured human trabecular meshwork cells express functional growth factor receptors. Investig. Ophthalmol. Vis. Sci. 1998, 39, 1575–1589. [Google Scholar]
- Mead, A.L.; Wong, T.T.; Cordeiro, M.F.; Anderson, I.K.; Khaw, P.T. Evaluation of anti-TGF-β2 antibody as a new postoperative anti-scarring agent in glaucoma surgery. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3394–3401. [Google Scholar] [CrossRef] [Green Version]
- Siriwardena, D.; Khaw, P.T.; King, A.J.; Donaldson, M.L.; Overton, B.M.; Migdal, C.; Cordeiro, M.F. Human antitransforming growth factor β(2) monoclonal antibody—A new modulator of wound healing in trabeculectomy: A randomized placebo controlled clinical study. Ophthalmology 2002, 109, 427–431. [Google Scholar] [CrossRef]
- Group, C.A.T.T.S.; Khaw, P.; Grehn, F.; Hollo, G.; Overton, B.; Wilson, R.; Vogel, R.; Smith, Z. A phase III study of subconjunctival human anti-transforming growth factor β(2) monoclonal antibody (CAT-152) to prevent scarring after first-time trabeculectomy. Ophthalmology 2007, 114, 1822–1830. [Google Scholar] [CrossRef]
- Group, C.A.T.T.S. CAT-152 Trabeculectomy Study. Ophthalmology 2007, 114, 1950. [Google Scholar] [CrossRef]
- Pfeiffer, N.; Voykov, B.; Renieri, G.; Bell, K.; Richter, P.; Weigel, M.; Thieme, H.; Wilhelm, B.; Lorenz, K.; Feindor, M.; et al. First-in-human phase I study of ISTH0036, an antisense oligonucleotide selectively targeting transforming growth factor β 2 (TGF-β2), in subjects with open-angle glaucoma undergoing glaucoma filtration surgery. PLoS ONE 2017, 12, e0188899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, D.M. Historic review of retinoblastoma. Ophthalmology 1987, 94, 654–662. [Google Scholar] [CrossRef]
- Honavar, S.G.; Singh, A.D. Management of advanced retinoblastoma. Ophthalmol. Clin. N. Am. 2005, 18, 65–73. [Google Scholar] [CrossRef]
- Kase, S.; Parikh, J.G.; Youssef, P.N.; Murphree, A.L.; Rao, N.A. Transforming growth factor β in retinoblastoma-related cataract. Arch. Ophthalmol. 2008, 126, 1539–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashiwagi, Y.; Horie, K.; Kanno, C.; Inomata, M.; Imamura, T.; Kato, M.; Yamamoto, T.; Yamashita, H. Trichostatin A-induced TGF-β type II receptor expression in retinoblastoma cell lines. Investig. Ophthalmol. Vis. Sci. 2010, 51, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Kimchi, A.; Wang, X.F.; Weinberg, R.A.; Cheifetz, S.; Massague, J. Absence of TGF-β receptors and growth inhibitory responses in retinoblastoma cells. Science 1988, 240, 196–199. [Google Scholar] [CrossRef]
- Horie, K.; Yamashita, H.; Mogi, A.; Takenoshita, S.; Miyazono, K. Lack of transforming growth factor-β type II receptor expression in human retinoblastoma cells. J. Cell Physiol. 1998, 175, 305–313. [Google Scholar] [CrossRef]
- DaCosta Byfield, S.; Major, C.; Laping, N.J.; Roberts, A.B. SB-505124 is a selective inhibitor of transforming growth factor-β type I receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2004, 65, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Asnaghi, L.; White, D.T.; Key, N.; Choi, J.; Mahale, A.; Alkatan, H.; Edward, D.P.; Elkhamary, S.M.; Al-Mesfer, S.; Maktabi, A.; et al. ACVR1C/SMAD2 signaling promotes invasion and growth in retinoblastoma. Oncogene 2019, 38, 2056–2075. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.D.; Bergman, L.; Seregard, S. Uveal melanoma: Epidemiologic aspects. Ophthalmol. Clin. N. Am. 2005, 18, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Laver, N.V.; McLaughlin, M.E.; Duker, J.S. Ocular melanoma. Arch. Pathol Lab. Med. 2010, 134, 1778–1784. [Google Scholar] [CrossRef]
- Esser, P.; Grisanti, S.; Bartz-Schmidt, K. TGF-β in uveal melanoma. Microsc. Res. Tech. 2001, 52, 396–400. [Google Scholar] [CrossRef]
- Ma, D.; Niederkorn, J.Y. Transforming growth factor-β down-regulates major histocompatibility complex class I antigen expression and increases the susceptibility of uveal melanoma cells to natural killer cell-mediated cytolysis. Immunology 1995, 86, 263–269. [Google Scholar] [PubMed]
- Pierce, D.F., Jr.; Gorska, A.E.; Chytil, A.; Meise, K.S.; Page, D.L.; Coffey, R.J., Jr.; Moses, H.L. Mammary tumor suppression by transforming growth factor β 1 transgene expression. Proc. Natl. Acad. Sci. USA 1995, 92, 4254–4258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, L.H.; Li, Y.; Chen, J.S.; Munoz, N.M.; Majumdar, A.; Chen, J.; Mishra, L. Targeting TGF-β signaling in cancer. Expert Opin. Ther. Targets 2013, 17, 743–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padua, D.; Massague, J. Roles of TGFβ in metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef]
- Meulmeester, E.; Ten Dijke, P. The dynamic roles of TGF-β in cancer. J. Pathol. 2011, 223, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Kang, Y. Targeting the transforming growth factor-β signalling pathway in metastatic cancer. Eur. J. Cancer 2010, 46, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Syed, V. TGF-β Signaling in Cancer. J. Cell Biochem. 2016, 117, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J. TGF-β antagonists: Why suppress a tumor suppressor? J. Clin. Investig. 2002, 109, 1533–1536. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Li, N.; Lv, N.; Huang, D. SMAD7: A timer of tumor progression targeting TGF-β signaling. Tumour. Biol. 2014, 35, 8379–8385. [Google Scholar] [CrossRef] [PubMed]
- de Caestecker, M.P.; Piek, E.; Roberts, A.B. Role of transforming growth factor-β signaling in cancer. J. Natl. Cancer Inst. 2000, 92, 1388–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, M. TGF-β and cancer. Microbes Infect. 1999, 1, 1327–1347. [Google Scholar] [CrossRef]
- Kim, K.S.; Park, J.M.; Kong, T.; Kim, C.; Bae, S.H.; Kim, H.W.; Moon, J. Retinal Angiogenesis Effects of TGF-β1 and Paracrine Factors Secreted From Human Placental Stem Cells in Response to a Pathological Environment. Cell Transplant. 2016, 25, 1145–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarranz-Ventura, J.; Fernandez-Robredo, P.; Recalde, S.; Salinas-Alaman, A.; Borras-Cuesta, F.; Dotor, J.; Garcia-Layana, A. Transforming growth factor-β inhibition reduces progression of early choroidal neovascularization lesions in rats: P17 and P144 peptides. PLoS ONE 2013, 8, e65434. [Google Scholar] [CrossRef] [Green Version]
- Lambert, V.; Munaut, C.; Jost, M.; Noel, A.; Werb, Z.; Foidart, J.M.; Rakic, J.M. Matrix metalloproteinase-9 contributes to choroidal neovascularization. Am. J. Pathol. 2002, 161, 1247–1253. [Google Scholar] [CrossRef] [Green Version]
- Ying, Y.; Ueta, T.; Jiang, S.; Lin, H.; Wang, Y.; Vavvas, D.; Wen, R.; Chen, Y.G.; Luo, Z. Metformin inhibits ALK1-mediated angiogenesis via activation of AMPK. Oncotarget 2017, 8, 32794–32806. [Google Scholar] [CrossRef]
- Awwad, K.; Hu, J.; Shi, L.; Mangels, N.; Abdel Malik, R.; Zippel, N.; Fisslthaler, B.; Eble, J.A.; Pfeilschifter, J.; Popp, R.; et al. Role of secreted modular calcium-binding protein 1 (SMOC1) in transforming growth factor β signalling and angiogenesis. Cardiovasc. Res. 2015, 106, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Fuchshofer, R.; Tamm, E.R. The role of TGF-β in the pathogenesis of primary open-angle glaucoma. Cell Tissue Res. 2012, 347, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Harris, A.; Prendes, M.A.; Alshawa, L.; Gross, J.C.; Wentz, S.M.; Rao, A.B.; Kim, N.J.; Synder, A.; Siesky, B. Targeting Transforming Growth Factor-β Signaling in Primary Open-Angle Glaucoma. J. Glaucoma 2017, 26, 390–395. [Google Scholar] [CrossRef]
- Saika, S. TGF-β signal transduction in corneal wound healing as a therapeutic target. Cornea 2004, 23, S25–S30. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.R.; Sharma, A.; Netto, M.V.; Sinha, S.; Wilson, S.E. Gene therapy in the cornea. Prog. Retin. Eye Res. 2005, 24, 537–559. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Kay, E.P.; Nakahara, M.; Hamuro, J.; Kinoshita, S.; Koizumi, N. Inhibition of TGF-β signaling enables human corneal endothelial cell expansion in vitro for use in regenerative medicine. PLoS ONE 2013, 8, e58000. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Foreman, D.M.; Ferguson, M.W. Control of scarring in adult wounds by neutralising antibody to transforming growth factor β. Lancet 1992, 339, 213–214. [Google Scholar] [CrossRef]
- Fukuda, K.; Chikama, T.; Takahashi, M.; Nishida, T. Long-term follow-up after lamellar keratoplasty in a patient with bilateral idiopathic corneal keloid. Cornea 2011, 30, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- Zahir-Jouzdani, F.; Soleimani, M.; Mahbod, M.; Mottaghitalab, F.; Vakhshite, F.; Arefian, E.; Shahhoseini, S.; Dinarvand, R.; Atyabi, F. Corneal chemical burn treatment through a delivery system consisting of TGF-β1 siRNA: In vitro and in vivo. Drug Deliv. Transl. Res. 2018, 8, 1127–1138. [Google Scholar] [CrossRef]
- Gurumurthy, S.; Iyer, G.; Srinivasan, B.; Agarwal, S.; Angayarkanni, N. Ocular surface cytokine profile in chronic Stevens-Johnson syndrome and its response to mucous membrane grafting for lid margin keratinisation. Br. J. Ophthalmol. 2018, 102, 169–176. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hachana, S.; Larrivée, B. TGF-β Superfamily Signaling in the Eye: Implications for Ocular Pathologies. Cells 2022, 11, 2336. https://doi.org/10.3390/cells11152336
Hachana S, Larrivée B. TGF-β Superfamily Signaling in the Eye: Implications for Ocular Pathologies. Cells. 2022; 11(15):2336. https://doi.org/10.3390/cells11152336
Chicago/Turabian StyleHachana, Soumaya, and Bruno Larrivée. 2022. "TGF-β Superfamily Signaling in the Eye: Implications for Ocular Pathologies" Cells 11, no. 15: 2336. https://doi.org/10.3390/cells11152336