
Inflammasome Activation in Retinal Pigment Epithelium from Human Donors with Age-Related Macular Degeneration

 ,
,  ,
,
Abstract
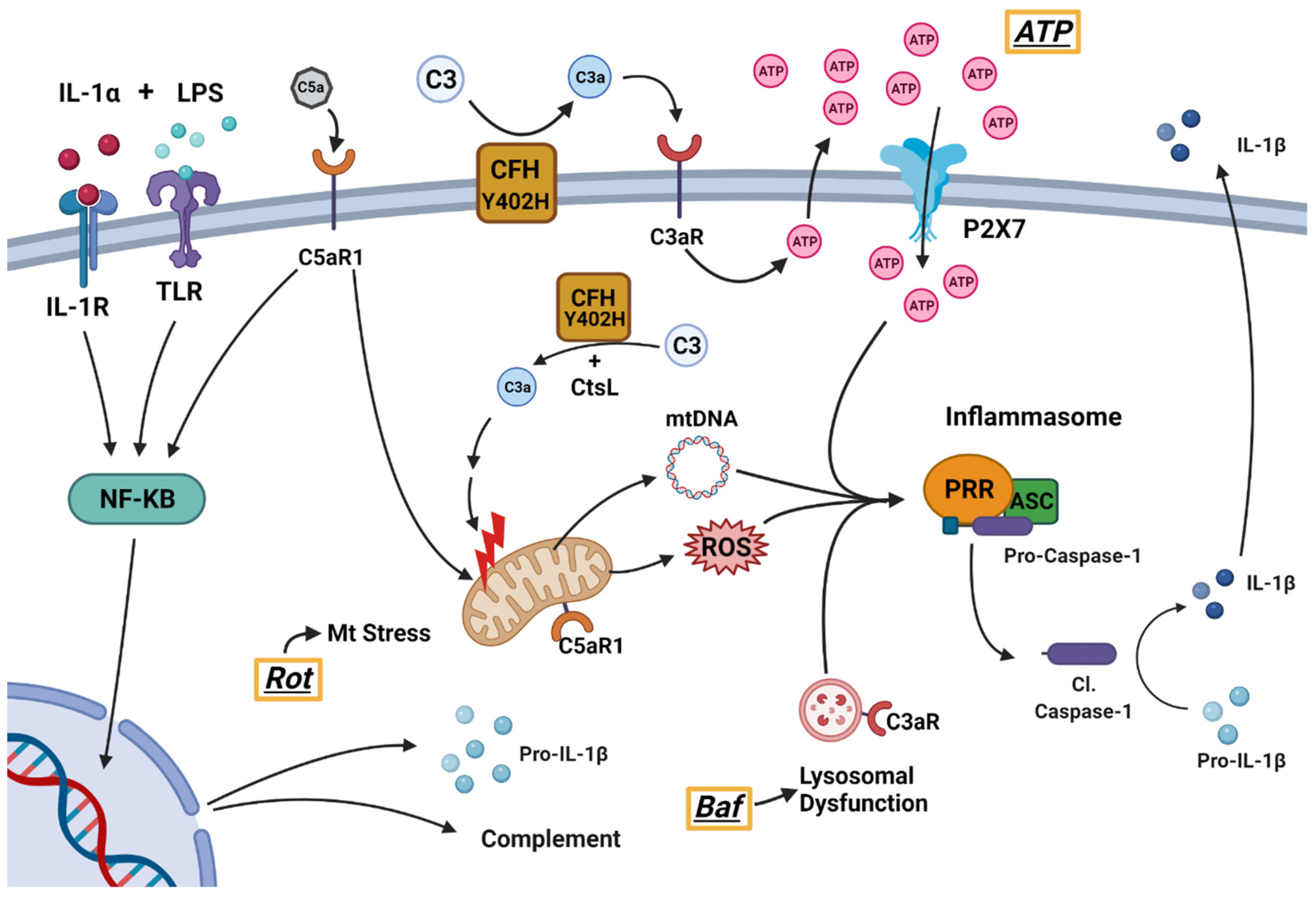
:1. Introduction
2. Materials and Methods
2.1. Human Eye Procurement and Grading for AMD
2.2. Complement Factor H Y402H Genotyping
2.3. Sample Preparation and Nano Liquid Chromatography (LC)-Mass Spectrometry (MS/MS) Analysis
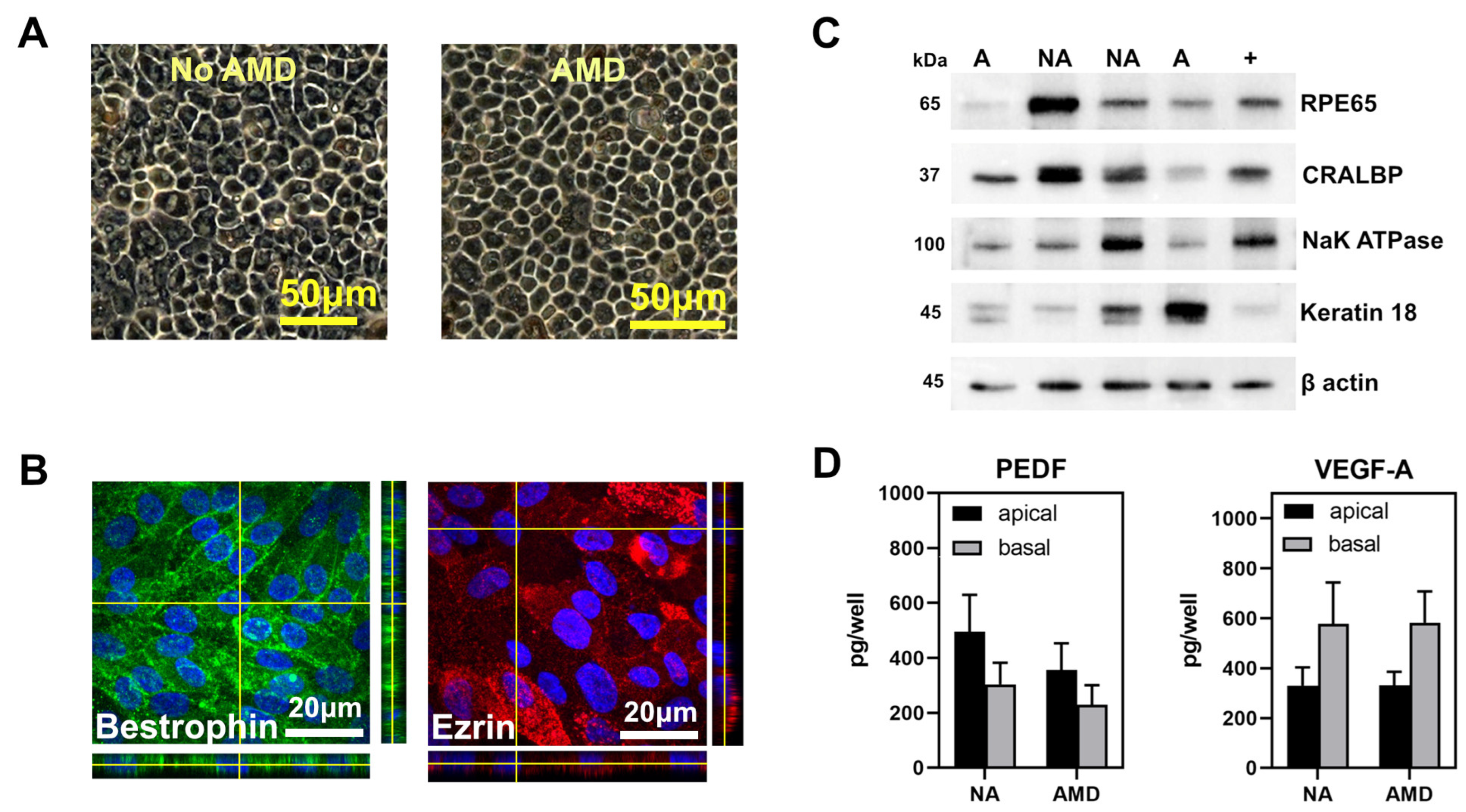
2.4. Cell Culture
2.5. Immunofluorescence
2.6. Western Blotting
2.7. ELISA
2.8. RNA Extraction, cDNA Synthesis, PCR
2.9. Live/Dead Assay
2.10. Detecting Cytosolic mtDNA
2.11. Statistical Analysis
3. Results
3.1. Identification of Complement and Inflammasome Proteins in RPE Tissue
3.2. haRPE Cultures to Investigate Inflammasome Activation
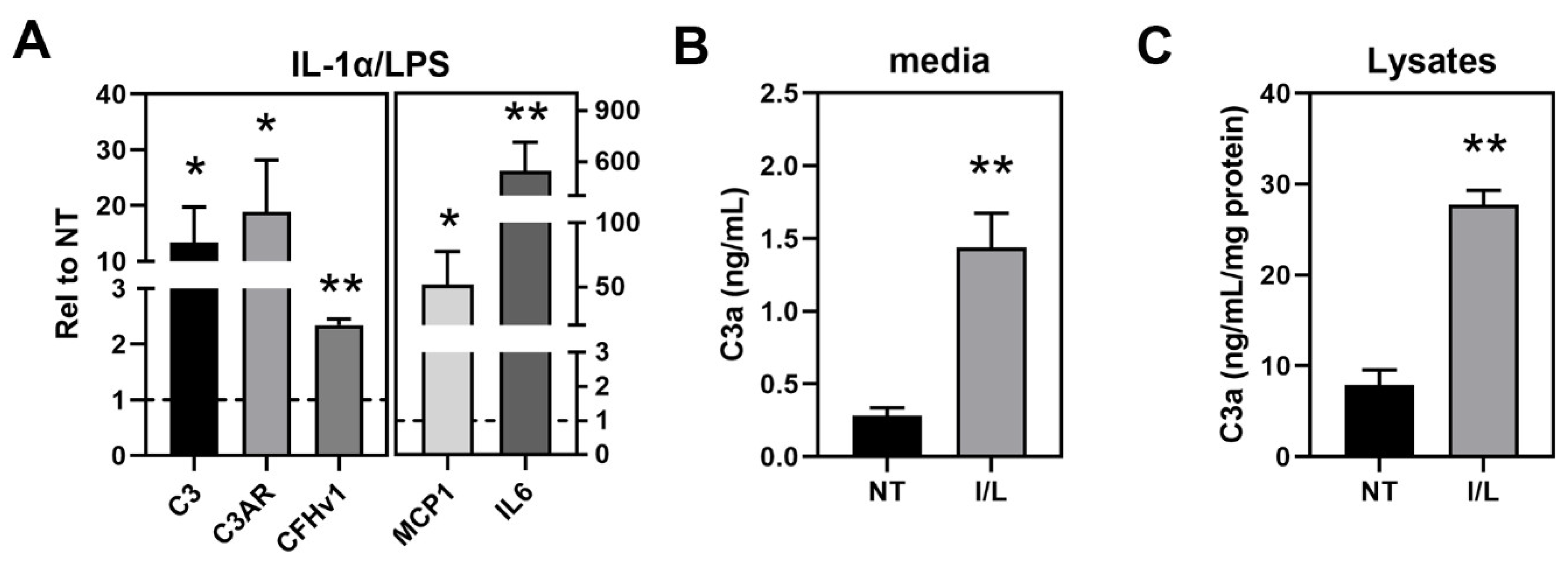
3.3. Investigation of Complement Pathway and Inflammasome Markers in haRPE Cells
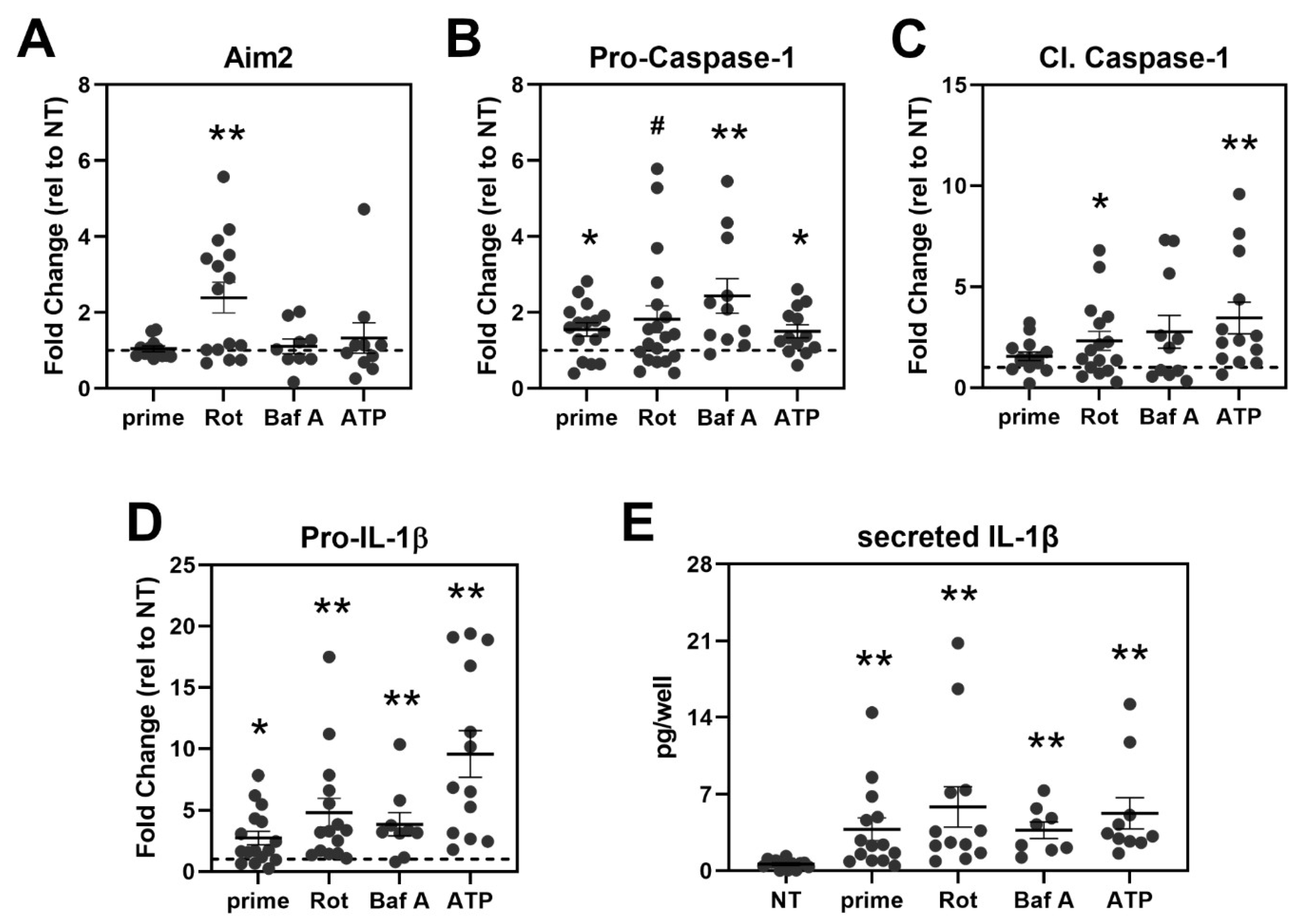
3.4. Inflammasome Assembly in haRPE Cells after Priming and Activation
3.5. Treatment Activates Caspase-1 and Triggers IL-1β Secretion in haRPE Cells
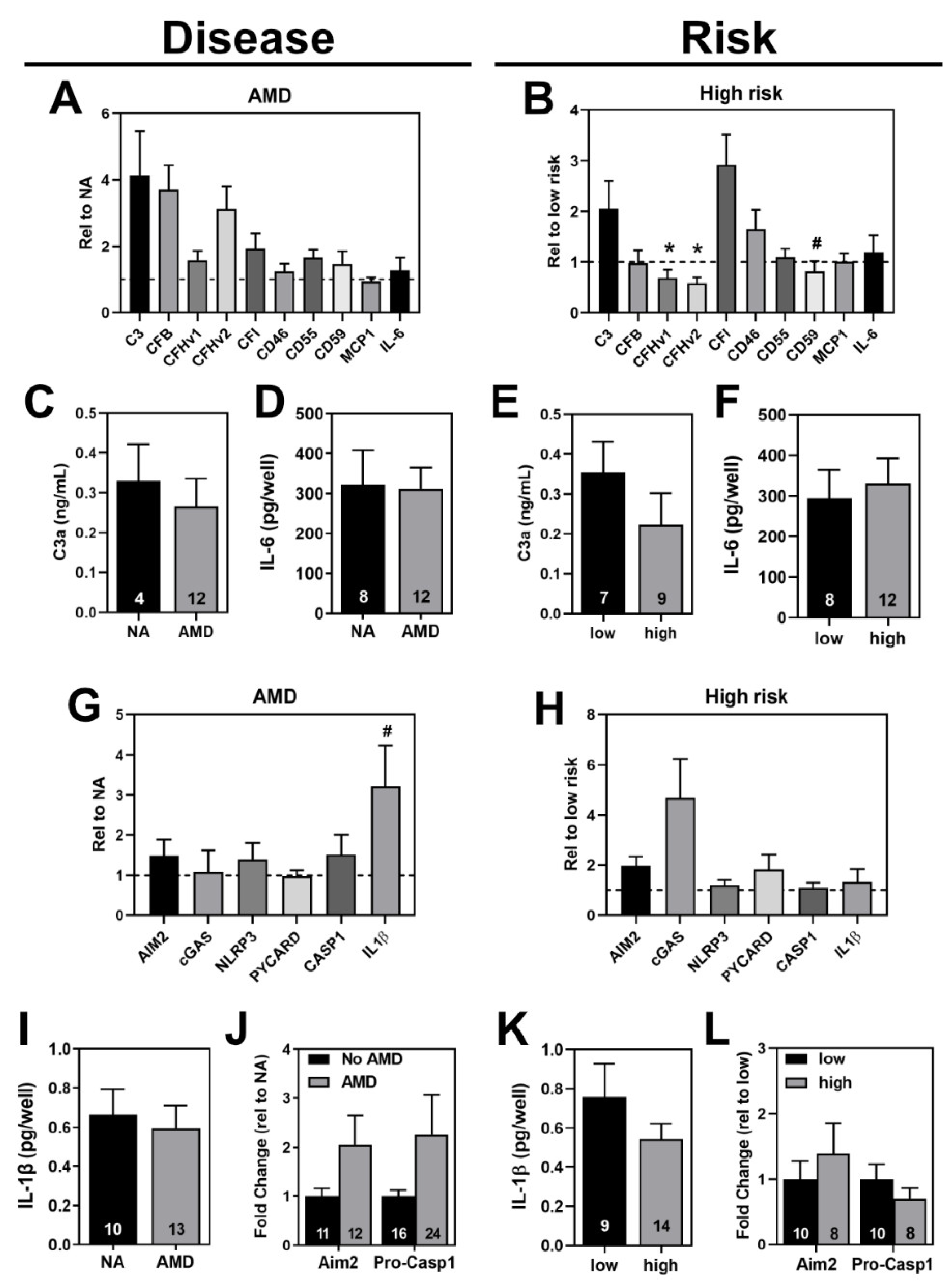
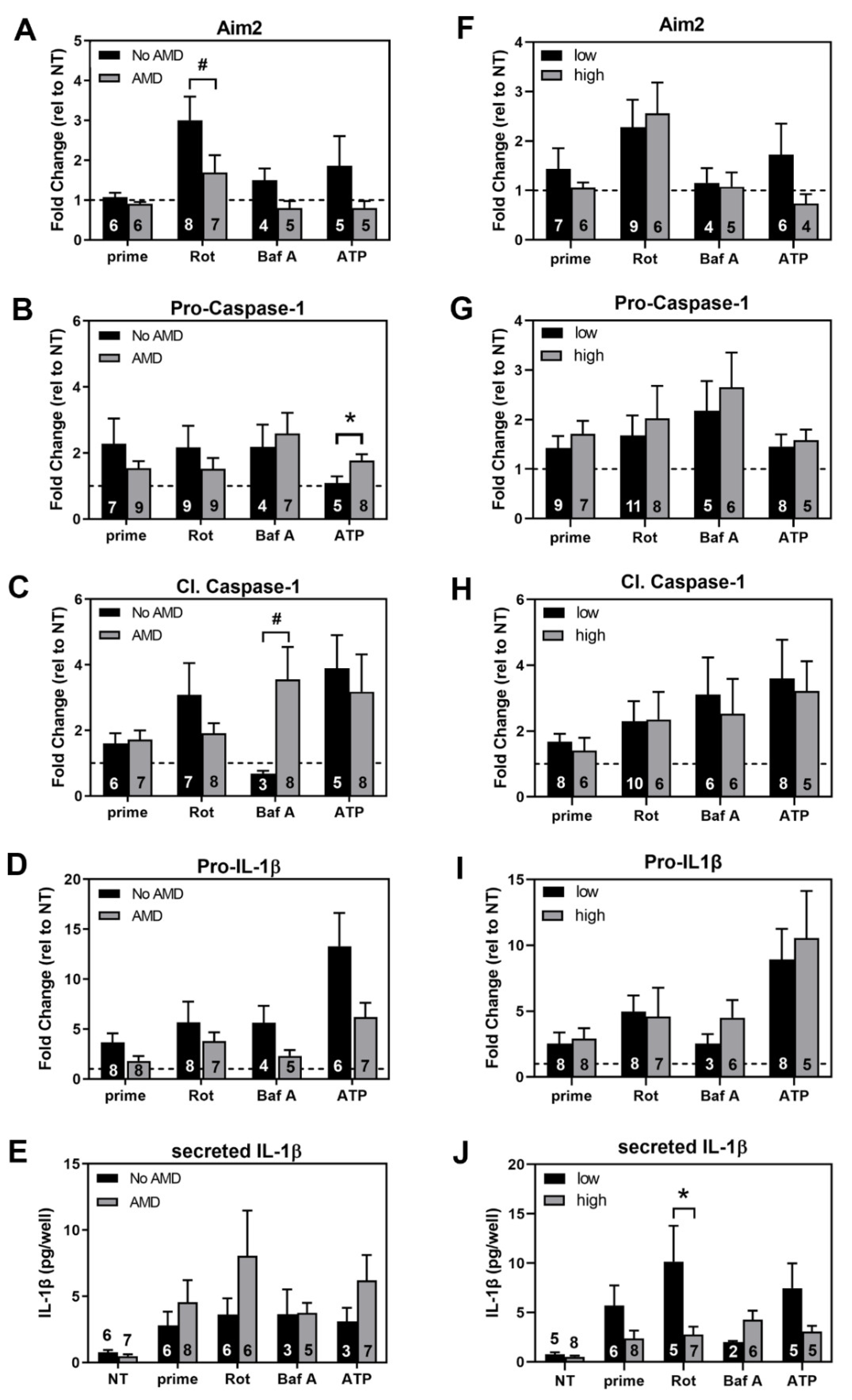
3.6. Comparing Inflammasome Activation in haRPE Cells by Disease State and Genotype
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bowes Rickman, C.; Farsiu, S.; Toth, C.A.; Klingeborn, M. Dry Age-Related Macular Degeneration: Mechanisms, Therapeutic Targets, and Imaging. Investig. Opthalmology Vis. Sci. 2013, 54, ORSF68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Layana, A.; Cabrera-López, F.; García-Arumí, J.; Arias-Barquet, L.; Ruiz-Moreno, J.M. Early and intermediate age-related macular degeneration: Update and clinical review. Clin. Interv. Aging 2017, 12, 1579–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [Green Version]
- Buschini, E.; Zola, M.; Fea, A.M.; Lavia, C.A.; Nassisi, M.; Pignata, G.; Zola, M.; Grignolo, F.M. Recent developments in the management of dry age-related macular degeneration. Clin. Ophthalmol. 2015, 9, 563. [Google Scholar] [CrossRef] [Green Version]
- Ross, R.J.; Verma, V.; Rosenberg, K.I.; Chan, C.C.; Tuo, J. Genetic markers and biomarkers for age-related macular degeneration. Expert Rev. Ophthalmol. 2007, 2, 443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangnon, R.E. Effect of the Y402H Variant in the complement factor h gene on the incidence and progression of age-related macular degeneration. Arch. Ophthalmol. 2012, 130, 1169–1176. [Google Scholar] [CrossRef] [Green Version]
- Sofat, R.; Casas, J.P.; Webster, A.R.; Bird, A.C.; Mann, S.S.; Yates, J.R.; Moore, A.T.; Sepp, T.; Cipriani, V.; Bunce, C.; et al. Complement factor H genetic variant and age-related macular degeneration: Effect size, modifiers and relationship to disease subtype. Int. J. Epidemiol. 2012, 41, 250–262. [Google Scholar] [CrossRef]
- Anderson, D.H.; Radeke, M.J.; Gallo, N.B.; Chapin, E.A.; Johnson, P.T.; Curletti, C.R.; Hancox, L.S.; Hu, J.; Ebright, J.N.; Malek, G.; et al. The pivotal role of the complement system in aging and age-related macular degeneration: Hypothesis re-visited. Prog. Retin. Eye Res. 2010, 29, 95–112. [Google Scholar] [CrossRef] [Green Version]
- Borras, C.; Canonica, J.; Jorieux, S.; Abache, T.; el Sanharawi, M.; Klein, C.; Delaunay, K.; Jonet, L.; Salvodelli, M.; Naud, M.C.; et al. CFH exerts anti-oxidant effects on retinal pigment epithelial cells independently from protecting against membrane attack complex. Sci. Rep. 2019, 9, 13873. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.; Leffler, J.; Smoląg, K.I.; Mytych, J.; Björk, A.; Chaves, L.D.; Alexander, J.J.; Quigg, R.J.; Blom, A.M. Factor H uptake regulates intracellular C3 activation during apoptosis and decreases the inflammatory potential of nucleosomes. Cell Death Differ. 2016, 23, 903–911. [Google Scholar] [CrossRef]
- Leffler, J.; Herbert, A.P.; Norström, E.; Schmidt, C.Q.; Barlow, P.N.; Blom, A.M.; Martin, M. Annexin-II, DNA, and histones serve as factor H ligands on the surface of apoptotic cells. J. Biol. Chem. 2010, 285, 3766–3776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, S.J.; Bishop, P.N.; Day, A.J. Complement factor H and age-related macular degeneration: The role of glycosaminoglycan recognition in disease pathology. Biochem. Soc. Trans. 2010, 38, 1342–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Ebeling, M.C.; Kapphahn, R.J.; Terluk, M.R.; Fisher, C.R.; Polanco, J.R.; Roehrich, H.; Leary, M.M.; Geng, Z.; Dutton, J.R.; et al. Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol. 2017, 13, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Terluk, M.R.; Kapphahn, R.J.; Soukup, L.M.; Gong, H.; Gallardo, C.; Montezuma, S.R.; Ferrington, D.A. Investigating mitochondria as a target for treating age-related macular degeneration. J. Neurosci. 2015, 35, 7304–7311. [Google Scholar] [CrossRef] [Green Version]
- Fisher, C.R.; Ebeling, M.C.; Geng, Z.; Kapphahn, R.J.; Roehrich, H.; Montezuma, S.R.; Dutton, J.R.; Ferrington, D.A. Human iPSC- and primary-retinal pigment epithelial cells for modeling age-related macular degeneration. Antioxidants 2022, 11, 605. [Google Scholar] [CrossRef] [PubMed]
- Golestaneh, N.; Chu, Y.; Xiao, Y.-Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2018, 8, e2537. [Google Scholar] [CrossRef]
- Guha, S.; Baltazar, G.C.; Coffey, E.E.; Tu, L.; Lim, J.C.; Beckel, J.M.; Patel, S.; Eysteinsson, T.; Lu, W.; O’Brien-Jenkins, A.; et al. Lysosomal alkalinization, lipid oxidation, and reduced phagosome clearance triggered by activation of the P2X7 receptor. FASEB J. 2013, 27, 4500–4509. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, C.H. Release of ATP by a human retinal pigment epithelial cell line: Potential for autocrine stimulation through subretinal space. J. Physiol. 2001, 534, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Liszewski, M.K.; Kolev, M.; Le Friec, G.; Leung, M.; Bertram, P.G.; Fara, A.F.; Subias, M.; Pickering, M.C.; Drouet, C.; Meri, S.; et al. Intracellular complement activation sustains t cell homeostasis and mediates effector differentiation. Immunity 2013, 39, 1143–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbore, G.; Kemper, C. A novel “complement–metabolism–inflammasome axis” as a key regulator of immune cell effector function. Eur. J. Immunol. 2016, 46, 1563–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarallo, V.; Hirano, Y.; Gelfand, B.D.; Dridi, S.; Kerur, N.; Kim, Y.; Cho, W.G.; Kaneko, H.; Fowler, B.J.; Bogdanovich, S.; et al. DICER1 Loss and Alu RNA Induce Age-Related Macular Degeneration via the NLRP3 Inflammasome and MyD88. Cell 2012, 149, 847–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, W.A.; Thein, T.; Kinnunen, K.; Lashkari, K.; Gregory, M.S.; D’Amore, P.A.; Ksander, B.R. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: Implications for age-related macular degeneration. Investig. Opthalmology Vis. Sci. 2013, 54, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Wang, J.C.C.; Gao, J.; Wong, M.; To, E.; White, V.A.; Cui, J.Z.; Matsubara, J.A. CFH Y402H polymorphism and the complement activation product C5a: Effects on NF-κB activation and inflammasome gene regulation. Br. J. Ophthalmol. 2016, 100, 713–718. [Google Scholar] [CrossRef]
- Doyle, S.L.; Campbell, M.; Ozaki, E.; Salomon, R.G.; Mori, A.; Kenna, P.F.; Farrar, G.J.; Kiang, A.S.; Humphries, M.M.; Lavelle, E.C.; et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat. Med. 2012, 18, 791–798. [Google Scholar] [CrossRef]
- Karunadharma, P.P.; Nordgaard, C.L.; Olsen, T.W.; Ferrington, D.A. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Investig. Opthalmol. Vis. Sci. 2010, 51, 5470–5479. [Google Scholar] [CrossRef] [Green Version]
- Olsen, T.W.; Feng, X. The Minnesota grading system of eye bank eyes for age-related macular degeneration. Investig. Opthalmol. Vis. Sci. 2004, 45, 4484–4490. [Google Scholar] [CrossRef]
- Ebeling, M.C.; Geng, Z.; Kapphahn, R.J.; Roehrich, H.; Montezuma, S.R.; Dutton, J.R.; Ferrington, D.A. Impaired mitochondrial function in iPSC-retinal pigment epithelium with the complement factor H polymorphism for age-related macular degeneration. Cells 2021, 10, 789. [Google Scholar] [CrossRef]
- Shen, S.; An, B.; Wang, X.; Hilchey, S.P.; Li, J.; Cao, J.; Tian, Y.; Hu, C.; Jin, L.; Ng, A.; et al. Surfactant cocktail-aided extraction/precipitation/on-pellet digestion strategy enables efficient and reproducible sample preparation for large-scale quantitative proteomics. Anal. Chem. 2018, 90, 10350–10359. [Google Scholar] [CrossRef]
- An, B.; Zhang, M.; Johnson, R.W.; Qu, J. Surfactant-aided precipitation/on-pellet-digestion (SOD) procedure provides robust and rapid sample preparation for reproducible, accurate and sensitive LC/MS quantification of therapeutic protein in plasma and tissues. Anal. Chem. 2015, 87, 4023–4029. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Shen, S.; Li, J.; Hu, Q.; Nie, L.; Tu, C.; Wang, X.; Poulsen, D.J.; Orsburn, B.C.; Wang, J.; et al. IonStar enables high-precision, low-missing-data proteomics quantification in large biological cohorts. Proc. Natl. Acad. Sci. USA 2018, 115, E4767–E4776. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Jin, L.; Hu, C.; Shen, S.; Qian, S.; Ma, M.; Zhu, X.; Li, F.; Wang, J.; Tian, Y.; et al. Ultra-high-resolution ionstar strategy enhancing accuracy and precision of MS1-based proteomics and an extensive comparison with state-of-the-art SWATH-MS in large-cohort quantification. Anal. Chem. 2021, 93, 4884–4893. [Google Scholar] [CrossRef]
- Bronner, D.N.; O’Riordan, M.X. Measurement of mitochondrial DNA release in response to ER stress. BioProtocol 2016, 6, e1839. [Google Scholar] [CrossRef] [PubMed]
- Ebeling, M.C.; Polanco, J.R.; Qu, J.; Tu, C.; Montezuma, S.R.; Ferrington, D.A. Improving retinal mitochondrial function as a treatment for age-related macular degeneration. Redox Biol. 2020, 34, 101552. [Google Scholar] [CrossRef] [PubMed]
- Kosmidou, C.; Efstathiou, N.E.; Hoang M v Notomi, S.; Konstantinou, E.K.; Hirano, M.; Takahashi, K.; Maidana, D.E.; Tsoka, P.; Young, L.; Gragoudas, E.S. Issues with the specificity of immunological reagents for NLRP3: Implications for age-related macular degeneration. Sci. Rep. 2018, 8, 461. [Google Scholar] [CrossRef] [PubMed]
- Cerniauskas, E.; Kurzawa-Akanbi, M.; Xie, L.; Hallam, D.; Moya-Molina, M.; White, K.; Steel, D.; Doherty, M.; Whitfield, P.; Al-Aama, J.; et al. Complement modulation reverses pathology in Y402H-retinal pigment epithelium cell model of age-related macular degeneration by restoring lysosomal function. Stem Cells Transl. Med. 2020, 9, 1585–1603. [Google Scholar] [CrossRef]
- Gong, J.; Cai, H.; Noggle, S.; Paull, D.; Rizzolo, L.J.; del Priore, L.V.; Fields, M.A. Stem cell-derived retinal pigment epithelium from patients with age-related macular degeneration exhibit reduced metabolism and matrix interactions. Stem Cells Transl. Med. 2020, 9, 364–376. [Google Scholar] [CrossRef] [Green Version]
- Brandstetter, C.; Holz, F.G.; Krohne, T.U. Complement component C5a primes retinal pigment epithelial cells for inflammasome activation by lipofuscin-mediated photooxidative damage. J. Biol. Chem. 2015, 290, 31189–31198. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, N.; Wolf, H.N.; Enzbrenner, A.; Schikora, J.; Reichenthaler, M.; Enzmann, V.; Pauly, D. Properdin modulates complement component production in stressed human primary retinal pigment epithelium cells. Antioxidants 2020, 9, 793. [Google Scholar] [CrossRef]
- Hallam, D.; Collin, J.; Bojic, S.; Chichagova, V.; Buskin, A.; Xu, Y.; Lafage, L.; Otten, E.; Anyfantis, G.; Mellough, C.; et al. An iPSC patient specific model of CFH (Y402H) polymorphism displays characteristic features of AMD and indicates a beneficial role for UV light exposure. Stem Cells 2017, 35, 2305–2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piippo, N.; Korkmaz, A.; Hytti, M.; Kinnunen, K.; Salminen, A.; Atalay, M.; Kaarniranta, K.; Kauppinen, A. Decline in cellular clearance systems induces inflammasome signaling in human ARPE-19 cells. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2014, 1843, 3038–3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandstetter, C.; Mohr, L.K.M.; Latz, E.; Holz, F.G.; Krohne, T.U. Light induces NLRP3 inflammasome activation in retinal pigment epithelial cells via lipofuscin-mediated photooxidative damage. J. Mol. Med. 2015, 93, 905–916. [Google Scholar] [CrossRef] [Green Version]
- Mohr, L.K.M.; Hoffmann, A.V.; Brandstetter, C.; Holz, F.G.; Krohne, T.U. Effects of inflammasome activation on secretion of inflammatory cytokines and vascular endothelial growth factor by retinal pigment epithelial cells. Investig. Opthalmology Vis. Sci. 2015, 56, 6404–6413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dib, B.; Lin, H.; Maidana, D.E.; Tian, B.; Miller, J.B.; Bouzika, P.; Miller, J.W.; Vavvas, D.G. Mitochondrial DNA has a pro-inflammatory role in, A.M.D. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2015, 1853, 2897–2906. [Google Scholar] [CrossRef] [Green Version]
- Gnanaguru, G.; Choi, A.R.; Amarnani, D.; D’Amore, P.A. Oxidized lipoprotein uptake through the cd36 receptor activates the nlrp3 inflammasome in human retinal pigment epithelial cells. Investig. Opthalmol. Vis. Sci. 2016, 57, 4704–4712. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, H.; Dridi, S.; Tarallo, V.; Gelfand, B.D.; Fowler, B.J.; Cho, W.G.; Kleinman, M.E.; Ponicsan, S.L.; Hauswirth, W.W.; Chiodo, V.A.; et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 2011, 471, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Hytti, M.; Korhonen, E.; Hongisto, H.; Kaarniranta, K.; Skottman, H.; Kauppinen, A. Differential expression of inflammasome-related genes in induced pluripotent stem-cell-derived retinal pigment epithelial cells with or without history of age-related macular degeneration. Int. J. Mol. Sci. 2021, 22, 6800. [Google Scholar] [CrossRef]
- Wang, Y.; Hanus, J.; Abu-Asab, M.; Shen, D.; Ogilvy, A.; Ou, J.; Chu, X.K.; Shi, G.; Li, W.; Wang, S.; et al. NLRP3 upregulation in retinal pigment epithelium in age-related macular degeneration. Int. J. Mol. Sci. 2016, 17, 73. [Google Scholar] [CrossRef]
- Cao, S.; Ko, A.; Partanen, M.; Pakzad-Vaezi, K.; Merkur, A.B.; Albiani, D.A.; Kirker, A.W.; Wang, A.; Cui, J.Z.; Forooghian, F.; et al. Relationship between systemic cytokines and complement factor H Y402H polymorphism in patients with dry age-related macular degeneration. Am. J. Ophthalmol. 2013, 156, 1176–1183. [Google Scholar] [CrossRef] [Green Version]
- Miyagishima, K.J.; Wan, Q.; Corneo, B.; Sharma, R.; Lotfi, M.R.; Boles, N.C.; Hua, F.; Maminishkis, A.; Zhang, C.; Blenkinsop, T.; et al. In pursuit of authenticity: Induced pluripotent stem cell-derived retinal pigment epithelium for clinical applications. Stem Cells Transl. Med. 2016, 5, 1562–1574. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Uniprot ID | # Unique Peptides B (Average) | % Sequence Coverage C (Average) |
|---|---|---|---|
| Complement component 1 Q subcomponent-binding protein (C1QBP) | Q07021 | 11/12 | 62/62 |
| Complement C1q subcomponent subunit B | P02746 | 0/4 | 0/22 |
| Complement C1q subcomponent subunit C (C1QC) | P02747 | 0/3 | 0/15 |
| Complement C1q tumor necrosis factor-related protein 5 (C1QTNF5) | Q9BXJ0 | 5/6 | 27/34 |
| Complement component 3 (C3) | P01024 | 59/51 | 48/44 |
| Complement component 4-A (C4A) | P0C0L4 | 54/56 | 40/43 |
| Complement component 4-B (C4B) | P0C0L5 | 55/57 | 42/45 |
| Complement component 5 (C5) | P01031 | N.D. | N.D. |
| Complement component 6 (C6) | P13671 | 26/28 | 34/41 |
| Complement component 7 (C7) | P10643 | 19/25 | 35/46 |
| Complement component 8 alpha chain (C8A) | P07357 | 11/9 | 27/27 |
| Complement component 8 Beta chain (C8B) | P07358 | 9/12 | 21/28 |
| Complement component 8 gamma chain (C8G) | P07360 | 8/8 | 49/49 |
| Complement component 9 (C9) | P02748 | 22/23 | 45/47 |
| Complement Factor B (CFB) | P00751 | 2/6 | 4/12 |
| Complement Factor H (CFH) | P08603 | 29/32 | 33/36 |
| Complement Factor H-related protein 1 (CFHR1) | Q03591 | 9/10 | 41/38 |
| Complement Factor H-related protein 2 (CFHR2) | P36980 | 5/5 | 31/27 |
| Complement Factor H-related protein 3 (CFHR3) | Q02985 | 2/0 | 6/0 |
| Complement Factor H-related protein 5 (CFHR5) | Q9BXR6 | 14/10 | 34/28 |
| Complement Factor I (CFI) | P05156 | 14/13 | 29/28 |
| Complement decay-accelerating factor (CD55) | P08247 | 8/7 | 29/29 |
| MAC inhibitory protein (CD59) | P13987 | 4/4 | 25/25 |
| Membrane cofactor protein (CD46) | P15529 | 9/8 | 21/21 |
| Interleukin-6 (IL6) | P05231 | N.D. | N.D. |
| Interleukin-6 receptor subunit beta (IL6ST) | P40189 | 11/11 | 21/21 |
| Monocyte chemoattractant protein-1 (MCP1) | Q6UZ82 | N.D. | N.D. |
| Interferon-inducible protein (AIM2) | O14862 | N.D. | N.D. |
| NLR family member X1 (NLRX1) | Q86UT6 | 17/21 | 26/33 |
| NACHT, LRR and PYD domains-containing protein 13 (NLRP13) | Q86W25 | 2/0 | 3/0 |
| NACHT, LRR and PYD domains-containing protein 2 (NLRP2) | Q9NX02 | N.D. | N.D. |
| NACHT, LRR and PYD domains-containing protein 3 (NLRP3) | Q96P20 | N.D. | N.D. |
| NLR family CARD domain-containing protein 4 (NLRC4) | Q9NPP4 | N.D. | N.D. |
| Apoptosis-associated speck-like protein containing a CARD (PYCARD) | Q9ULZ3 | N.D. | N.D. |
| Caspase-1 | P29466 | N.D. | N.D. |
| Interleukin-1 beta (IL1B) | P01584 | N.D. | N.D. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebeling, M.C.; Fisher, C.R.; Kapphahn, R.J.; Stahl, M.R.; Shen, S.; Qu, J.; Montezuma, S.R.; Ferrington, D.A. Inflammasome Activation in Retinal Pigment Epithelium from Human Donors with Age-Related Macular Degeneration. Cells 2022, 11, 2075. https://doi.org/10.3390/cells11132075
Ebeling MC, Fisher CR, Kapphahn RJ, Stahl MR, Shen S, Qu J, Montezuma SR, Ferrington DA. Inflammasome Activation in Retinal Pigment Epithelium from Human Donors with Age-Related Macular Degeneration. Cells. 2022; 11(13):2075. https://doi.org/10.3390/cells11132075
Chicago/Turabian StyleEbeling, Mara C., Cody R. Fisher, Rebecca J. Kapphahn, Madilyn R. Stahl, Shichen Shen, Jun Qu, Sandra R. Montezuma, and Deborah A. Ferrington. 2022. "Inflammasome Activation in Retinal Pigment Epithelium from Human Donors with Age-Related Macular Degeneration" Cells 11, no. 13: 2075. https://doi.org/10.3390/cells11132075