
PKC-Mediated Orai1 Channel Phosphorylation Modulates Ca2+ Signaling in HeLa Cells

 ,
,
Abstract
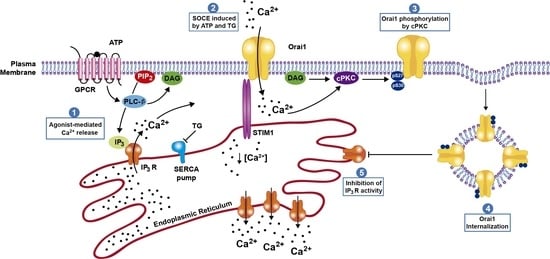
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. The Orai1 Channel Overexpression Resulted in a Higher Agonist-Induced Ca2+ Entry and Reduced ER Ca2+ Depletion
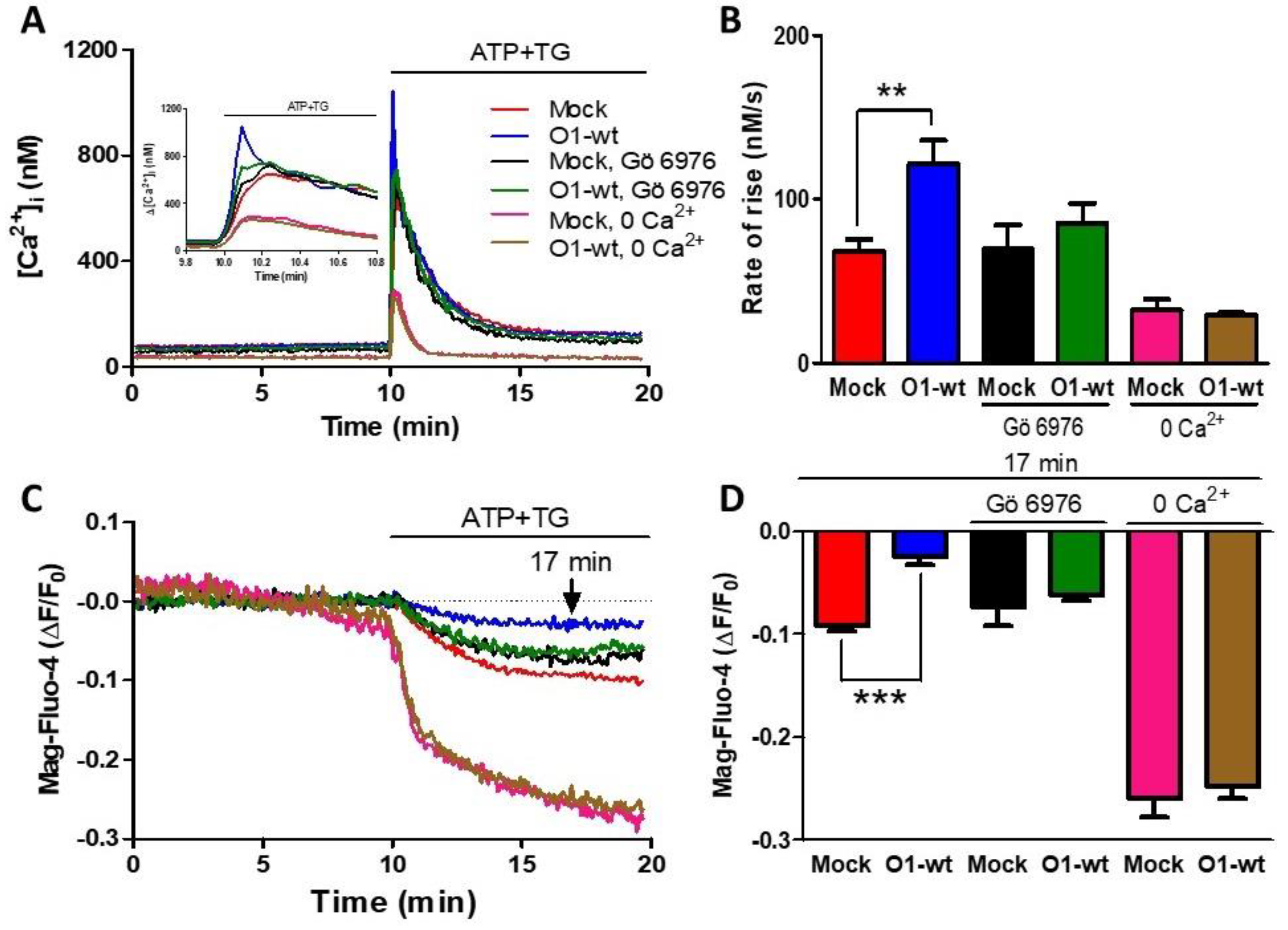
2.2. The Effects of the Orai1 Channel on Ca2+ Entry and the [Ca2+]ER Required the Participation of Ca2+-Dependent PKCs
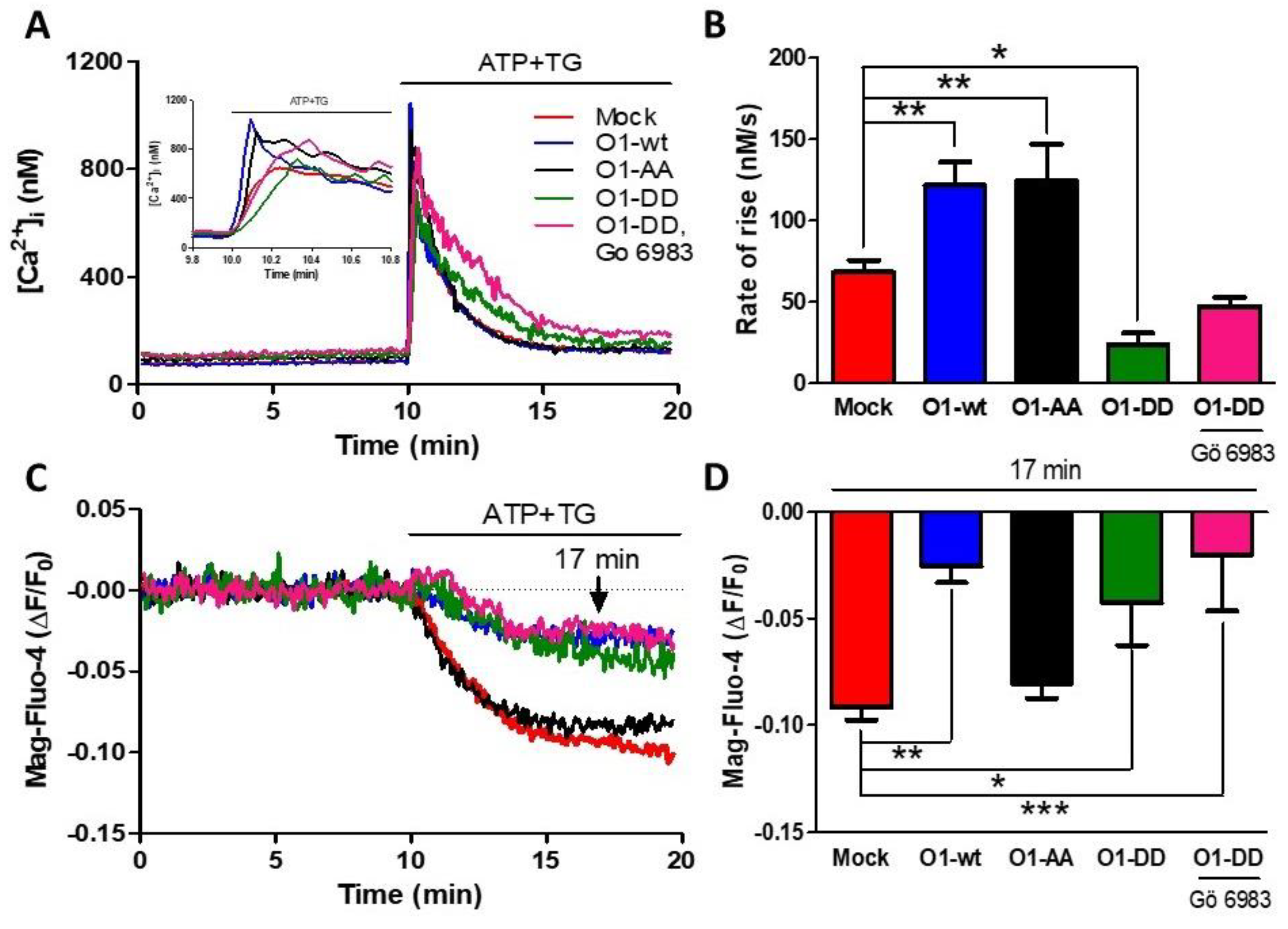
2.3. The Mutant Orai1 S27/30A Channel (O1-AA) Did Not Inhibit ER Ca2+ Release, While the Phosphomimetic S27/30D Channel (O1-DD) Inhibited ER Ca2+ Release without Any Participation of PKC
2.4. The Phosphorylation of the Orai1 Channel Enhanced Its Interaction with IP3Rs
2.5. The O1-DD Channel Has a Higher Intracellular Localization Than O1-Wt and O1-AA Channels and a Lower SOCE
2.6. O1-DD Channel Inhibited While O1-AA Stimulated the Frequency of ATP-Induced [Ca2+]i Oscillations without TG
3. Discussion
4. Material and Methods
4.1. Materials
4.2. Cell Culture and Transfection
4.3. [Ca2+]i and [Ca2+]ER Determination in Cell Populations by Spectrofluorometry
4.4. Point Mutations of Orai1 Channel
4.5. Immunoprecipitation and Western Blot for Orai1 Channel
4.6. Proximity Ligation Assay to Assess the Association between Orai1 and IP3R
4.7. Confocal Imaging of Transfected HeLa Cells
4.8. Data Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Catterall, W.A. Voltage-Gated Calcium Channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef] [PubMed]
- Salido, G.M.; Sage, S.O.; Rosado, J.A. TRPC Channels and Store-Operated Ca2+ Entry. Biochim. Biophys. Acta 2009, 1793, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Kirber, M.T.; Guerrero-Hernández, A.; Bowman, D.S.; Fogarty, K.E.; Tuft, R.A.; Singer, J.J.; Fay, F.S. Multiple Pathways Responsible for the Stretch-Induced Increase in Ca2+ Concentration in Toad Stomach Smooth Muscle Cells. J. Physiol. 2000, 524, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Trebak, M.; Putney, J.W., Jr. ORAI Calcium Channels. Physiology 2017, 32, 332–342. [Google Scholar] [CrossRef]
- Gwack, Y.; Srikanth, S.; Feske, S.; Cruz-Guilloty, F.; Oh-hora, M.; Neems, D.S.; Hogan, P.G.; Rao, A. Biochemical and Functional Characterization of Orai Proteins. J. Biol. Chem. 2007, 282, 16232–16243. [Google Scholar] [CrossRef] [Green Version]
- Smyth, J.T.; Hwang, S.Y.; Tomita, T.; DeHaven, W.I.; Mercer, J.C.; Putney, J.W. Activation and Regulation of Store-Operated Calcium Entry. J. Cell. Mol. Med. 2010, 14, 2337–2349. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.B.; Putney, J.W., Jr. Store-Operated Calcium Channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [Green Version]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an Essential and Conserved Component of Store-Operated Ca2+ Channel Function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.L.; Yeromin, A.V.; Zhang, X.H.-F.; Yu, Y.; Safrina, O.; Penna, A.; Roos, J.; Stauderman, K.A.; Cahalan, M.D. Genome-Wide RNAi Screen of Ca(2+) Influx Identifies Genes That Regulate Ca(2+) Release-Activated Ca(2+) Channel Activity. Proc. Natl. Acad. Sci. USA 2006, 103, 9357–9362. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 Is an Essential Pore Subunit of the CRAC Channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.-H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A Mutation in Orai1 Causes Immune Deficiency by Abrogating CRAC Channel Function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Gudlur, A.; Zeraik, A.E.; Hirve, N.; Rajanikanth, V.; Bobkov, A.A.; Ma, G.; Zheng, S.; Wang, Y.; Zhou, Y.; Komives, E.A.; et al. Calcium Sensing by the STIM1 ER-Luminal Domain. Nat. Commun. 2018, 9, 4536. [Google Scholar] [CrossRef] [PubMed]
- Grabmayr, H.; Romanin, C.; Fahrner, M. STIM Proteins: An Ever-Expanding Family. Int. J. Mol. Sci. 2021, 22, 378. [Google Scholar] [CrossRef] [PubMed]
- Smyth, J.T.; DeHaven, W.I.; Bird, G.S.; Putney, J.W. Ca2+-Store-Dependent and -Independent Reversal of Stim1 Localization and Function. J. Cell Sci. 2008, 121, 762–772. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.-J.; Worley, P.F.; Muallem, S. SOAR and the Polybasic STIM1 Domains Gate and Regulate Orai Channels. Nat. Cell Biol. 2009, 11, 337–343. [Google Scholar] [CrossRef]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 Clusters and Activates CRAC Channels via Direct Binding of a Cytosolic Domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Lange, I.; Feske, S. A Minimal Regulatory Domain in the C Terminus of STIM1 Binds to and Activates ORAI1 CRAC Channels. Biochem. Biophys. Res. Commun. 2009, 385, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Soboloff, J.; Spassova, M.A.; Tang, X.D.; Hewavitharana, T.; Xu, W.; Gill, D.L. Orai1 and STIM Reconstitute Store-Operated Calcium Channel Function. J. Biol. Chem. 2006, 281, 20661–20665. [Google Scholar] [CrossRef] [Green Version]
- Yen, M.; Lewis, R.S. Numbers Count: How STIM and Orai Stoichiometry Affect Store-Operated Calcium Entry. Cell Calcium 2019, 79, 35–43. [Google Scholar] [CrossRef]
- Hogan, P.G.; Rao, A. Store-Operated Calcium Entry: Mechanisms and Modulation. Biochem. Biophys. Res. Commun. 2015, 460, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Jousset, H.; Frieden, M.; Demaurex, N. STIM1 Knockdown Reveals That Store-Operated Ca2+ Channels Located Close to Sarco/Endoplasmic Ca2+ ATPases (SERCA) Pumps Silently Refill the Endoplasmic Reticulum. J. Biol. Chem. 2007, 282, 11456–11464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampieri, A.; Zepeda, A.; Asanov, A.; Vaca, L. Visualizing the Store-Operated Channel Complex Assembly in Real Time: Identification of SERCA2 as a New Member. Cell Calcium 2009, 45, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Manjarrés, I.M.; Alonso, M.T.; García-Sancho, J. Calcium Entry-Calcium Refilling (CECR) Coupling between Store-Operated Ca2+ Entry and Sarco/Endoplasmic Reticulum Ca2+-ATPase. Cell Calcium 2011, 49, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, M.T.; Manjarrés, I.M.; García-Sancho, J. Privileged Coupling between Ca2+ Entry through Plasma Membrane Store-Operated Ca2+ Channels and the Endoplasmic Reticulum Ca2+ Pump. Mol. Cell. Endocrinol. 2012, 353, 37–44. [Google Scholar] [CrossRef]
- Courjaret, R.J.; Machaca, K. Expanding the Store-Operated Ca2+ Entry Microdomain through Ca2+ Tunneling. Curr. Opin. Physiol. 2020, 17, 158–162. [Google Scholar] [CrossRef]
- Béliveau, É.; Lessard, V.; Guillemette, G. STIM1 Positively Regulates the Ca2+ Release Activity of the Inositol 1,4,5-Trisphosphate Receptor in Bovine Aortic Endothelial Cells. PLoS ONE 2014, 9, e114718. [Google Scholar] [CrossRef]
- Decuypere, J.P.; Monaco, G.; Kiviluoto, S.; Oh-hora, M.; Luyten, T.; De Smedt, H.; Parys, J.B.; Missiaen, L.; Bultynck, G. STIM1, but Not STIM2, Is Required for Proper Agonist-Induced Ca2+ Signaling. Cell Calcium 2010, 48, 161–167. [Google Scholar] [CrossRef]
- Emrich, S.M.; Yoast, R.E.; Xin, P.; Arige, V.; Wagner, L.E.; Hempel, N.; Gill, D.L.; Sneyd, J.; Yule, D.I.; Trebak, M. Omnitemporal Choreographies of All Five STIM/Orai and IP3Rs Underlie the Complexity of Mammalian Ca2+ Signaling. Cell Rep. 2021, 34, 108760. [Google Scholar] [CrossRef]
- Woodard, G.E.; López, J.J.; Jardín, I.; Salido, G.M.; Rosado, J.A. TRPC3 Regulates Agonist-Stimulated Ca2+ Mobilization by Mediating the Interaction between Type I Inositol 1,4,5-Trisphosphate Receptor, RACK1, and Orai1. J. Biol. Chem. 2010, 285, 8045–8053. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.H.; Li, Q.; Kim, M.S.; Shin, D.M.; Feske, S.; Birnbaumer, L.; Cheng, K.T.; Ambudkar, I.S.; Muallem, S. Polarized but Differential Localization and Recruitment of STIM1, Orai1 and TRPC Channels in Secretory Cells. Traffic 2011, 12, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Lur, G.; Sherwood, M.W.; Ebisui, E.; Haynes, L.; Feske, S.; Robert, S.; Burgoyne, R.D.; Mikoshiba, K.; Petersen, O.H.; Tepikin, A.V. InsP3 Receptors and Orai Channels in Pancreatic Acinar Cells: Co-Localization and Its Consequences. Biochem. J. 2011, 436, 231–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, A.C. Protein Kinase C: Perfectly Balanced. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 208–230. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, M.; Tomita, T.; Janoshazi, A.; Putney, J.W. Alternative Translation Initiation Gives Rise to Two Isoforms of Orai1 with Distinct Plasma Membrane Mobilities. J. Cell Sci. 2012, 125, 4354–4361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Collado, J.; Lopez, J.J.; Jardin, I.; Camello, P.J.; Falcon, D.; Regodon, S.; Salido, G.M.; Smani, T.; Rosado, J.A. Adenylyl Cyclase Type 8 Overexpression Impairs Phosphorylation-Dependent Orai1 Inactivation and Promotes Migration in MDA-MB-231 Breast Cancer Cells. Cancers 2019, 11, 1624. [Google Scholar] [CrossRef] [Green Version]
- Desai, P.N.; Zhang, X.; Wu, S.; Janoshazi, A.; Bolimuntha, S.; Putney, J.W.; Trebak, M. Multiple Types of Calcium Channels Arising from Alternative Translation Initiation of the Orai1 Message. Sci. Signal. 2015, 8, ra74. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Ueyama, T.; Lange, I.; Feske, S.; Saito, N. Protein Kinase C-Induced Phosphorylation of Orai1 Regulates the Intracellular Ca2+ Level via the Store-Operated Ca2+ Channel. J. Biol. Chem. 2010, 285, 25720–25730. [Google Scholar] [CrossRef] [Green Version]
- Hooper, R.; Zhang, X.; Webster, M.; Go, C.; Kedra, J.; Marchbank, K.; Gill, D.L.; Weeraratna, A.T.; Trebak, M.; Soboloff, J. Novel Protein Kinase C-Mediated Control of Orai1 Function in Invasive Melanoma. Mol. Cell. Biol. 2015, 35, 2790–2798. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Pathak, T.; Yoast, R.; Emrich, S.; Xin, P.; Nwokonko, R.M.; Johnson, M.; Wu, S.; Delierneux, C.; Gueguinou, M.; et al. A Calcium/CAMP Signaling Loop at the ORAI1 Mouth Drives Channel Inactivation to Shape NFAT Induction. Nat. Commun. 2019, 10, 1971. [Google Scholar] [CrossRef]
- Willoughby, D.; Everett, K.L.; Halls, M.L.; Pacheco, J.; Skroblin, P.; Vaca, L.; Klussmann, E.; Cooper, D.M.F. Direct Binding between Orai1 and AC8 Mediates Dynamic Interplay between Ca2+ and CAMP Signaling. Sci. Signal. 2012, 5, ra29. [Google Scholar] [CrossRef]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell Jr, J.E.; Meyer, T. STIM Is a Ca2+ Sensor Essential for Ca2+-Store-Depletion-Triggered Ca2+ Influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [Green Version]
- Gallegos-Gómez, M.-L.; Greotti, E.; López-Méndez, M.-C.; Sánchez-Vázquez, V.-H.; Arias, J.-M.; Guerrero-Hernández, A. The The Trans Golgi Region Is a Labile Intracellular Ca2+ Store Sensitive to Emetine. Sci. Rep. 2018, 8, 17143. [Google Scholar] [CrossRef]
- Navas-Navarro, P.; Rojo-Ruiz, J.; Rodriguez-Prados, M.; Ganfornina, M.D.; Looger, L.L.; Alonso, M.T.; García-Sancho, J. GFP-Aequorin Protein Sensor for Ex Vivo and In Vivo Imaging of Ca2+ Dynamics in High-Ca2+ Organelles. Cell Chem. Biol. 2016, 23, 738–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martiny-Baron, G.; Kazanietz, M.G.; Mischak, H.; Blumberg, P.M.; Kochs, G.; Hug, H.; Marmé, D.; Schächtele, C. Selective Inhibition of Protein Kinase C Isozymes by the Indolocarbazole Gö 6976*. J. Biol. Chem. 1993, 268, 9194–9197. [Google Scholar] [CrossRef]
- Young, L.H.; Balin, B.J.; Weis, M.T. Gö 6983: A Fast Acting Protein Kinase C Inhibitor That Attenuates Myocardial Ischemia/Reperfusion Injury. Cardiovasc. Drug Rev. 2005, 23, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Sun, L.; Machaca, K. Constitutive Recycling of the Store-Operated Ca2+ Channel Orai1 and Its Internalization during Meiosis. J. Cell Biol. 2010, 191, 523–535. [Google Scholar] [CrossRef] [Green Version]
- Leon Aparicio, D.; Pacheco, J.; Chavez-Reyes, J.; Galindo, J.M.; Valdes, J.; Vaca, L.; Guerrero-Hernandez, A. Orai3 Channel Is the 2-APB-Induced Endoplasmic Reticulum Calcium Leak. Cell Calcium 2017, 65, 91–101. [Google Scholar] [CrossRef]
- Rossi, A.M.; Taylor, C.W. Reliable Measurement of Free Ca2+ Concentrations in the ER Lumen Using Mag-Fluo-4. Cell Calcium 2020, 87, 102188. [Google Scholar] [CrossRef]
- Hofer, A.M.; Machen, T.E. Technique for in Situ Measurement of Calcium in Intracellular Inositol 1,4,5-Trisphosphate-Sensitive Stores Using the Fluorescent Indicator Mag-Fura-2 (Gastric Glands/Thapsigargin/Heparin). Proc. Natl. Acad. Sci. USA 1993, 90, 2598–2602. [Google Scholar] [CrossRef] [Green Version]
- Salter, M.W.; Hicks, J.L. ATP Causes Release of Intracellular Ca2+ via the Phospholipase Cβ/IP3 Pathway in Astrocytes from the Dorsal Spinal Cord. J. Neurosci. 1995, 15, 2961–2971. [Google Scholar] [CrossRef]
- Sampieri, A.; Santoyo, K.; Asanov, A.; Vaca, L. Association of the IP3R to STIM1 Provides a Reduced Intraluminal Calcium Microenvironment, Resulting in Enhanced Store-Operated Calcium Entry. Sci. Rep. 2018, 8, 13252. [Google Scholar] [CrossRef]
- Yoast, R.E.; Emrich, S.M.; Zhang, X.; Xin, P.; Johnson, M.T.; Fike, A.J.; Walter, V.; Hempel, N.; Yule, D.I.; Sneyd, J.; et al. The Native ORAI Channel Trio Underlies the Diversity of Ca2+ Signaling Events. Nat. Commun. 2020, 11, 2444. [Google Scholar] [CrossRef] [PubMed]
- Scrimgeour, N.; Litjens, T.; Ma, L.; Barritt, G.J.; Rychkov, G.Y. Properties of Orai1 Mediated Store-Operated Current Depend on the Expression Levels of STIM1 and Orai1 Proteins. J. Physiol. 2009, 587, 2903–2918. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.L.; De Souza, L.B.; Zheng, C.; Cheng, K.T.; Liu, X.; Goldsmith, C.M.; Feske, S.; Ambudkar, I.S. STIM2 Enhances Receptor-Stimulated Ca2+ Signaling by Promoting Recruitment of STIM1 to the Endoplasmic Reticulum-Plasma Membrane Junctions. Sci. Signal. 2015, 8, ra3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leon-Aparicio, D.; Chavez-Reyes, J.; Guerrero-Hernandez, A. Activation of Endoplasmic Reticulum Calcium Leak by 2-APB Depends on the Luminal Calcium Concentration. Cell Calcium 2017, 65, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Frischauf, I.; Zayats, V.; Deix, M.; Hochreiter, A.; Jardin, I.; Muik, M.; Lackner, B.; Svobodová, B.; Pammer, T.; Litviňuková, M.; et al. A Calcium-Accumulating Region, CAR, in the Channel Orai1 Enhances Ca2+ Permeation and SOCE-Induced Gene Transcription. Sci. Signal. 2015, 8, ra131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A New Generation of Ca2+ Indicators with Greatly Improved Fluorescence Properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]
- Poenie, M. Alteration of Intracellular Fura-2 Fluorescence by Viscosity: A Simple Correction. Cell Calcium 1990, 11, 85–91. [Google Scholar] [CrossRef]
- Heckman, K.L.; Pease, L.R. Gene Splicing and Mutagenesis by PCR-Driven Overlap Extension. Nat. Protoc. 2007, 2, 924–932. [Google Scholar] [CrossRef]
- Albarran, L.; Lopez, J.J.; Amor, N.B.; Martin-cano, F.E.; Berna-erro, A.; Smani, T.; Salido, G.M.; Rosado, J.A. Dynamic Interaction of SARAF with STIM1 and Orai1 to Modulate Store-Operated Calcium Entry. Sci. Rep. 2016, 6, 24452. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Martínez, E.; Sánchez-Vázquez, V.H.; León-Aparicio, D.; Sanchez-Collado, J.; Gallegos-Gómez, M.-L.; Rosado, J.A.; Arias, J.M.; Guerrero-Hernández, A. PKC-Mediated Orai1 Channel Phosphorylation Modulates Ca2+ Signaling in HeLa Cells. Cells 2022, 11, 2037. https://doi.org/10.3390/cells11132037
Martínez-Martínez E, Sánchez-Vázquez VH, León-Aparicio D, Sanchez-Collado J, Gallegos-Gómez M-L, Rosado JA, Arias JM, Guerrero-Hernández A. PKC-Mediated Orai1 Channel Phosphorylation Modulates Ca2+ Signaling in HeLa Cells. Cells. 2022; 11(13):2037. https://doi.org/10.3390/cells11132037
Chicago/Turabian StyleMartínez-Martínez, Ericka, Víctor Hugo Sánchez-Vázquez, Daniel León-Aparicio, Jose Sanchez-Collado, Martín-Leonardo Gallegos-Gómez, Juan A. Rosado, Juan M. Arias, and Agustin Guerrero-Hernández. 2022. "PKC-Mediated Orai1 Channel Phosphorylation Modulates Ca2+ Signaling in HeLa Cells" Cells 11, no. 13: 2037. https://doi.org/10.3390/cells11132037