
Compensatory Upregulation of LPA2 and Activation of the PI3K-Akt Pathway Prevent LPA5-Dependent Loss of Intestinal Epithelial Cells in Intestinal Organoids


Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Intestinal Crypt Isolation and 3-D Culture of Enteroids
2.3. Treatment of Enteroids with Inhibitors
2.4. Treatment of Mice with LPA2 Antagonist
2.5. Immunofluorescence Staining of Mouse Intestine
2.6. Immunofluorescence Staining of Enteroids
2.7. Quantitative RT-PCR (qRT-PCR)
2.8. Statistical Analysis
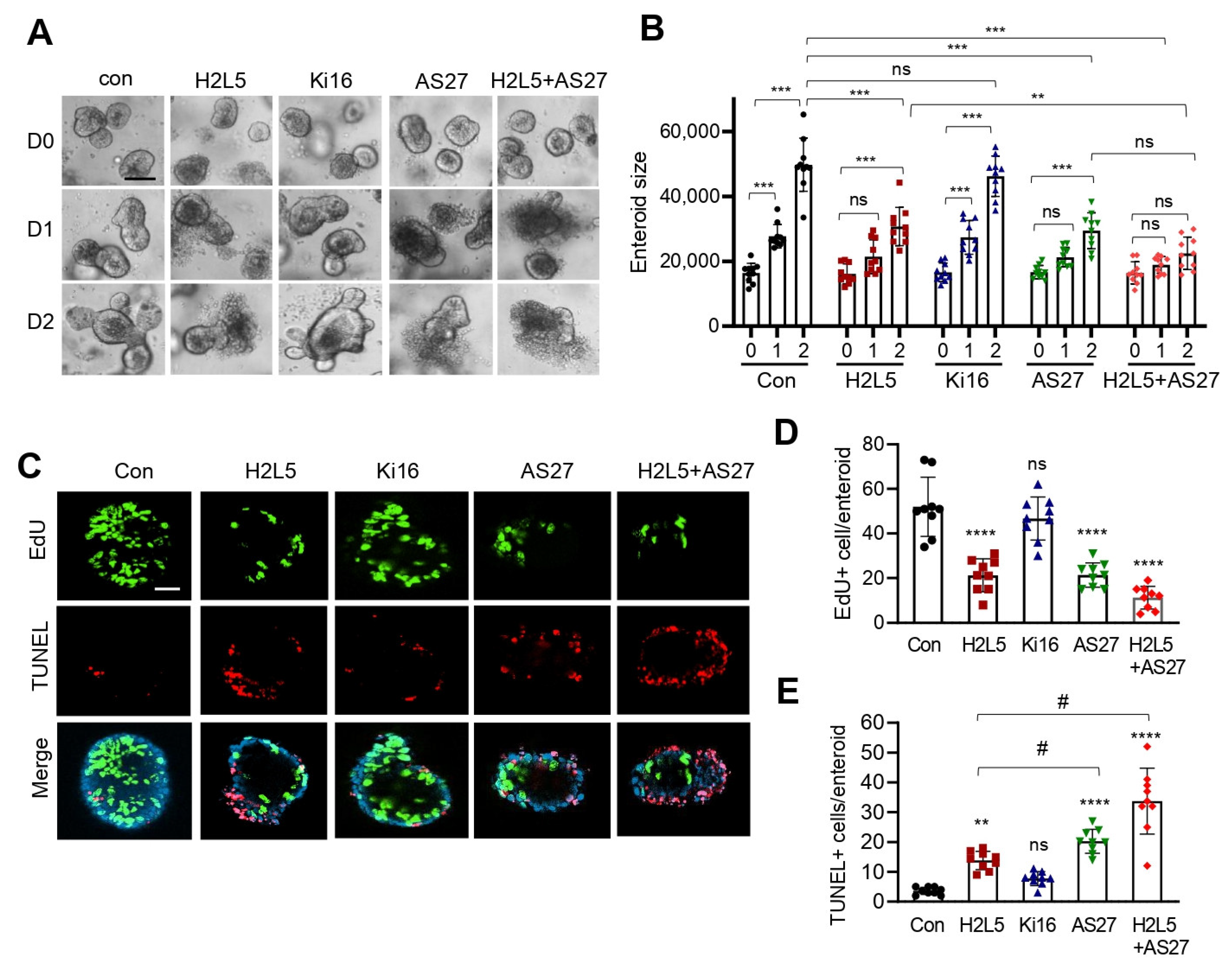
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Van der Flier, L.G.; Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.P.; Hellmich, M.R.; Townsend, C.M., Jr.; Evers, B.M. Role of gastrointestinal hormones in the proliferation of normal and neoplastic tissues. Endocr. Rev. 2003, 24, 571–599. [Google Scholar] [CrossRef]
- Drozdowski, L.; Thomson, A.B. Intestinal hormones and growth factors: Effects on the small intestine. World J. Gastroenterol. 2009, 15, 385–406. [Google Scholar] [CrossRef] [Green Version]
- Omi, J.; Kano, K.; Aoki, J. Current Knowledge on the Biology of Lysophosphatidylserine as an Emerging Bioactive Lipid. Cell Biochem. Biophys. 2021, 79, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Herr, D.R.; Noguchi, K.; Yung, Y.C.; Lee, C.W.; Mutoh, T.; Lin, M.E.; Teo, S.T.; Park, K.E.; Mosley, A.N.; et al. LPA receptors: Subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 157–186. [Google Scholar] [CrossRef] [Green Version]
- Tigyi, G. Aiming drug discovery at lysophosphatidic acid targets. Br. J. Pharmacol. 2010, 161, 241–270. [Google Scholar] [CrossRef] [Green Version]
- Contos, J.J.A.; Fukushima, N.; Weiner, J.A.; Kaushal, D.; Chun, J. Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior. Proc. Natl. Acad. Sci. USA 2000, 97, 13384–13389. [Google Scholar] [CrossRef] [Green Version]
- Contos, J.J.A.; Ishii, I.; Fukushima, N.; Kingsbury, M.A.; Ye, X.; Kawamura, S.; Brown, J.H.; Chun, J. Characterization of lpa2 (Edg4) and lpa1/lpa2 (Edg2/Edg4) Lysophosphatidic Acid Receptor Knockout Mice: Signaling Deficits without Obvious Phenotypic Abnormality Attributable to Lpamol. Cell. Biol. 2002, 22, 6921–6929. [Google Scholar]
- Ye, X.; Hama, K.; Contos, J.J.A.; Anliker, B.; Inoue, A.; Skinner, M.K.; Suzuki, H.; Amano, T.; Kennedy, G.; Arai, H.; et al. LPA3-mediated lysophosphatidic acid signalling in embryo implantation and spacing. Nature 2005, 435, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Sumida, H.; Noguchi, K.; Kihara, Y.; Abe, M.; Yanagida, K.; Hamano, F.; Sato, S.; Tamaki, K.; Morishita, Y.; Kano, M.R.; et al. LPA4 regulates blood and lymphatic vessel formation during mouse embryogenesis. Blood 2010, 116, 5060–5070. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, D.; Kobayashi, D.; Akahoshi, N.; Ohto-Nakanishi, T.; Yoshioka, K.; Takuwa, Y.; Mizuno, S.; Takahashi, S.; Ishii, S. Lysophosphatidic acid-induced YAP/TAZ activation promotes developmental angiogenesis by repressing Notch ligand Dll4. J. Clin. Investig. 2019, 129, 4332–4349. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.E.; Rivera, R.R.; Chun, J. Targeted deletion of LPA5 identifies novel roles for lysophosphatidic acid signaling in development of neuropathic pain. J. Biol. Chem. 2012, 287, 17608–17617. [Google Scholar] [CrossRef] [Green Version]
- Callaerts-Vegh, Z.; Leo, S.; Vermaercke, B.; Meert, T.; D’Hooge, R. LPA5 receptor plays a role in pain sensitivity, emotional exploration and reversal learning. Genes Brain Behav. 2012, 11, 1009–1019. [Google Scholar]
- Wang, M.; He, P.; Han, Y.; Dong, L.; Yun, C.C. Control of Intestinal Epithelial Permeability by Lysophosphatidic Acid Receptor. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1073–1092. [Google Scholar] [CrossRef]
- Liang, Z.; He, P.; Han, Y.; Yun, C.C. Survival of Stem Cells and Progenitors in the Intestine Is Regulated by LPA5-Dependent Signaling. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 129–150. [Google Scholar] [CrossRef]
- Jenkin, K.A.; He, P.; Yun, C.C. Expression of lysophosphatidic acid receptor 5 is necessary for the regulation of intestinal Na(+)/H(+) exchanger 3 by lysophosphatidic acid in vivo. Am. J. Physiol. Gastrointest Liver Physiol. 2018, 315, G433–G442. [Google Scholar] [CrossRef]
- Fells, J.I.; Tsukahara, R.; Liu, J.; Tigyi, G.; Parrill, A.L. Structure-based drug design identifies novel LPA3 antagonists. Bioorg. Med. Chem. 2009, 17, 7457–7464. [Google Scholar] [CrossRef] [Green Version]
- Volden, P.A.; Skor, M.N.; Johnson, M.B.; Singh, P.; Patel, F.N.; McClintock, M.K.; Brady, M.J.; Conzen, S.D. Mammary Adipose Tissue-Derived Lysophospholipids Promote Estrogen Receptor-Negative Mammary Epithelial Cell Proliferation. Cancer Prev. Res. 2016, 9, 367–378. [Google Scholar] [CrossRef] [Green Version]
- Ohta, H.; Sato, K.; Murata, N.; Damirin, A.; Malchinkhuu, E.; Kon, J.; Kimura, T.; Tobo, M.; Yamazaki, Y.; Watanabe, T.; et al. Ki16425, a Subtype-Selective Antagonist for EDG-Family Lysophosphatidic Acid Receptors. Mol. Pharmacol. 2003, 64, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Murai, N.; Hiyama, H.; Kiso, T.; Sekizawa, T.; Watabiki, T.; Oka, H.; Aoki, T. Analgesic effects of novel lysophosphatidic acid receptor 5 antagonist AS2717638 in rodents. Neuropharmacology 2017, 126, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Wang, D.; Iyer, S.; Ghaleb, A.M.; Shim, H.; Yang, V.W.; Chun, J.; Yun, C.C. The absence of LPA2 attenuates tumor formation in an experimental model of colitis-associated cancer. Gastroenterology 2009, 136, 1711–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.; Balazs, L.; Wang, D.A.; Van Middlesworth, L.; Tigyi, G.; Johnson, L.R. Lysophosphatidic acid protects and rescues intestinal epithelial cells from radiation- and chemotherapy-induced apoptosis. Gastroenterology 2002, 123, 206–216. [Google Scholar] [CrossRef]
- Rusovici, R.; Ghaleb, A.; Shim, H.; Yang, V.W.; Yun, C.C. Lysophosphatidic acid prevents apoptosis of Caco-2 colon cancer cells via activation of mitogen-activated protein kinase and phosphorylation of Bad. Biochim. Biophys. Acta 2007, 1770, 1194–1203. [Google Scholar] [CrossRef] [Green Version]
- Konno, T.; Kotani, T.; Setiawan, J.; Nishigaito, Y.; Sawada, N.; Imada, S.; Saito, Y.; Murata, Y.; Matozaki, T. Role of lysophosphatidic acid in proliferation and differentiation of intestinal epithelial cells. PLoS ONE 2019, 14, e0215255. [Google Scholar] [CrossRef] [Green Version]
- Sheng, H.; Shao, J.; Townsend, C.M., Jr.; Evers, B.M. Phosphatidylinositol 3-kinase mediates proliferative signals in intestinal epithelial cells. Gut 2003, 52, 1472–1478. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Bialkowska, A.; Rusovici, R.; Chanchevalap, S.; Shim, H.; Katz, J.P.; Yang, V.W.; Yun, C.C. Lysophosphatidic acid facilitates proliferation of colon cancer cells via induction of Kruppel-like factor. J. Biol. Chem. 2007, 282, 15541–15549. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Shuyu, E.; Tsukahara, R.; Valentine, W.J.; Durgam, G.; Gududuru, V.; Balazs, L.; Manickam, V.; Arsura, M.; Vanmiddlesworth, L.; et al. The Lysophosphatidic Acid Type 2 Receptor Is Required for Protection Against Radiation-Induced Intestinal Injury. Gastroenterology 2007, 132, 1834–1851. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Kimura, Y.; Gududuru, V.; Wu, W.; Balogh, A.; Szabo, E.; Thompson, K.E.; Yates, C.R.; Balazs, L.; Johnson, L.R.; et al. Mitigation of the hematopoietic and gastrointestinal acute radiation syndrome by octadecenyl thiophosphate, a small molecule mimic of lysophosphatidic acid. Radiat. Res. 2015, 183, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Kiss, G.N.; Lee, S.C.; Fells, J.I.; Liu, J.; Valentine, W.J.; Fujiwara, Y.; Thompson, K.E.; Yates, C.R.; Sumegi, B.; Tigyi, G. Mitigation of radiation injury by selective stimulation of the LPA(2) receptor. Biochim. Biophys. Acta 2013, 1831, 117–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.K.; Riederer, B.; Krabbenhoft, A.; Rausch, B.; Bonhagen, J.; Lehmann, U.; de Jonge, H.R.; Donowitz, M.; Yun, C.; Weinman, E.J.; et al. Differential roles of NHERF1, NHERF2, and PDZK1 in regulating CFTR-mediated intestinal anion secretion in mice. J. Clin. Investig. 2009, 119, 540–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, B.; Szabo, E.; Lee, S.C.; Balogh, A.; Norman, D.; Inoue, A.; Ono, Y.; Aoki, J.; Tigyi, G. The LPA2 receptor agonist Radioprotectin-1 spares Lgr5-positive intestinal stem cells from radiation injury in murine enteroids. Cell. Signal. 2018, 51, 23–33. [Google Scholar] [CrossRef]
- Lin, S.; Lee, S.J.; Shim, H.; Chun, J.; Yun, C.C. The Absence of LPA receptor 2 Reduces the Tumorigenesis by ApcMin Mutation in the Intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G1128–G1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Leoni, G.; Neumann, P.A.; Chun, J.; Nusrat, A.; Yun, C.C. Distinct phospholipase C-beta isozymes mediate lysophosphatidic acid receptor 1 effects on intestinal epithelial homeostasis and wound closure. Mol. Cell. Biol. 2013, 33, 2016–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedraza, C.; Sanchez-Lopez, J.; Castilla-Ortega, E.; Rosell-Valle, C.; Zambrana-Infantes, E.; Garcia-Fernandez, M.; Rodriguez de Fonseca, F.; Chun, J.; Santin, L.J.; Estivill-Torrus, G. Fear extinction and acute stress reactivity reveal a role of LPA(1) receptor in regulating emotional-like behaviors. Brain Struct Funct. 2014, 219, 1659–1672. [Google Scholar] [CrossRef]
- Moreno-Fernandez, R.D.; Nieto-Quero, A.; Gomez-Salas, F.J.; Chun, J.; Estivill-Torrus, G.; Rodriguez de Fonseca, F.; Santin, L.J.; Perez-Martin, M.; Pedraza, C. Effects of genetic deletion versus pharmacological blockade of the LPA1 receptor on depression-like behaviour and related brain functional activity. Dis. Models Mech. 2018, 11, dmm035519. [Google Scholar] [CrossRef] [Green Version]
- El-Brolosy, M.A.; Stainier, D.Y.R. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017, 13, e1006780. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, M.N.; Schreiber, K.H.; Zhang, Y.; Duc, A.C.; Rao, S.; Hale, J.S.; Academia, E.C.; Shah, S.R.; Morton, J.F.; Holstein, C.A.; et al. The ribosomal protein Rpl22 controls ribosome composition by directly repressing expression of its own paralog, Rpl22l. PLoS Genet. 2013, 9, e1003708. [Google Scholar]
- Dayton, T.L.; Gocheva, V.; Miller, K.M.; Israelsen, W.J.; Bhutkar, A.; Clish, C.B.; Davidson, S.M.; Luengo, A.; Bronson, R.T.; Jacks, T.; et al. Germline loss of PKM2 promotes metabolic distress and hepatocellular carcinoma. Genes Dev. 2016, 30, 1020–1033. [Google Scholar] [CrossRef] [Green Version]
- Dignass, A.U.; Sturm, A. Peptide growth factors in the intestine. Eur. J. Gastroenterol. Hepatol. 2001, 13, 763–770. [Google Scholar] [CrossRef]
- Wetzker, R.; Bohmer, F.D. Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat. Rev. Mol. Cell Biol. 2003, 4, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kawai, T.; O’Brien, S.; Thomas, W.; Harris, R.C.; Eguchi, S. Epidermal Growth Factor Receptor Transactivation: Mechanisms, Pathophysiology, and Potential Therapies in the Cardiovascular System. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 627–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tigyi, G.; Miledi, R. Lysophosphatidates bound to serum albumin activate membrane currents in Xenopus oocytes and neurite retraction in PC12 pheochromocytoma cells. J. Biol. Chem. 1992, 267, 21360–21367. [Google Scholar] [CrossRef]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Han, Y.; Jenkin, K.; Lee, S.J.; Sasaki, M.; Klapproth, J.M.; He, P.; Yun, C.C. Lysophosphatidic Acid Receptor 1 Is Important for Intestinal Epithelial Barrier Function and Susceptibility to Colitis. Am. J. Pathol. 2018, 188, 353–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellinghaus, D.; Folseraas, T.; Holm, K.; Ellinghaus, E.; Melum, E.; Balschun, T.; Laerdahl, J.K.; Shiryaev, A.; Gotthardt, D.N.; Weismuller, T.J.; et al. Genome-wide association analysis in primary sclerosing cholangitis and ulcerative colitis identifies risk loci at GPR35 and TCF. Hepatology 2013, 58, 1074–1083. [Google Scholar] [CrossRef]
- Imielinski, M.; Baldassano, R.N.; Griffiths, A.; Russell, R.K.; Annese, V.; Dubinsky, M.; Kugathasan, S.; Bradfield, J.P.; Walters, T.D.; Sleiman, P.; et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat. Genet. 2009, 41, 1335–1340. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, A.E.; Lappin, J.E.; Taylor, D.L.; Nicklin, S.A.; Milligan, G. GPR35 as a Novel Therapeutic Target. Front. Endocrinol. 2011, 2, 68. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 5′-3′ | ||
|---|---|---|
| Lpar1 | Forward | CACAGTCAGCAAGCTGGTGATG |
| Reverse | TCTCCGAGTATTGGGTCCTG | |
| Lpar2 | Forward | TCACTGGTCAATGCAGTGGT |
| Reverse | AAGGGTGGAGTCCATCAGTG | |
| Lpar3 | Forward | ACGGCTCCCATGAAGCTAAT |
| Reverse | TTCATGACGGAGTTGAGGAG | |
| Lpar4 | Forward | TGCATCAGTGTGGATGGTTT |
| Reverse | GAAGCCTTCAAAGCAAGTCG | |
| Lpar5 | Forward | GCTCCAGTGCCCTGACTATC |
| Reverse | GGGAAGTGACAGGGTGAAGA | |
| Lpar6 | Forward | TGACTGTGAACCACAGAGCA |
| Reverse | ACTTTGCAACACGGAATTGG | |
| ACTB | Forward | AGCCATGTACGTAGCCATCC |
| Reverse | TCTCAGCTGTGGTGGTGAAG | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, Z.; Yun, C.C. Compensatory Upregulation of LPA2 and Activation of the PI3K-Akt Pathway Prevent LPA5-Dependent Loss of Intestinal Epithelial Cells in Intestinal Organoids. Cells 2022, 11, 2243. https://doi.org/10.3390/cells11142243
Liang Z, Yun CC. Compensatory Upregulation of LPA2 and Activation of the PI3K-Akt Pathway Prevent LPA5-Dependent Loss of Intestinal Epithelial Cells in Intestinal Organoids. Cells. 2022; 11(14):2243. https://doi.org/10.3390/cells11142243
Chicago/Turabian StyleLiang, Zhongxing, and C. Chris Yun. 2022. "Compensatory Upregulation of LPA2 and Activation of the PI3K-Akt Pathway Prevent LPA5-Dependent Loss of Intestinal Epithelial Cells in Intestinal Organoids" Cells 11, no. 14: 2243. https://doi.org/10.3390/cells11142243