
Indirect Negative Effect of Mutant Ataxin-1 on Short- and Long-Term Synaptic Plasticity in Mouse Models of Spinocerebellar Ataxia Type 1

 ,
,  ,
,
Abstract
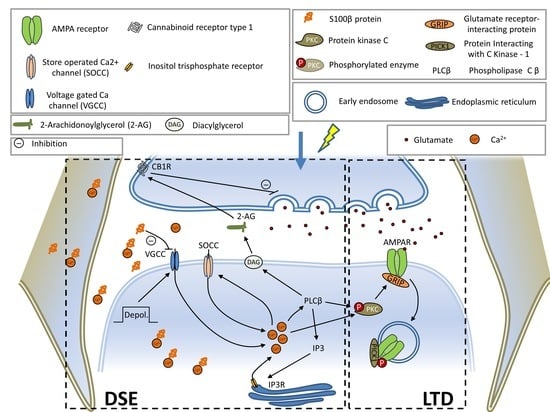
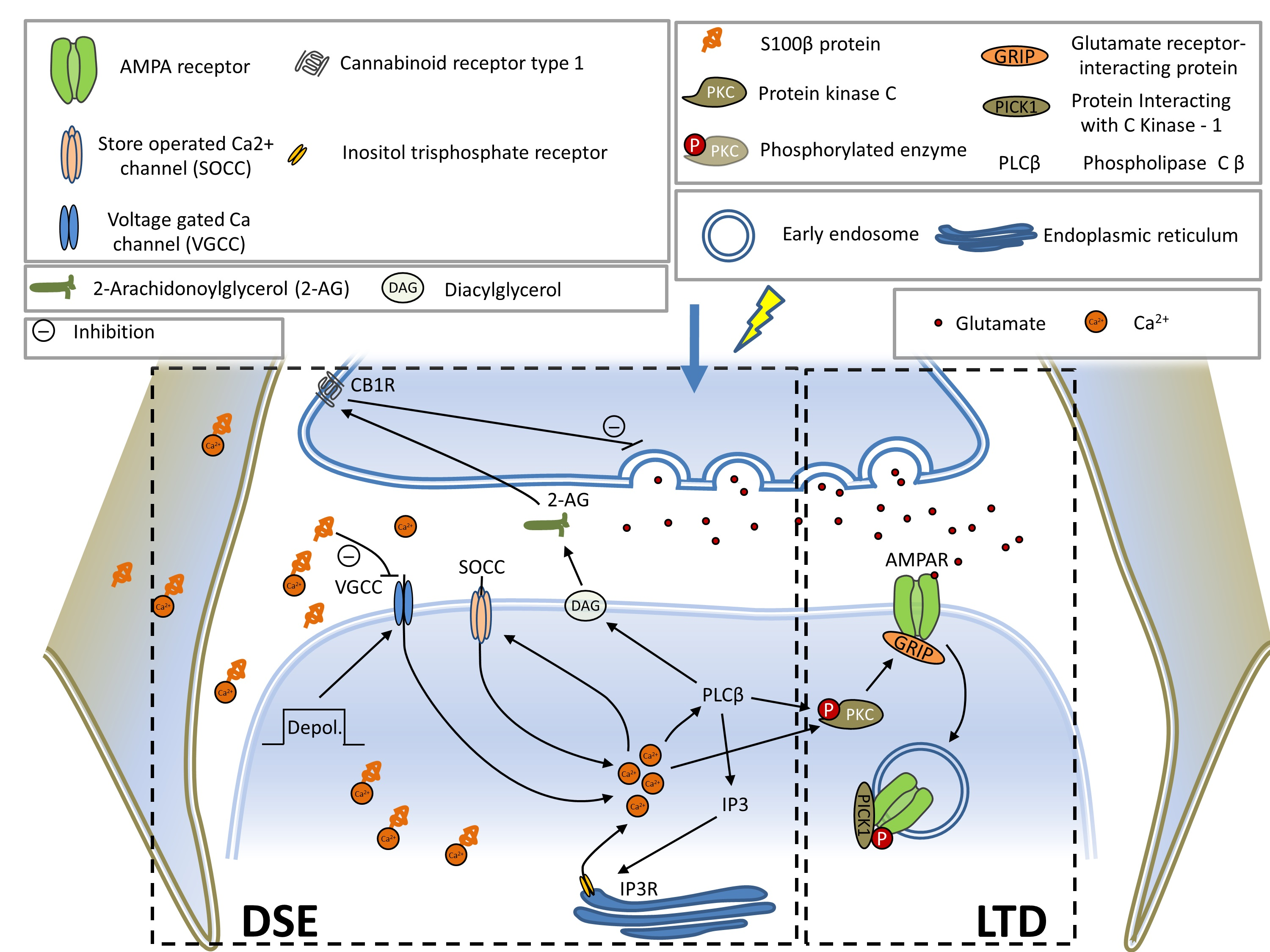
:
1. Introduction
2. Materials and Methods
2.1. LVV Production and Amplification
2.2. LVV Injections
2.3. Rotarod Test
2.4. Acute Slice Preparation
2.5. Electrophysiology
2.6. Immunohistochemistry
2.7. Data Analysis
3. Results
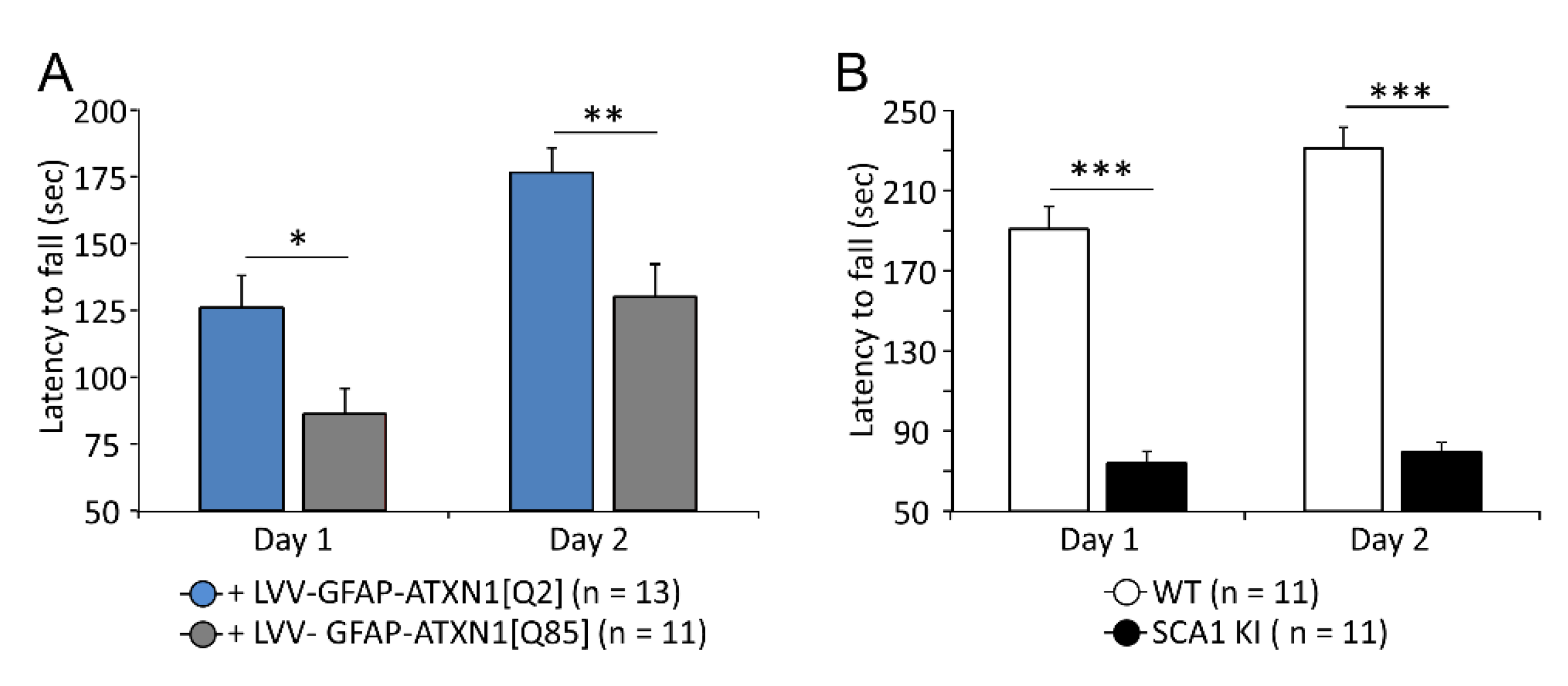
3.1. Expression of Mutant ATXN1 Disrupts Motor Coordination
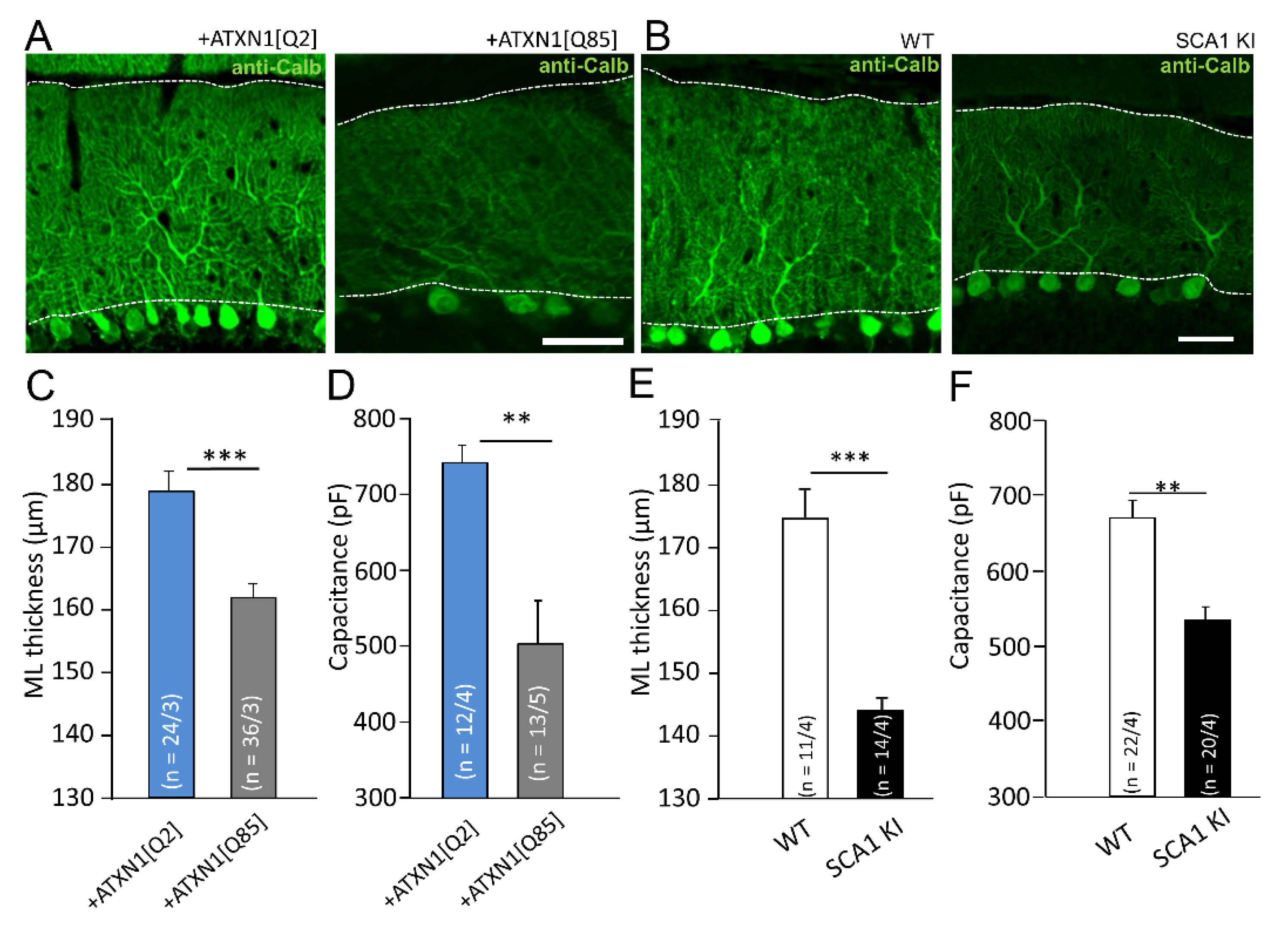
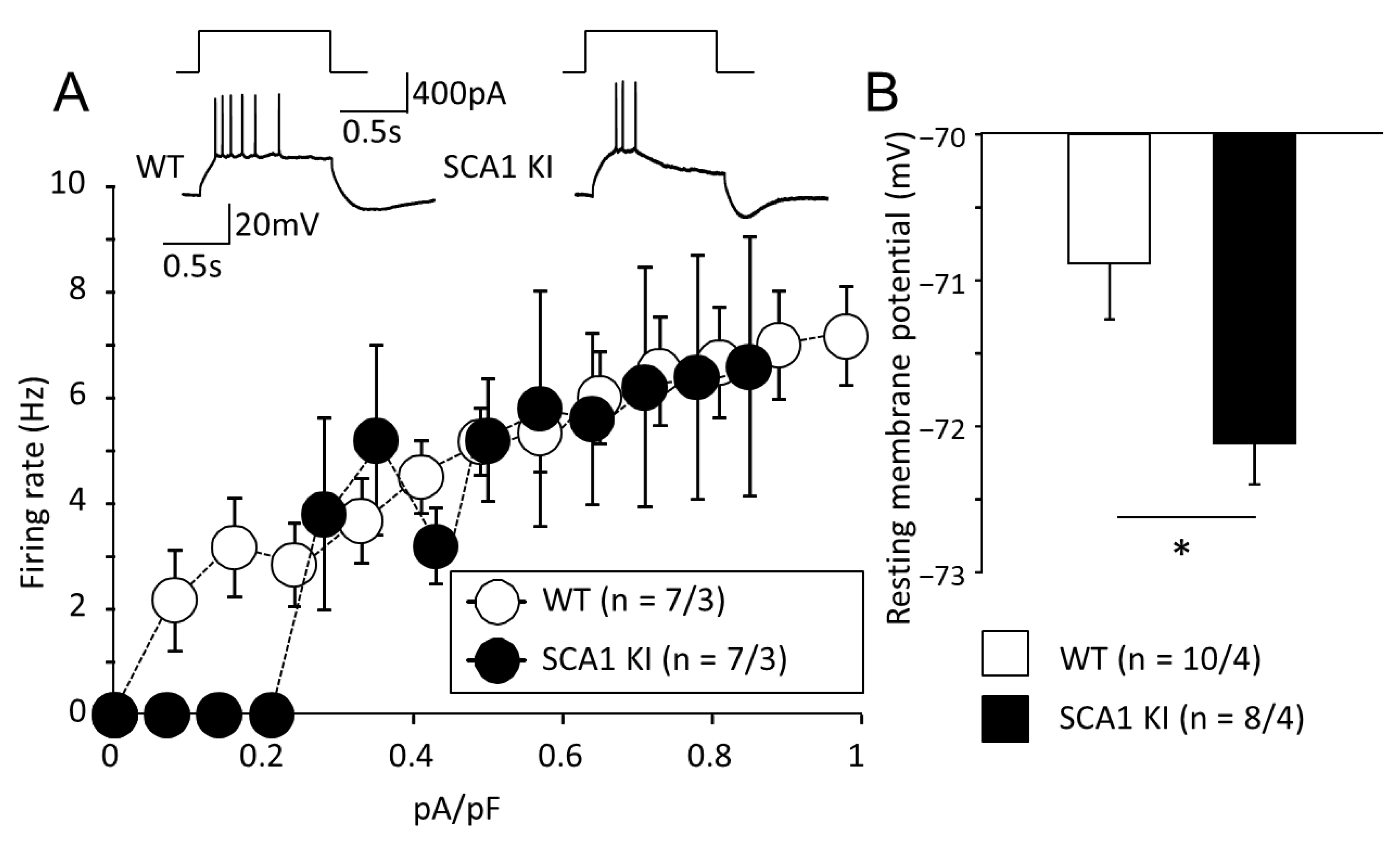
3.2. Expression of Mutant ATXN1 Selectively in BG and Non-Selectively as in SCA1 KI Model Affects Morphology and Electrophysiological Properties of PCs
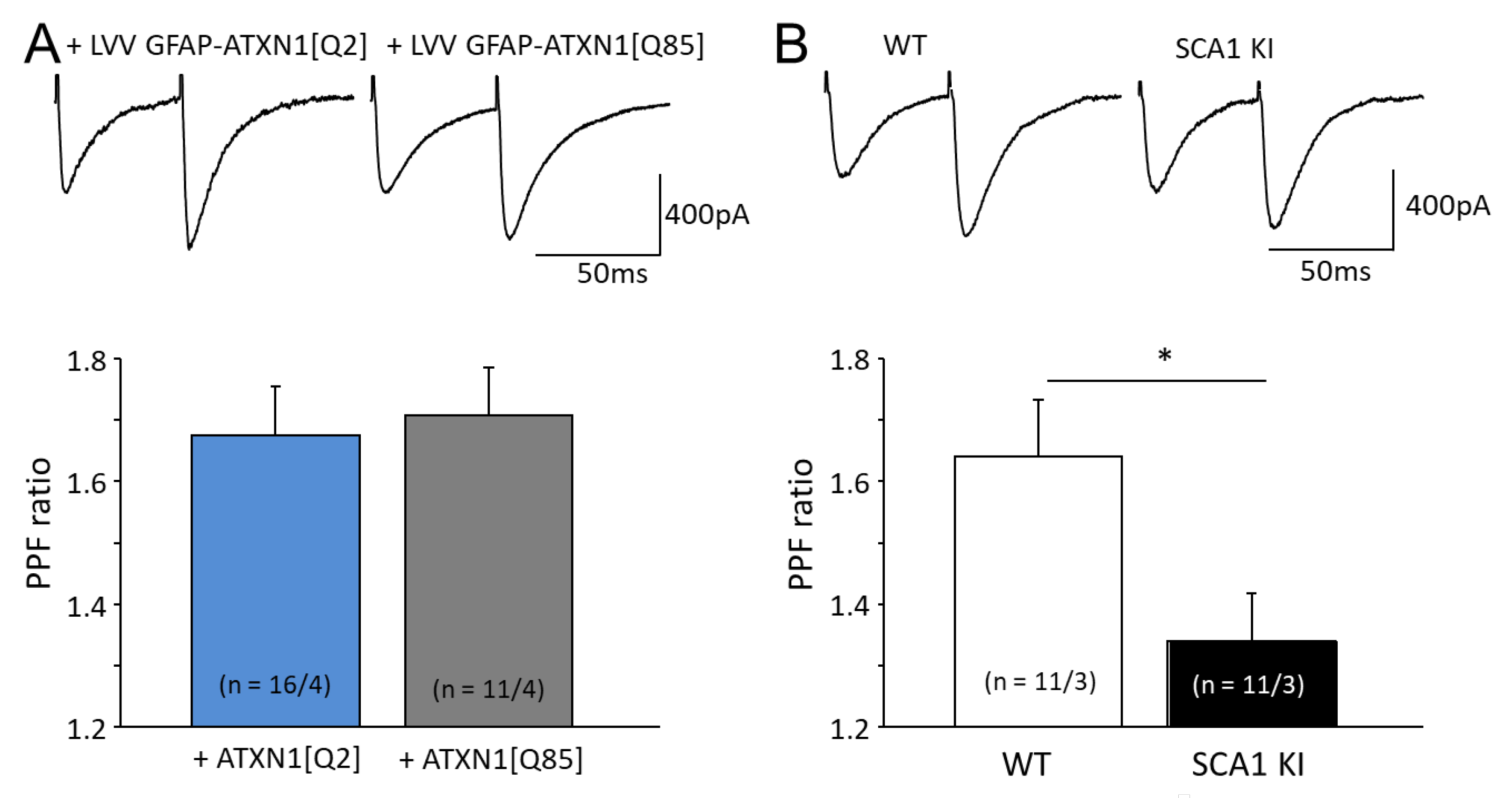
3.3. Expression of Mutant ATXN1 Effects Short Term Synaptic Plasticity
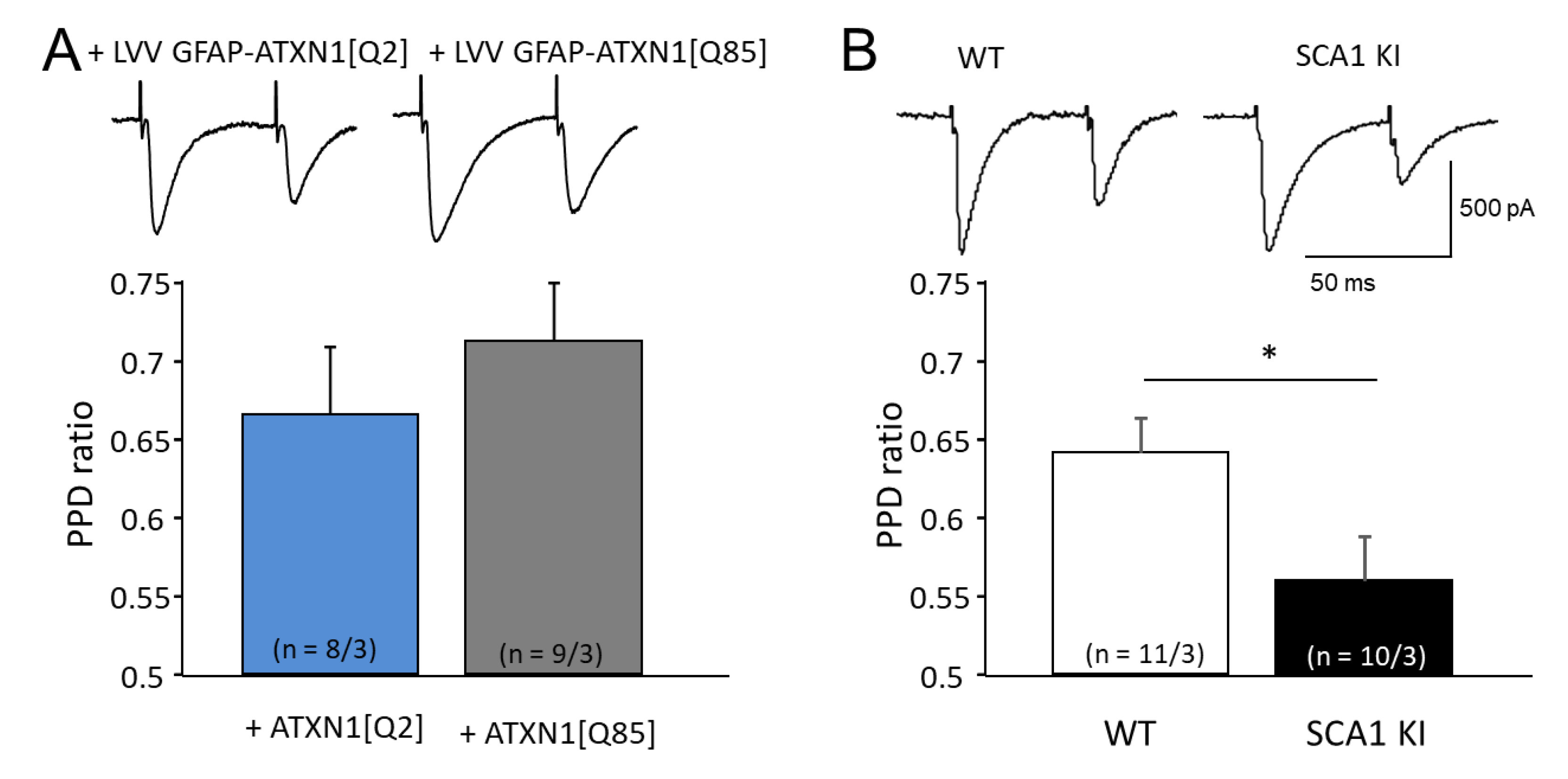
3.4. Mutant Ataxin-1 in BG Compromises Postsynaptic Short-Term Plasticity in PCs
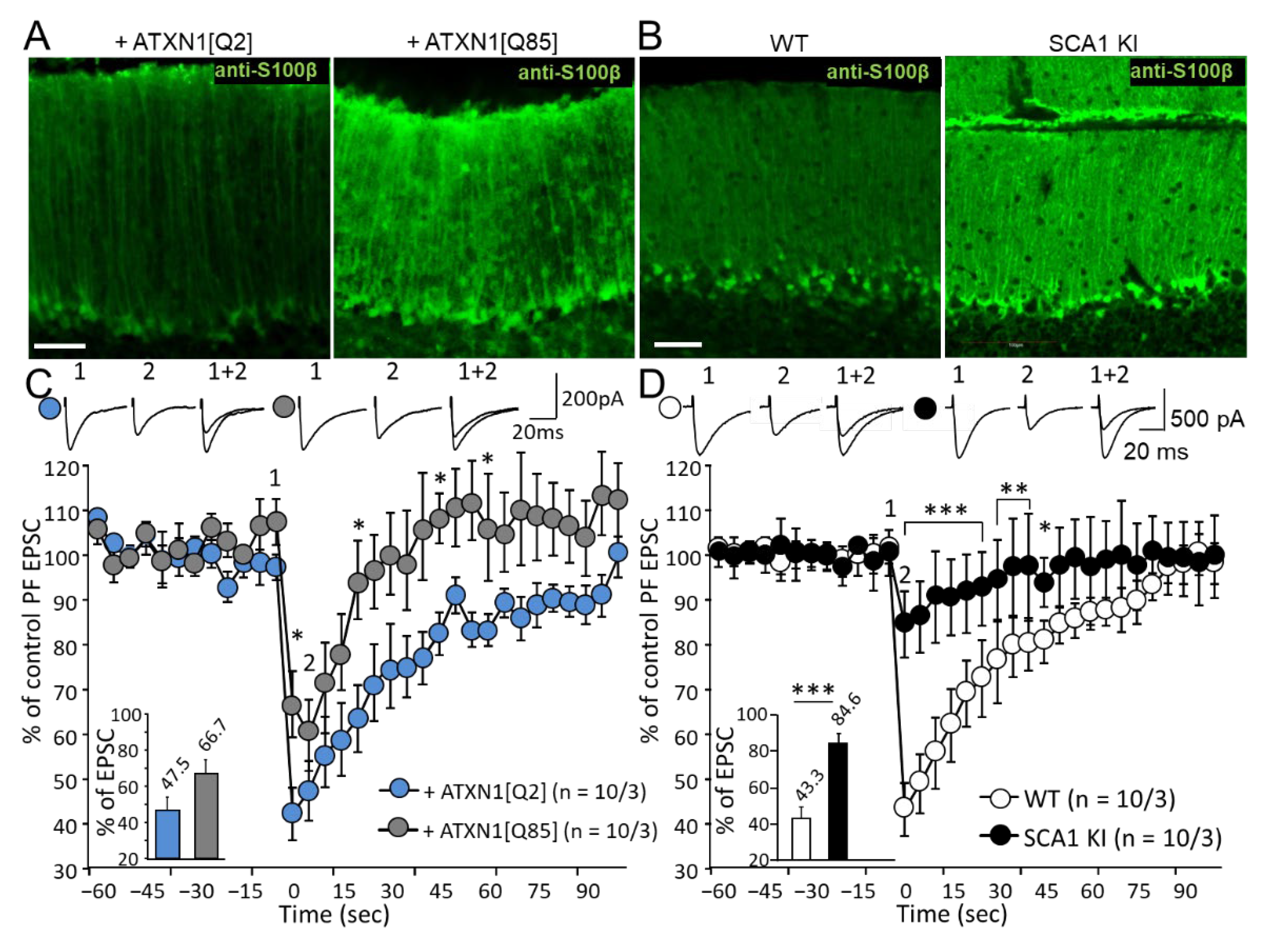
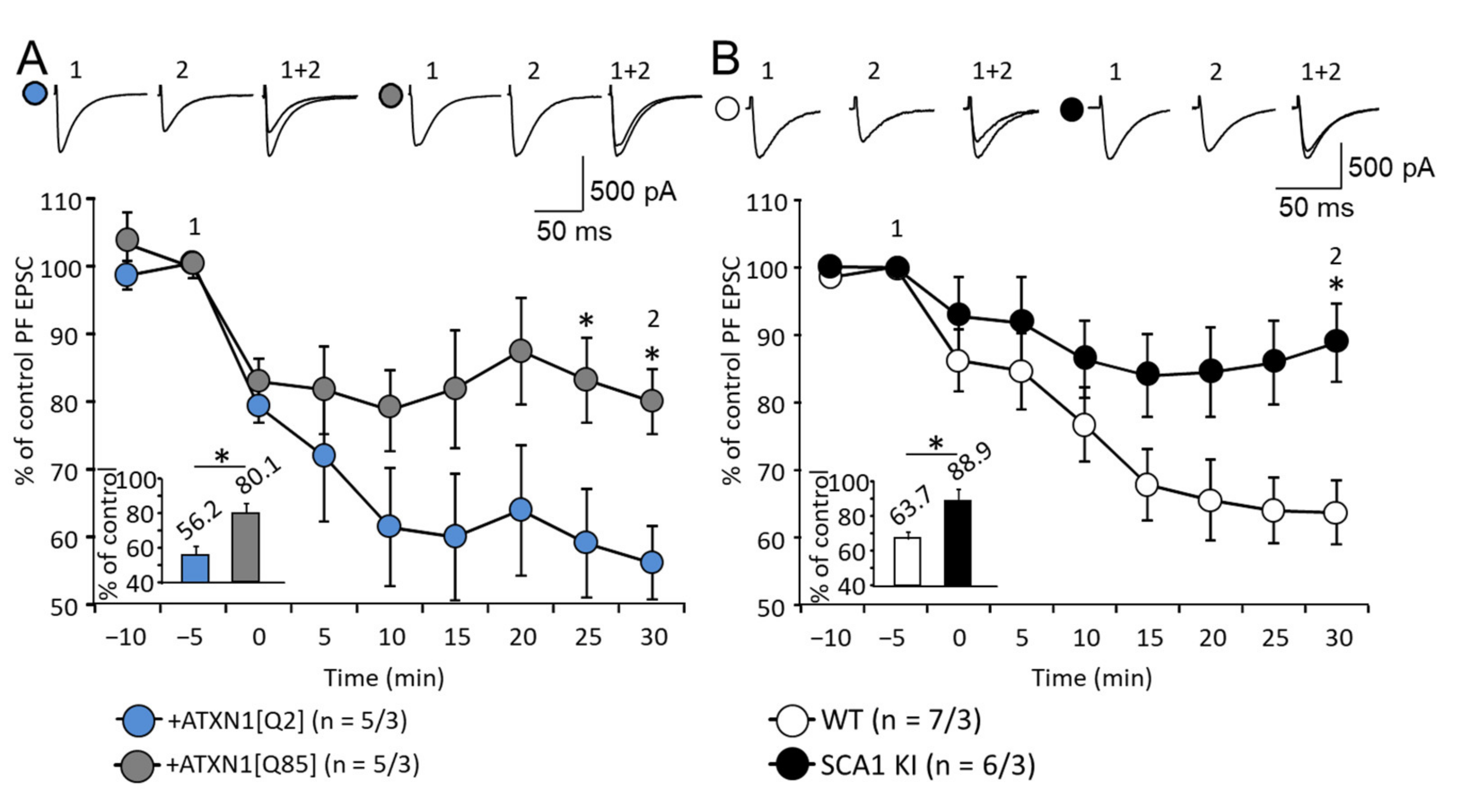
3.5. Expression of Mutant ATXN1 Affects Long-Term Synaptic Plasticity in PF-PC Synapses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ranum, L.P.W.; Duvick, L.A.; Rich, S.S.; Schut, L.J.; Litt, M.; Orr, H.T. Localization of the autosomal dominant HLA-linked spinocerebellar ataxia (SCA1) locus, in two kindreds, with an 8-cM subregion of chromosome 6p. Am. J. Hum. Genet. 1991, 49, 31–41. [Google Scholar] [PubMed]
- Zoghbi, H.Y.; Jodice, C.; Sandkuyl, L.A.; Kwiatkowski, T.J., Jr.; McCall, A.E.; Huntoon, S.A.; Lulli, P.; Spadaro, M.; Litt, M.; Cann, H.M. The gene for autosomal dominant spinocerebellar ataxia (SCA1) maps telomeric to the HLA complex and is closely linked to the D6S89 locus in three kindreds. Am. J. Hum. Genet. 1991, 49, 23–30. [Google Scholar]
- Di Donato, S.; Mariotti, C.; Taroni, F. Spinocerebellar ataxia type 1. Handb. Clin. Neurol. 2012, 103, 399–421. [Google Scholar] [PubMed]
- Serra, H.G.; Duvick, L.; Zu, T.; Carlson, K.; Stevens, S.; Jorgensen, N.; Lysholm, A.; Burright, E.; Zoghbi, H.Y.; Clark, H.B.; et al. RORalpha-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell 2006, 127, 697–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burright, E.N.; Clark, H.B.; Servadio, A.; Matilla, T.; Feddersen, R.M.; Yunis, W.S.; Duvick, L.A.; Zoghbi, H.Y.; Orr, H.T. SCA1 transgenic mice: A model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell 1995, 82, 937–948. [Google Scholar] [CrossRef] [Green Version]
- Shuvaev, A.N.; Hosoi, N.; Sato, Y.; Yanagihara, D.; Hirai, H. Progressive impairment of cerebellar mGluR signalling and its therapeutic potential for cerebellar ataxia in spinocerebellar ataxia type 1 model mice. J. Physiol. 2017, 595, 141–164. [Google Scholar] [CrossRef] [Green Version]
- Shiwaku, H.; Yagishita, S.; Eishi, Y.; Okazawa, H. Bergmann glia are reduced in spinocerebellar ataxia type 1. Neuroreport 2013, 24, 620–625. [Google Scholar] [CrossRef]
- Cvetanovic, M.; Ingram, M.; Orr, H.; Opal, P. Early activation of microglia and astrocytes in mouse models of spinocerebellar ataxia type 1. Neuroscience 2015, 289, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Shuvaev, A.N.; Belozor, O.S.; Mozhei, O.; Yakovleva, D.A.; Potapenko, I.V.; Shuvaev, A.N.; Smolnikova, M.V.; Salmin, V.V.; Salmina, A.B.; Hirai, H.; et al. Chronic optogenetic stimulation of Bergman glia leads to dysfunction of EAAT1 and Purkinje cell death, mimicking the events caused by expression of pathogenic ataxin-1. Neurobiol. Dis. 2021, 154, 105340. [Google Scholar] [CrossRef]
- Watase, K.; Weeber, E.J.; Xu, B.; Antalffy, B.; Yuva-Paylor, L.; Hashimoto, K.; Kano, M.; Atkinson, R.; Sun, Y.; Armstrong, D.L.; et al. A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron 2002, 34, 905–919. [Google Scholar] [CrossRef] [Green Version]
- Shiwaku, H.; Yoshimura, N.; Tamura, T.; Sone, M.; Ogishima, S.; Watase, K.; Tagawa, K.; Okazawa, H. Suppression of the novel ER protein Maxer by mutant ataxin-1 in Bergman glia contributes to non-cell-autonomous toxicity. EMBO J. 2010, 29, 2446–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Paton, J.F.; Kasparov, S. Viral vectors based on bidirectional cell-specific mammalian promoters and transcriptional amplification strategy for use in vitro and in vivo. BMC Biotechnol. 2008, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewinson, J.; Paton, J.F.; Kasparov, S. Viral gene delivery: Optimized protocol for production of high titer lentiviral vectors. Methods Mol. Biol. 2013, 998, 65–75. [Google Scholar] [PubMed]
- Deacon, R.M. Measuring motor coordination in mice. J. Vis. Exp. 2013, 29, e2609. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Shuvaev, A.N.; Konno, A.; Matsuzaki, Y.; Watanave, M.; Hirai, H. Regulatory connection between the expression level of classical protein kinase C and pruning of climbing fibers from cerebellar Purkinje cells. J. Neurochem. 2017, 143, 6. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.; Henning, H.A.; Konnerth, A. mGluR1/TRPC3-mediated synaptic transmission and calcium signaling in mammalian central neurons. Cold Spring Harb. Perspect. Biol. 2011, 3, a006726. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Rojas, F.; Sakaba, T.; Kawaguchi, S.Y. Ca(2+) current facilitation determines short-term facilitation at inhibitory synapses between cerebellar Purkinje cells. J. Physiol. 2015, 593, 4889–4904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucker, R.S.; Regehr, W.G. Short-Term Synaptic Plasticity. Annu. Rev. Physiol. 2002, 64, 355–405. [Google Scholar] [CrossRef] [Green Version]
- O’Donovan, M.J.; Rinzel, J. Synaptic depression: A dynamic regulator of synaptic communication with varied functional roles. Trends Neurosci. 1997, 20, 431–433. [Google Scholar] [CrossRef]
- Hablitz, L.M.; Gunesch, A.N.; Cravetchi, O.; Moldavan, M.; Allen, C.N. Cannabinoid Signaling Recruits Astrocytes to Modulate Presynaptic Function in the Suprachiasmatic Nucleus. eNeuro 2020, 7. [Google Scholar] [CrossRef]
- Gómez-Gonzalo, M.; Navarrete, M.; Perea, G.; Covelo, A.; Martín-Fernández, M.; Shigemoto, R.; Luján, R.; Araque, A. Endocannabinoids Induce Lateral Long-Term Potentiation of Transmitter Release by Stimulation of Gliotransmission. Cerebral Cortex 2015, 25, 3699–3712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarrete, M.; Diez, A.; Araque, A. Astrocytes in endocannabinoid signalling. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130599. [Google Scholar] [CrossRef] [PubMed]
- Edamakanti, C.R.; Do, J.; Didonna, A.; Martina, M.; Opal, P. Mutant ataxin1 disrupts cerebellar development in spinocerebellar ataxia type 1. J. Clin. Investig. 2018, 128, 2252–2265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rancz, E.A.; Häusser, M. Dendritic calcium spikes are tunable triggers of cannabinoid release and short-term synaptic plasticity in cerebellar Purkinje neurons. J. Neurosci. 2006, 26, 5428–5437. [Google Scholar] [CrossRef]
- Ito, M. Long-term depression. Annu. Rev. Neurosci. 1989, 12, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Kano, M.; Hashimoto, K.; Tabata, T. Type-1 metabotropic glutamate receptor in cerebellar Purkinje cells: A key molecule responsible for long-term depression, endocannabinoid signalling and synapse elimination. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 2173–2186. [Google Scholar] [CrossRef]
- Alvarez, V.A.; Sabatini, B.L. Anatomical and physiological plasticity of dendritic spines. Annu. Rev. Neurosci. 2007, 30, 79–97. [Google Scholar] [CrossRef] [Green Version]
- Borczyk, M.; Śliwińska, M.A.; Caly, A.; Bernas, T.; Radwanska, K. Neuronal plasticity affects correlation between the size of dendritic spine and its postsynaptic density. Sci. Rep. 2019, 9, 1693. [Google Scholar] [CrossRef]
- Qiu, D.L.; Knopfel, T. Presynaptically expressed long-term depression at cerebellar parallel fiber synapses. Pflugers Arch. 2009, 457, 865–875. [Google Scholar] [CrossRef] [Green Version]
- Kreitzer, A.C.; Regehr, W.G. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 2001, 29, 717–727. [Google Scholar] [CrossRef] [Green Version]
- Belozor, O.S.; Yakovleva, D.A.; Potapenko, I.V.; Shuvaev, A.N.; Smolnikova, M.V.; Vasilev, A.; Pozhilenkova, E.A.; Shuvaev, A.N. Extracellular S100β Disrupts Bergman Glia Morphology and Synaptic Transmission in Cerebellar Purkinje Cells. Brain Sci. 2019, 9, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konno, A.; Shuvaev, A.N.; Miyake, N.; Miyake, K.; Iizuka, A.; Matsuura, S.; Huda, F.; Nakamura, K.; Yanagi, S.; Shimada, T.; et al. Mutant ataxin-3 with an abnormally expanded polyglutamine chain disrupts dendritic development and metabotropic glutamate receptor signaling in mouse cerebellar Purkinje cells. Cerebellum 2014, 13, 29–41. [Google Scholar] [CrossRef]
- Bushart, D.D.; Chopra, R.; Singh, V.; Murphy, G.G.; Wulff, H.; Shakkottai, V.G. Targeting potassium channels to treat cerebellar ataxia. Ann. Clin. Transl. Neurol. 2018, 5, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Vig, P.J.S.; Lopez, M.E.; Wei, J.; D’Souza, D.R.; Subramony, S.H.; Henegar, J.; Fratkin, J.D. Glial S100B Positive Vacuoles In Purkinje Cells: Earliest Morphological Abnormality In SCA1 Transgenic Mice. J. NeurolSci. Turk. 2006, 23, 166–174. [Google Scholar]
- Vig, P.J.S.; Shao, Q.; Subramony, S.H.; Lopez, M.E.; Safaya, E. Bergmann Glial S100B Activates Myo-inositol Monophosphatase 1 and Co-localizes to Purkinje Cell Vacuoles in SCA1 Transgenic Mice. Cerebellum 2009, 8, 231–244. [Google Scholar] [CrossRef] [Green Version]
- Cerrato, V.; Mercurio, S.; Leto, K.; Fuca, E.; Hoxha, E.; Bottes, S.; Pagin, M.; Milanese, M.; Ngan, C.-Y.; Concina, G.; et al. Sox2 conditional mutation in mouse causes ataxic symptoms,cerebellar vermis hypoplasia, and postnatal defects of Bergmannglia. Glia 2018, 66, 1929–1946. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Cm (pF) | Ra (mΩ) | Rm (mΩ) |
|---|---|---|---|
| WT (n = 20/8) | 670.4 ± 26.7 | 11.6 ± 0.6 | 239.4 ± 25.0 |
| SCA1 KI (n = 22/8) | 534.1 ± 27.6 ** | 10.7 ± 0.8 | 253.0 ± 30.7 |
| +LVV-GFAP-Flag-ATXN1[Q2] (n = 12/3) | 748.4 ± 22.6 | 11.1 ± 0.5 | 236.6 ± 13.2 |
| +LVV-GFAP-Flag-ATXN1[Q85] (n = 13/3) | 508.6 ± 57.5 ††† | 11.4 ± 0.8 | 259.7 ± 18.0 |
| Genotype | PF EPSC (pA) | Rise Time (ms) | Decay Time (ms−1) | CF EPSC (pA) | Rise Time (ms) | Decay Time (ms−1) |
|---|---|---|---|---|---|---|
| WT (n = 13/3) | 844.8 ± 95.5 | 2.7 ± 0.2 | 10.3 ± 0.9 | 855.0 ± 67.7 | 1.3 ± 0.1 | 8.2 ± 0.5 |
| SCA1 KI (n = 15/3) | 743.2 ± 99.3 | 2.5 ± 0.4 | 12.2 ± 1.3 | 877.7 ± 110.1 | 1.2 ± 0.1 | 8.6 ± 0.3 |
| +LVV-GFAP-Flag-ATXN1[Q2] (n = 9/3) | 714.6 ± 84.1 | 2.4 ± 0.2 | 10.7 ± 2.0 | 767.9 ± 112.1 | 1.3 ± 0.3 | 11.4 ± 1.1 |
| +LVV-GFAP-Flag-ATXN1[Q85] (n = 8/3) | 801.9 ± 94.8 | 2.3 ± 0.2 | 13.6 ± 1.2 | 676.0 ± 86.5 | 1.4 ± 0.2 | 10.1 ± 1.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shuvaev, A.N.; Belozor, O.S.; Mozhei, O.I.; Shuvaev, A.N.; Fritsler, Y.V.; Khilazheva, E.D.; Mosyagina, A.I.; Hirai, H.; Teschemacher, A.G.; Kasparov, S. Indirect Negative Effect of Mutant Ataxin-1 on Short- and Long-Term Synaptic Plasticity in Mouse Models of Spinocerebellar Ataxia Type 1. Cells 2022, 11, 2247. https://doi.org/10.3390/cells11142247
Shuvaev AN, Belozor OS, Mozhei OI, Shuvaev AN, Fritsler YV, Khilazheva ED, Mosyagina AI, Hirai H, Teschemacher AG, Kasparov S. Indirect Negative Effect of Mutant Ataxin-1 on Short- and Long-Term Synaptic Plasticity in Mouse Models of Spinocerebellar Ataxia Type 1. Cells. 2022; 11(14):2247. https://doi.org/10.3390/cells11142247
Chicago/Turabian StyleShuvaev, Anton N., Olga S. Belozor, Oleg I. Mozhei, Andrey N. Shuvaev, Yana V. Fritsler, Elena D. Khilazheva, Angelina I. Mosyagina, Hirokazu Hirai, Anja G. Teschemacher, and Sergey Kasparov. 2022. "Indirect Negative Effect of Mutant Ataxin-1 on Short- and Long-Term Synaptic Plasticity in Mouse Models of Spinocerebellar Ataxia Type 1" Cells 11, no. 14: 2247. https://doi.org/10.3390/cells11142247