1. Introduction
Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae), commonly known as the sweetpotato whitefly, is widely distributed in the subtropics/tropics, where it can be an agricultural pest [
1,
2,
3]. Feeding by this pest induces physiological symptoms and results in the transmission of a devastating group of plant viruses belonging to the genus
Begomovirus [
4,
5,
6,
7,
8].
Bemisia tabaci comprises several cryptic species with distinct phylogeographical distributions [
9,
10,
11,
12,
13]. These cryptic species and their haplotypes are morphologically indistinguishable from one another, and some have been found to possess distinct biological traits such as fecundity, endosymbiont complement, host range, the propensity to develop insecticide resistance, ecological adaptation, and, importantly, transmission specificity involving begomoviruses [
1,
14,
15,
16,
17,
18].
Begomoviruses are small non-enveloped viruses with single-stranded circular DNA genomes of about 2800 (monopartite) to 5200 (bipartite) nt each, and they possess a unique morphology consisting of twinned or paired icosahedral particles [
5]. Monopartite viruses such as TYLCV consist of a single DNA component, and bipartite viruses such as SiGMV and CuLCrV include both DNA-A and DNA-B components [
5,
19,
20,
21]. For the successful transmission of begomoviruses, whiteflies must ingest viral particles with their stylets while feeding. Virions pass through the food canal and reach the esophagus and midgut, where they traverse into the hemolymph. Virions are then endocytosed into the primary salivary glands and are egested with the saliva into the plant phloem. This circulative translocation of begomoviruses in
B. tabaci is dependent upon the viral coat protein interactions with probable receptors and other whitefly proteins [
22]. Few receptors and begomovirus-interacting proteins such as the GroEL chaperone protein, heat shock proteins, midgut proteins, peptidyl-prolyl isomerase protein genes, and the peptidoglycan recognition protein gene have been identified [
23,
24,
25,
26,
27]. However, there may be many others involved, and unravelling them is critical to understanding virus transmission by whiteflies.
Among the members of the
B. tabaci species complex, the Middle East-Asia Minor 1 (MEAM1), also known as the North Africa-Middle East mitotype or B biotype, and the Mediterranean (MED), also known as the North Africa-Mediterranean mitotype or Q biotype, are invasive and widely distributed [
2,
10,
11,
12].
Bemisia tabaci MEAM1 was first reported in the United States in the mid-1980s and has since become the predominant cryptic species on field crops after displacing the native New World 1 species, also known as the American Tropics (AMTROP) cryptic species or A biotype, whereas
B. tabaci MED was first reported in 2004 and is primarily confined to greenhouse-grown ornamentals [
1,
15,
28]. Recently,
B.
tabaci MED was reported on open-field crops in Florida and Georgia [
29,
30]. In the southeastern United States,
B. tabaci MEAM1 occurs on field-grown vegetable crops such as tomato, eggplant, snap bean, and cucurbits and on cotton [
30,
31]. In these cropping systems,
B. tabaci MEAM1 is known to transmit tomato yellow leaf curl virus (TYLCV), sida golden mosaic virus (SiGMV), and cucurbit leaf crumple virus (CuLCrV), which cause diseases [
32,
33]. In laboratory studies,
B. tabaci MED tissues accumulated reduced amounts of the two New World begomoviruses compared to
B. tabaci MEAM1 and did not transmit SiGMV and CuLCrV, albeit it was an efficient vector of TYLCV [
34,
35,
36]. Molecular and cellular factors underlying the differential transmission of these and several other begomoviruses by the
B. tabaci MEAM1 and MED have not been elucidated, and studies identifying their primary transmission determinants are critical.
Plant viruses can alter the phenotype and physiological traits of their hosts, which in turn modulates vector preference and fitness, at times favoring their spread [
37,
38]. Begomovirus infections were reported to influence vector preference and fitness in different ways depending on the host plant [
39,
40,
41,
42]. However, the mechanisms involved in such begomoviruses-induced macro-effects on their whitefly vectors are not well understood, and high throughput sequencing platforms have been utilized to understand virus-associated macro-effects on whiteflies [
43].
Using various omics platforms, including transcriptomics, the virus-associated micro-effects of begomoviruses and criniviruses have been examined for
B. tabaci (MEAM1 and MED) [
44,
45,
46,
47,
48,
49,
50,
51,
52,
53,
54]. However, what remains unclear is whether micro-effects are due to the direct effect of the virus on the vector or to the indirect effects resulting from the modulation of the physiological changes in the infected host plant following virus infection. To minimize the host-modulated indirect effects of plant viruses on their vectors through feeding on the plant sap of virus-infected hosts, it is crucial to transfer viruliferous whiteflies from a virus-infected host plant to a virus non-host plant for gut clearing prior to RNA extraction [
44].
In this study, differential gene expression in B. tabaci MEAM1 and MED was assessed by transcriptome analysis post acquisition of three begomoviruses, the Old World monopartite TYLCV, and the New World bipartite SiGMV and CuLCrV. Libraries were prepared for viruliferous and non-viruliferous whiteflies given a 72 h acquisition access period (AAP) on the respective begomovirus-infected or non-infected host plants, followed by a 72 h feeding access on cotton, a non-host of the three begomoviruses. The objectives of this study were to (i) assess the differences in gene expression between B. tabaci MEAM1 and MED upon the acquisition of Old and New World begomoviruses, and (ii) locate putative hub genes and co-expressed genes (modules) responsive to the acquisition of Old and New World begomoviruses using weighted gene correlation network analysis (WGCNA).
4. Discussion
This study assessed the differences in gene expression in
B. tabaci MEAM1 and MED whiteflies in response to the acquisition of Old and New World begomoviruses. Both the Old and New World viruses examined in this study were transmitted by the
B. tabaci MEAM1 cryptic species, and only the Old World virus (TYLCV) was transmitted by the
B. tabaci MED cryptic species [
36]. Gene expression differences were compared between the two cryptic species following the acquisition of TYLCV, SiGMV, or CuLCrV. Across all three virus treatments, more transcriptional changes were recorded with
B. tabaci MEAM1 adults (881 genes) than with
B. tabaci MED adults (559 genes), which represented only 5.6% and 3.6%, respectively, of the overall number of genes (15,668) reported for the
B. tabaci MEAM1 genome (
http://www.whiteflygenomics.org/cgi-bin/bta/index.cgi accessed on 15 December 2021). The high number of transcriptional responses occurring in
B. tabaci MEAM1 whitefly as opposed to the
B. tabaci MED whiteflies could be due to the reduced interactions of the two New World begomoviruses in
B. tabaci MED adults.
The findings indicated that begomovirus species induced variable transcriptional changes in whiteflies upon acquisition. In agreement, previous studies have also reported variable transcriptional responses in whiteflies upon virus acquisition. For instance, 236 genes were differentially expressed in
B. tabaci MEAM1 adults that acquired tomato yellow leaf curl China virus (TYLCCNV) compared with non-viruliferous whiteflies [
71]. Two other studies using the same whitefly cryptic species and virus species reported different numbers of differentially expressed genes in
B. tabaci MEAM1 that acquired TYLCCNV when compared with their non-viruliferous counterparts. There were 457 genes in the study of Luan et al. [
50], whereas there were 1606 genes in the study of Luan et al. [
44]. Similarly, in the TYLCV pathosystem, two independent studies reported a contrasting number of differentially expressed genes (79 genes versus 1347 genes) in viruliferous
B. tabaci MEAM1 compared with non-viruliferous, while another study identified 78 differentially expressed genes in viruliferous
B. tabaci MED, compared with the non-viruliferous treatment [
47,
48,
49]. Further, contrasting transcriptional changes in the whitefly have been observed in semi-persistently transmitted criniviruses. For example, for the tomato chlorosis virus (ToCV) pathosystem, one study reported 221 differentially expressed genes in viruliferous
B. tabaci MED adults compared with non-viruliferous adults, whereas another study reported 1155 differentially expressed genes in viruliferous
B. tabaci MEAM1 adults compared with non-viruliferous adults [
45,
49]. In another crinivirus pathosystem, cucurbit yellow stunting disorder virus, 262 differentially expressed genes were reported in viruliferous
B. tabaci MEAM1 compared with non-viruliferous counterparts [
46]. The differences in the experimental design and analysis, such as: (i) AAP from 1–7 days, (ii) viruliferous whiteflies with no gut clearing to minimize potential indirect host plant effects, (iii) low throughput compared with high throughput sequencing platforms, (iv) the different number of libraries sequenced, and (v) bioinformatics analyses, could have partly contributed to the observed variations in the number of transcriptional changes observed in whiteflies in different studies.
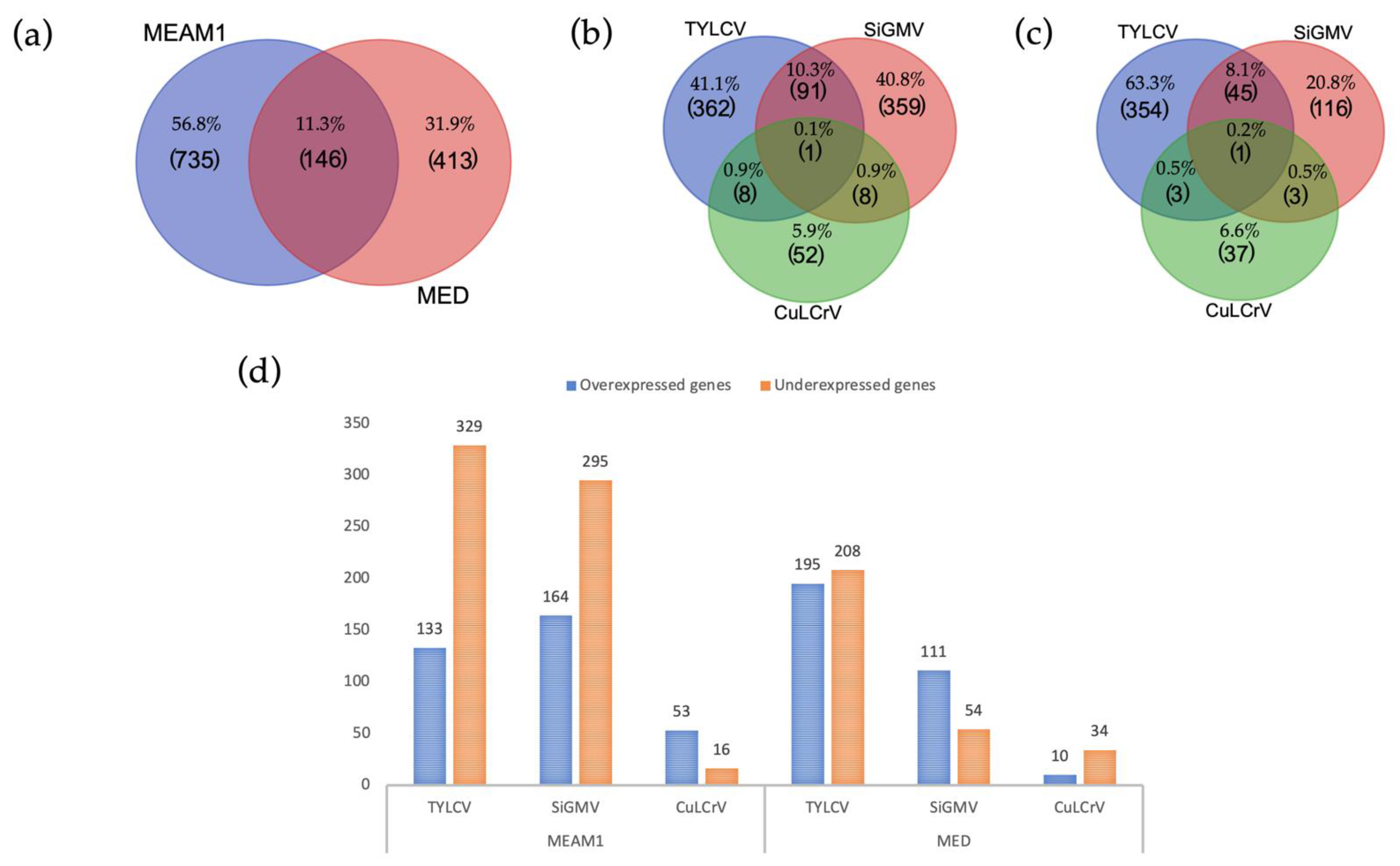
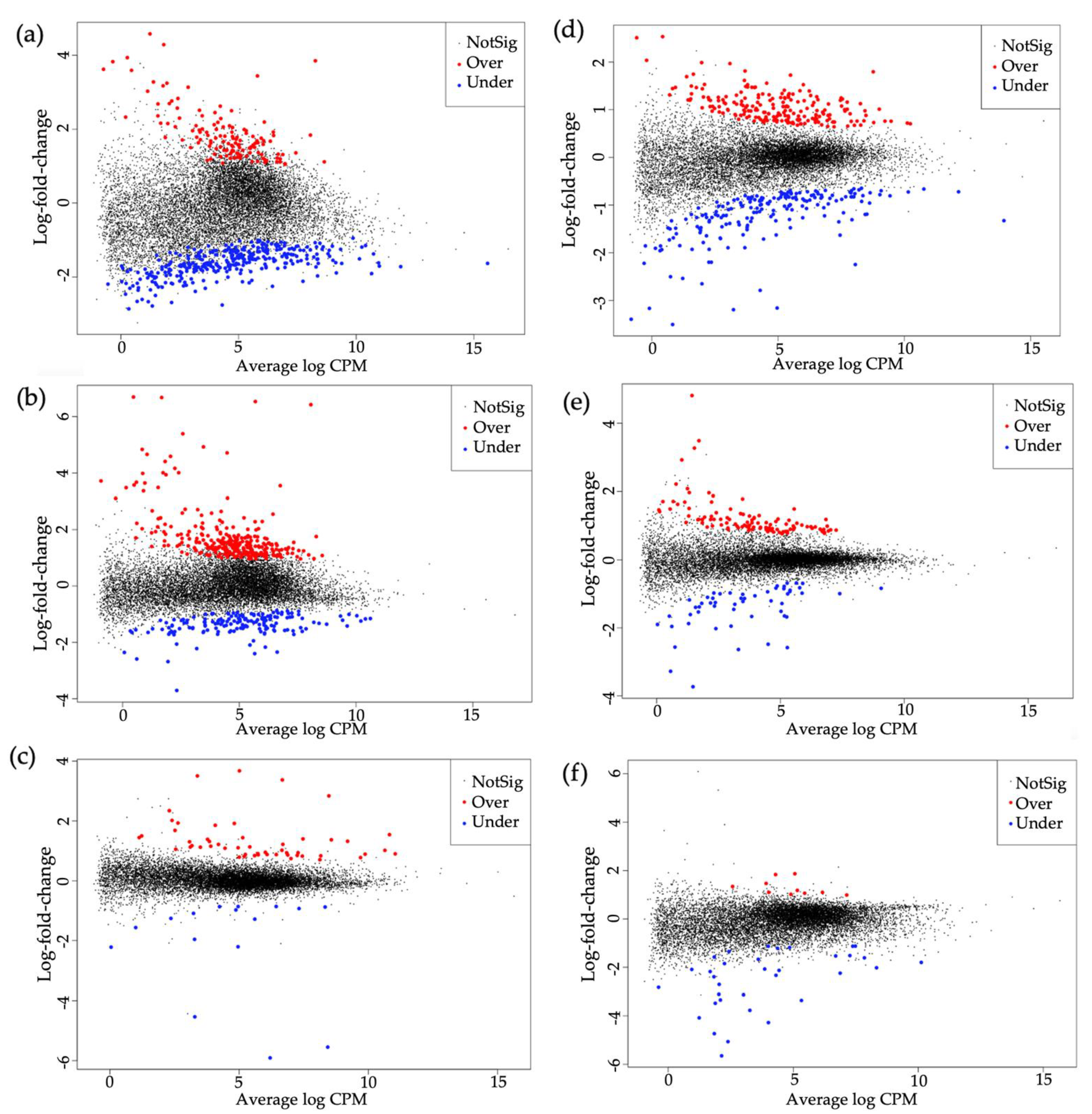
This study investigated the transcriptional changes in
B. tabaci MEAM1 and MED induced by three plant viruses belonging to the same genus,
Begomovirus, and differential gene expression patterns varied between them. More transcriptional changes were associated with TYLCV acquisition (MEAM1–462 genes and MED–413 genes), followed by SiGMV acquisition (MEAM1–459 genes and MED–165 genes), whereas CuLCrV acquisition (MEAM1–69 genes and MED–44 gene) induced the least number of transcriptional changes. Both TYLCV and SiGMV accumulated at significantly higher levels (copies per ng DNA) in both their host plants and vector’s tissues (midgut, hemolymph, and salivary glands), while CuLCrV accumulated at significantly lower amounts in its host plant and vector’s tissues [
30,
36].
Bemisia tabaci MEAM1 transmits SiGMV and CuLCrV, and their circulative tropism in whiteflies could have facilitated binding with different whitefly proteins and putative receptors, leading to increased transcriptional changes [
22,
72,
73].
Bemisia tabaci MED did not transmit the two New World viruses and accumulated at reduced amounts in its tissues; hence, the reduced circulative tropism of New World viruses could have influenced the lower transcriptional responses compared to TYLCV. The differences in
B. tabaci MEAM1 and MED in terms of acquisition and the competency to inoculate different begomoviruses observed here and in previous studies [
74,
75] support previous hypotheses surrounding
B.
tabaci-begomovirus transmission specificity.
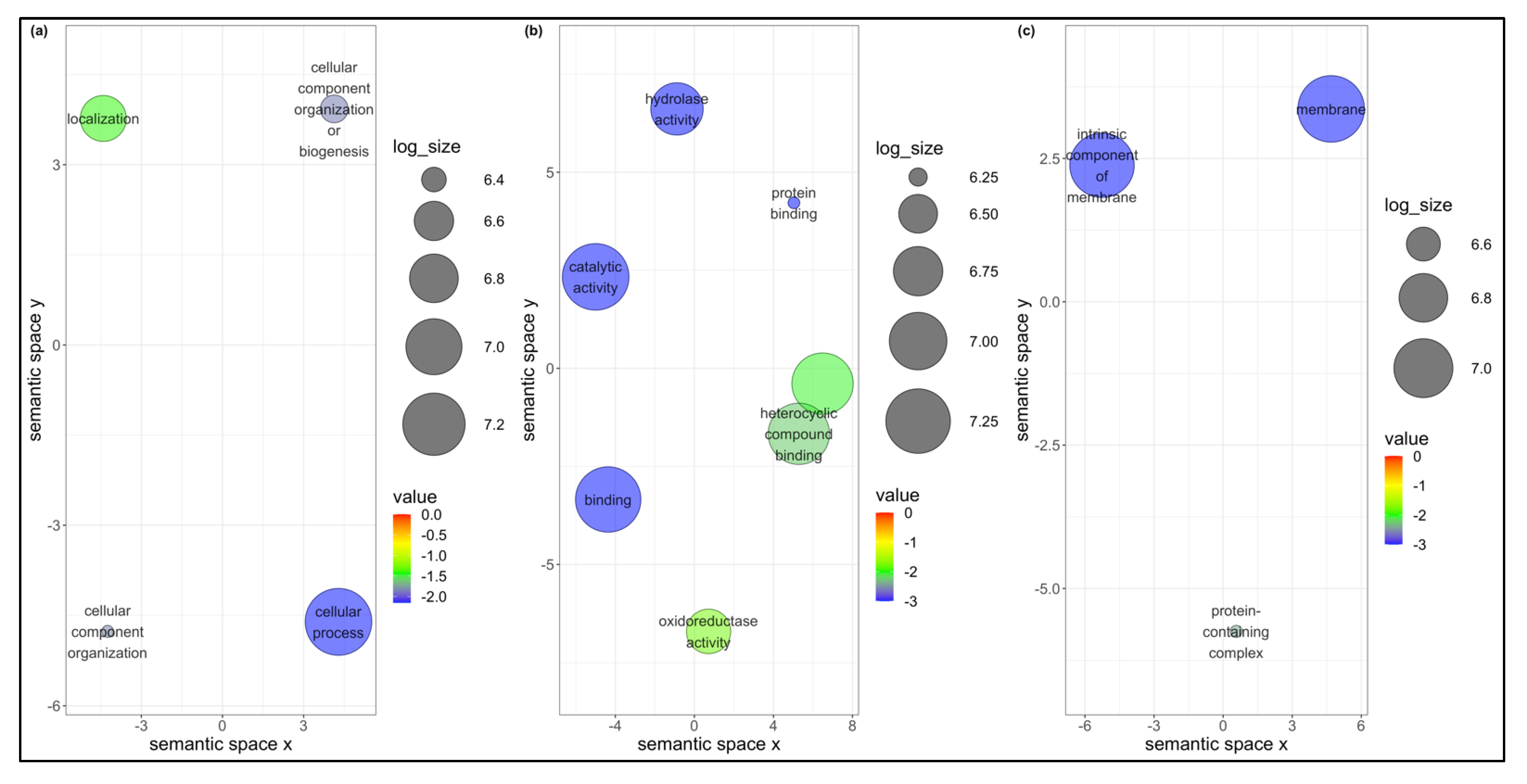
The acquisition of three begomoviruses by
B. tabaci MEAM1 and MED resulted in transcriptional changes, of which 146 genes were differentially expressed in common between the two invasive
B. tabaci species. KEGG analysis annotated only 42 of the 146 genes, and the most represented pathways were classified under metabolic pathways involving carbohydrate and lipid metabolism. Virus (hepatitis C virus and HIV) infection and replication have been reported to increase carbohydrate and lipid metabolism in infected CD4
+ T cells [
76,
77]. The majority of the genes associated with lipid metabolism in both
B. tabaci MEAM1 and MED were underexpressed, and those associated with carbohydrate metabolism were underexpressed in
B. tabaci MEAM1 and partly overexpressed in
B. tabaci MED. The results herein are consistent with previous reports of the downregulation of genes associated with lipid metabolism in
B. tabaci MEAM1 or MED post acquisition of ToCV, TYLCCNV, and TYLCV, or of TYLCV and ToCV from co-infected plants [
44,
49]. In contrast, some studies have reported the upregulation of genes associated with lipid metabolism in a circulative and propagative virus study system (tomato spotted wilt orthotospovirus-thrips) [
78,
79]. Evidence for reduced gene expression associated with carbohydrate and lipid metabolism in viruliferous
B. tabaci MEAM1 and MED may offer another line of evidence to counter the hypothesis that begomoviruses replicate in their whitefly vector [
80,
81]. Nevertheless, the temporary replication of at least one begomovirus (TYLCV) in
B. tabaci MEAM1 salivary glands has been reported in a recent study [
82]. The replication affected TYLCV accumulation in the salivary glands minimally given the overall virus accumulation within whiteflies.
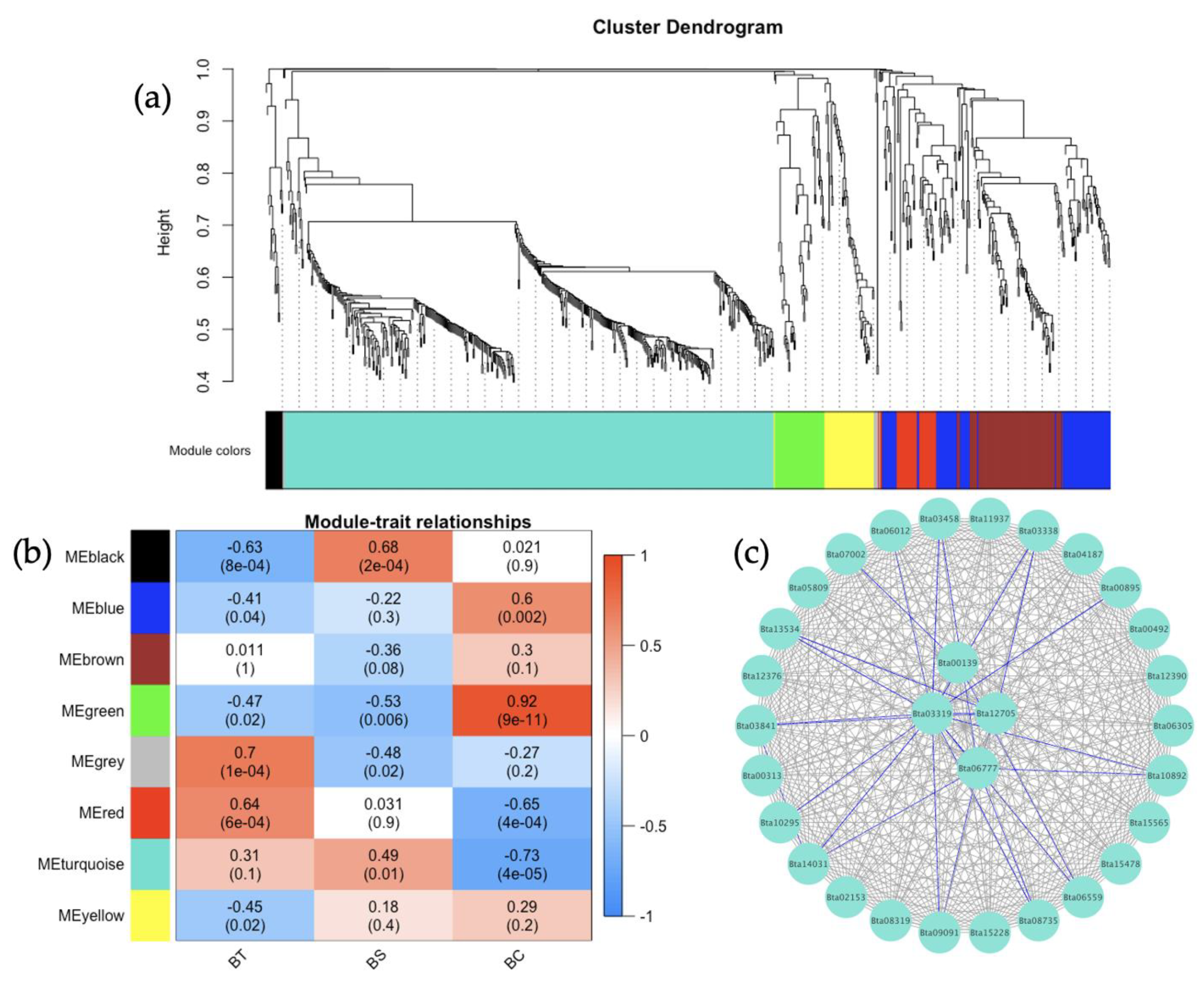
The weighted gene correlation network analysis (WGCNA) has been applied to analyze various biological processes for different organisms [
83,
84]. In this study, the gene co-expression network analysis of
B. tabaci MEAM1 and MED adults that acquired TYLCV, SiGMV, or CuLCrV identified several candidate genes for studying the vector competence of two invasive whiteflies. WGCNA provided scale-free networks between genes within modules, and these modules aided in identifying genes that make
B. tabaci MEAM1 a more competent vector of New World begomoviruses (SiGMV and CuLCrV) than its sister cryptic species
B. tabaci MED. For instance, among the top four interacting genes identified in the largest module for
B. tabaci MEAM1 was the gene
Bta15228 (ubiquitin-conjugating enzyme E2 L3, putative). The ubiquitin-conjugating enzyme was required for Notch signaling activation during
Drosophila wing development. In addition, this gene was involved in the endocytic trafficking of the Notch protein [
85]. This gene could also be involved in the endocytic trafficking of virus particles in
B. tabaci MEAM1. In
B. tabaci MED, three serine protease genes (
Bta03155,
Bta03156, and
Bta03265) were identified in the largest module among the top four interacting genes. Serine proteases are proteolytic enzymes responsible for digestion, larval-pupal molting, signal transduction, and immune responses in insects [
86,
87,
88,
89]. The identification of serine protease genes among the top interacting genes in
B. tabaci MED adults could be signatures of heightened innate immune responses such as melanization and the production of antimicrobial peptides triggered by the acquisition of begomoviruses. The ubiquitin-conjugating enzyme and serine protease genes identified in
B. tabaci MEAM1 and MED adults, respectively, could be important targets for future investigation.
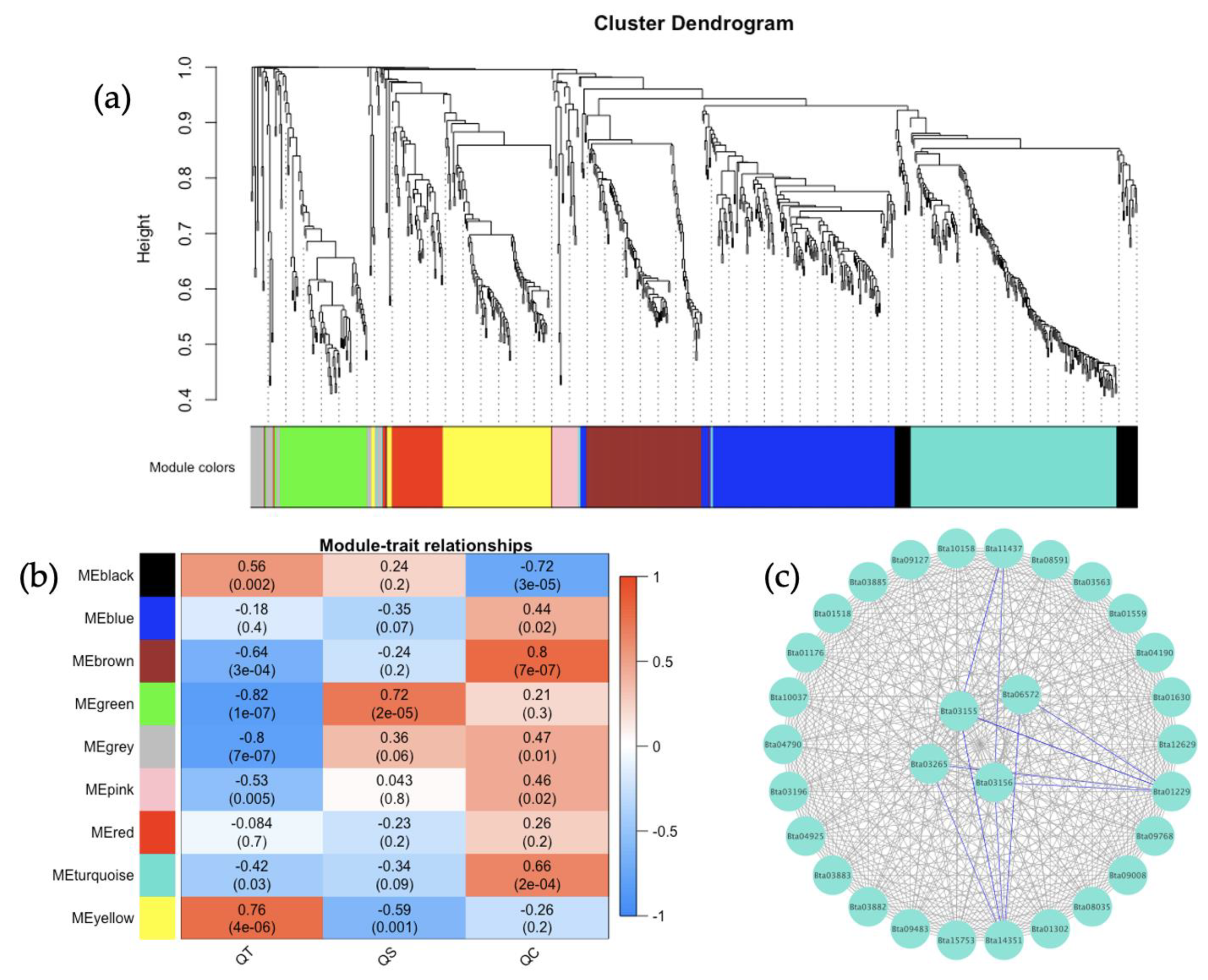
Similarly, the gene co-expression network analysis for TYLCV, SiGMV, or CuLCrV acquired by
B. tabaci MEAM1 and MED adults was examined with the purpose of identifying candidate genes involved in the acquisition of Old and New World begomoviruses. None of the top three interacting genes identified in the largest module for TYLCV, an Old World begomovirus, were functionally annotated. In contrast, a majority of the top three/four interacting genes identified in the largest module for New World begomoviruses—namely, SiGMV and CuLCrV—were functionally annotated. For instance, among the top four interacting genes identified in the largest module for SiGMV was the gene
Bta13010—phosphatidylethanolamine-binding protein (PEBP). The downregulation of PEBP genes in the
Bombyx mori strain resistant to
Bombyx mori nucleopolyhedrovirus (BmNPV) upon infection induced enhanced apoptosis, thereby repressing the ability of BmNPV to infect other cells [
90]. In
B. tabaci MED adults that transmitted TYLCV, the relative expression of PEBP4 was increased significantly and was shown to interact with the TYLCV coat protein, putatively influencing apoptosis and autophagy mechanisms in the whitefly [
91]. The potential for the direct role of PEBP4 in
B. tabaci MEAM1 capable of transmitting SiGMV remains to be investigated; however, based on the increased relative expression observed in this study and the prior inference, PEBP4 may be conferring a similar function across multiple begomovirus-vector systems. Among the three interacting genes identified in the largest module for CuLCrV was the gene
Bta02748 (AP-3 complex subunit mu-1). Adaptin proteins (AP) are membrane-bound heterotetrameric complexes localized in cellular buds and vesicles, and their main role is intracellular trafficking in the trans-Golgi network [
92]. The interaction of AP with the human respiratory syncytial virus matrix protein was essential for the latter’s trafficking in the host cells [
93]. The regulation of the AP-3 complex subunit mu-1 in the whiteflies that acquired a New World begomovirus could play a role in virus tropism, although this requires functional validation.
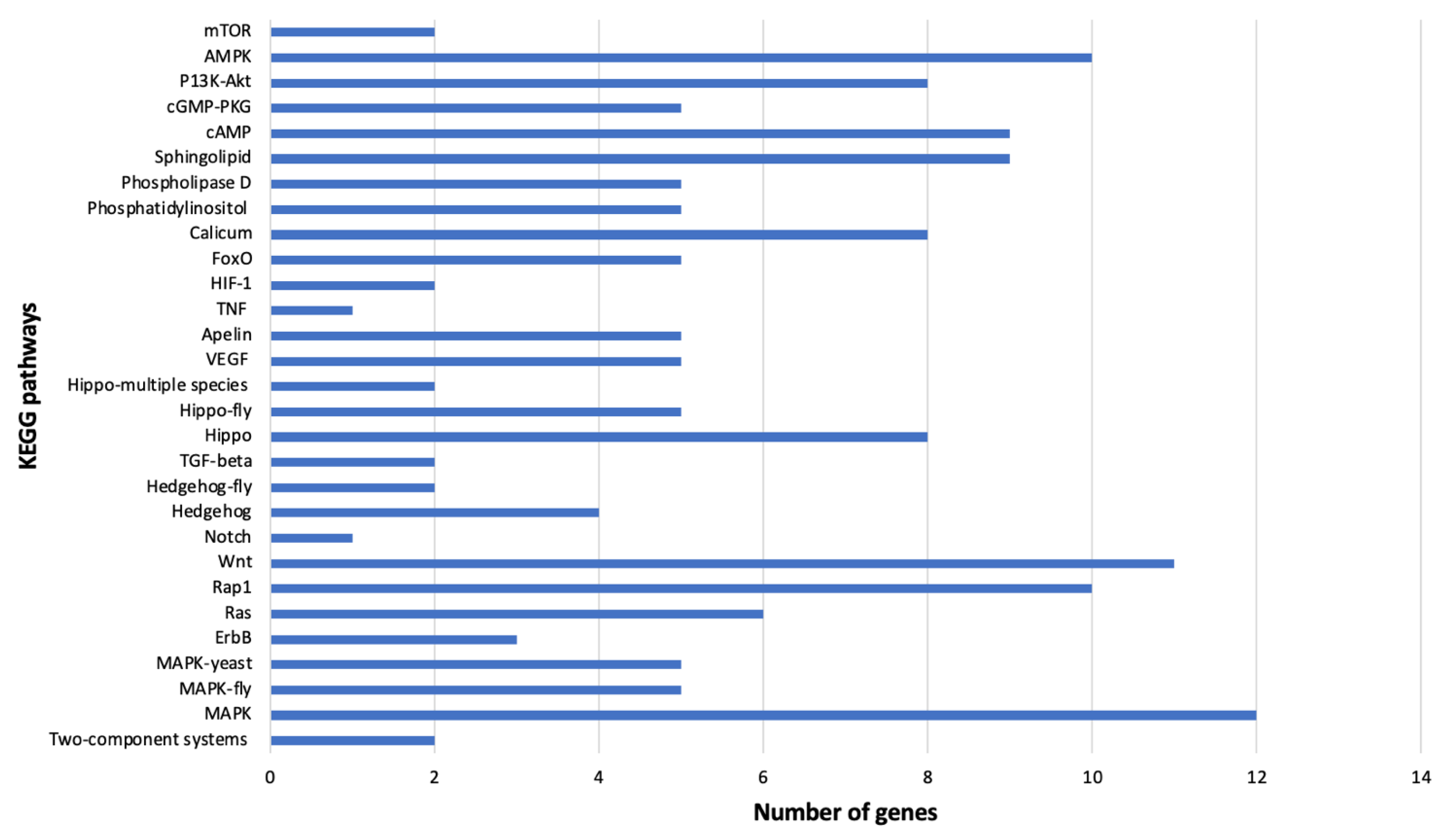
KEGG analysis identified many genes implicated in human virus infection that were differentially regulated in viruliferous
B. tabaci MEAM1 (21 overexpressed and 9 underexpressed genes) and MED (5 overexpressed and 5 underexpressed genes) adults compared with non-viruliferous counterparts. For example, in
B. tabaci MEAM1 adults that acquired SiGMV,
Bta14253—protein kinase C was overexpressed compared with non-viruliferous adults. This gene was implicated in the infection of human immunodeficiency virus 1 (HIV), hepatitis B virus, coronavirus, influenza A virus, and human cytomegalovirus by KEGG analysis. Protein kinase C agonists are reported to induce latent HIV expression from viral reservoirs and protect primary CD4
+ T cells from HIV infection through the down-modulation of HIV coreceptor expression [
94]. Another class of genes, zinc finger proteins (
Bta01535 and
Bta03437) was underexpressed in
B. tabaci MEAM1 adults that acquired TYLCV compared with their non-viruliferous counterparts. In addition, these genes (zinc finger proteins) were also modulated in both directions (up and down) in
B. tabaci MED adults that acquired SiGMV compared with their non-viruliferous counterparts. Zinc finger proteins were also implicated in herpes simplex virus 1 infection (HSV-1). The upregulation of zinc finger proteins was reported to play a role in HSV-1 replication and binding to promoter proteins [
95]. Five heat shock proteins (
Bta00008,
Bta02903,
Bta03000,
Bta06076, and
Bta14532) were all, except in one instance, underexpressed in viruliferous whiteflies compared with non-viruliferous counterparts. The increased expression of heat shock proteins was reported after measles virus infection, resulting in increased cytopathic effects [
96]. The role of the above-selected genes in both
B. tabaci MEAM1 and MED adults following the acquisition of begomoviruses is unknown and warrants further investigation.
For the successful circulative translocation of begomoviruses in whiteflies, the viral coat protein must interact with putative receptors at the midgut, in the hemolymph, and at the primary salivary glands [
22]. A few putative receptors such as the GroEL chaperone protein, heat shock proteins, midgut proteins, peptidyl-prolyl isomerase protein, and peptidoglycan recognition protein gene were identified in previous studies [
23,
24,
25,
26,
27]. In addition to those, this study identified other possible putative receptors in the cytokine–cytokine receptor interaction and cell adhesion molecule pathways that may play a role in the circulative movement of begomoviruses in their vectors, and they include
Bta04818—type I serine/threonine kinase receptor and
Bta07388—syndecan. Serine/threonine kinase, an anchored protein-associated kinase in mammalian cells, was reported as an important cellular component in regulating the entry or clathrin-mediated endocytosis of rabies virus (RABV) [
97]. Syndecan is a cell surface heparan sulphate proteoglycan (HSPGs), which plays several roles including virus (hepatitis E virus and herpes simplex virus) attachment and entry [
98]. Hepatitis E virus, human papillomavirus, and HIV are some of the viruses reported to bind to cell surface HSPGs for their initial attachment to host cells [
99,
100,
101]. The upregulation of the type I serine/threonine kinase receptor and syndecan genes in
B. tabaci MEAM1 adults that acquired SiGMV could be associated with their binding to viral DNA, thereby enabling circulative translocation. Aminopeptidase N is another gene that was differentially expressed (underexpressed) in both
B. tabaci MEAM1 and MED adults that acquired TYLCV. This gene, a plant virus receptor, was overexpressed in aphids and thrips that acquired pea enation mosaic virus and tomato spotted wilt virus (TSWV), respectively [
78,
102]. Whether the aminopeptidase N gene plays any role in begomovirus (TYLCV) reception in either
B. tabaci MEAM1 or MED adults is unknown at this juncture.
Whiteflies have an innate immune system that has been documented against pathogens [
103,
104]. The RNA interference (RNAi) pathway is the major mechanism insects use against virus infection [
43]. In addition, other innate antimicrobial pathways such as Imd, Toll, JAK-STAT, phagocytosis, apoptosis, and proteolysis were reported to play crucial roles in insects’ (Diptera and Lepidoptera) antiviral responses [
105,
106,
107]. A majority of the genes associated with innate immune pathways in the viruliferous
B. tabaci MEAM1 adults were overexpressed, as opposed to the viruliferous
B. tabaci MED adults, where they were underexpressed (
Table 4 and
Table 5). For instance, several cathepsin B genes were identified in both
B. tabaci MEAM1 and MED adults. These genes were associated with apoptosis and immune system pathways such as the NOD-like receptor signaling pathway and antigen processing and presentation pathway. Further, cathepsins are implicated to play a role in virus transmission [
108,
109]. All of the nine genes identified in
B. tabaci MED were underexpressed, while four genes were overexpressed and three were underexpressed in
B. tabaci MEAM1 adults (
Table 4). Similar to our findings, several studies have identified the differential expression (both up- and downregulation) of genes such as cathepsins associated with innate immune pathways in whiteflies following begomovirus or crinivirus acquisition [
44,
45,
46,
47,
49,
51]. The contrasting differential expression of genes under the innate immune pathways such as cathepsins in
B. tabaci MEAM1 and MED identified in this study possibly highlights differences in the immune responses to virus acquisition or other vector-virus interactions between the two
B. tabaci cryptic species. Ras-like GTP-binding proteins were other immune response genes identified only in
B. tabaci MEAM1 adults that acquired TYLCV or SIGMV. Like
B. tabaci MEAM1 adults that acquired TYLCV or SiGMV, ras-like GTP-binding proteins were overexpressed in thrips that were exposed to TSWV [
78]. The upregulation of these genes in viruliferous
B. tabaci MEAM1 adults could be another mechanism evolved by whiteflies to counter begomoviruses.
The exposure to begomoviruses, especially TYLCV and CuLCrV, affected the expression of several fitness-related genes in
B. tabaci MEAM1 and MED. This study identified the up- and downregulation of genes associated with egg production, spermatogenesis, and longevity in whiteflies that acquired begomoviruses. For example,
Bta06648—gametocyte-specific factor 1 (GTSF1) gene was overexpressed in
B. tabaci MEAM1 adults that acquired TYLCV. In
Drosophila, this gene was essential for P-element-induced wimpy testis-interacting RNA-mediated transcriptional repression, the histone mediated repression of transposons, and their neighboring genes in the ovary [
110]. Similarly, in mice, this gene was essential for spermatogenesis and transposon suppression in mouse testes [
111]. The upregulation of the GTSF1 gene in
B. tabaci MEAM1 adults that acquired TYLCV may increase the development of sperm cells in their male reproductive organs, thereby influencing reproduction. Two egg production associated genes—vitellogenin (
Bta07852 and
Bta11903) were underexpressed in
B. tabaci MEAM1 and MED adults that acquired TYLCV, whereas one vitellogenin gene (
Bta11903) was overexpressed in
B. tabaci MEAM1 adults that acquired CuLCrV. Further, most genes associated with aging pathways in viruliferous
B. tabaci MEAM1 and MED adults were underexpressed. Positive, neutral, and negative effects of begomovirus infection on whitefly fecundity and longevity have been reported [
42,
112,
113,
114]. Evidence at the transcriptome level for the underexpression of the egg production gene in
B. tabaci MEAM1 and MED adults that acquired TYLCV, as indicated in this study, is consistent with the Pan et al. [
114] study. That study reported that
B. tabaci MEAM1 and MED that acquired TYLCV produced fewer eggs on cotton than their non-viruliferous counterparts [
114]. In contrast, the overexpression of oviposition-related genes in
B. tabaci MEAM1 that acquired CuLCrV may indicate higher fecundity, but this assumption was not supported by Gautam et al. [
42]. There were no significant differences in the fecundity of
B. tabaci MEAM1 females that acquired CuLCrV and their non-viruliferous counterparts [
42]. Gautam et al. [
36] demonstrated that the fitness effects of both
B. tabaci MEAM1 and MED did not vary within each host species, and in this study, host effects were eliminated by gut clearing for 72 h. Consequently, this study predominantly provides evidence at the transcriptome level for some of the begomoviruses-induced (predominantly direct) macro-level fitness effects on their vectors—
B. tabaci MEAM1 and MED.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}