
The Role of AlphαSynuclein in Mouse Models of Acute, Inflammatory and Neuropathic Pain

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
| 19403 | 5′- TCA GCC ACG ATA AAA CTG AGG -3′ |
| 19404 | 5′- GTG AGG GCT GTG GGT ATC TG -3′ |
| oIMR8444 | 5′- GCC TGA AGA ACG AGA TCA GC -3′ |
2.2. Nerve Injury
2.3. Behavioral Testing
2.3.1. Rotarod Test
2.3.2. Mechanical Sensitivity
2.3.3. Hot/Cold Plate Test
2.3.4. Formalin Test
2.3.5. Zymosan-Induced Paw Inflammation
2.3.6. Cold Allodynia
2.4. Western Blot Analysis
2.5. Immunofluorescence
2.6. Glutamate Measurement in the Cerebrospinal Fluid (CSF)
2.7. Real-Time PCR
| CD11b | FW 5′-CTGCCTCAGGGATCCGGAAAG-3′ |
| RV 5′-TGTCTGCCTCGGGGATGACATC-3′ | |
| COX-2 | FW 5′-AGACACTCAGGTAGACATGATCTACCCT-3′ |
| RV 5′-GGCACCAGACCAAAGACTTCC-3′ | |
| GFAP | FW 5′-AGAACAACCTGGCTGCGTAT-3′ |
| RV 5′-TCCTCCTCCAGCGATTCAAC-3′ | |
| IL1β | FW 5′-CTGGTGTGTGACGTTCCCATTA-3′ |
| RV 5′-CCGACAGCACGAGGCTTT-3′ | |
| TNFα | FW 5′-GCTGAGCTCAAACCCTGGTA-3′ |
| RV 5′-CGGACTCCGCAAAGTCTAAG-3′ | |
| GAPDH | FW 5′-CAATGTGTCCGTCGTGGATCT-3′ |
| RV 5′-GTCCTCAGTGTAGCCCAAGATG-3′ |
2.8. Data Analysis
3. Results
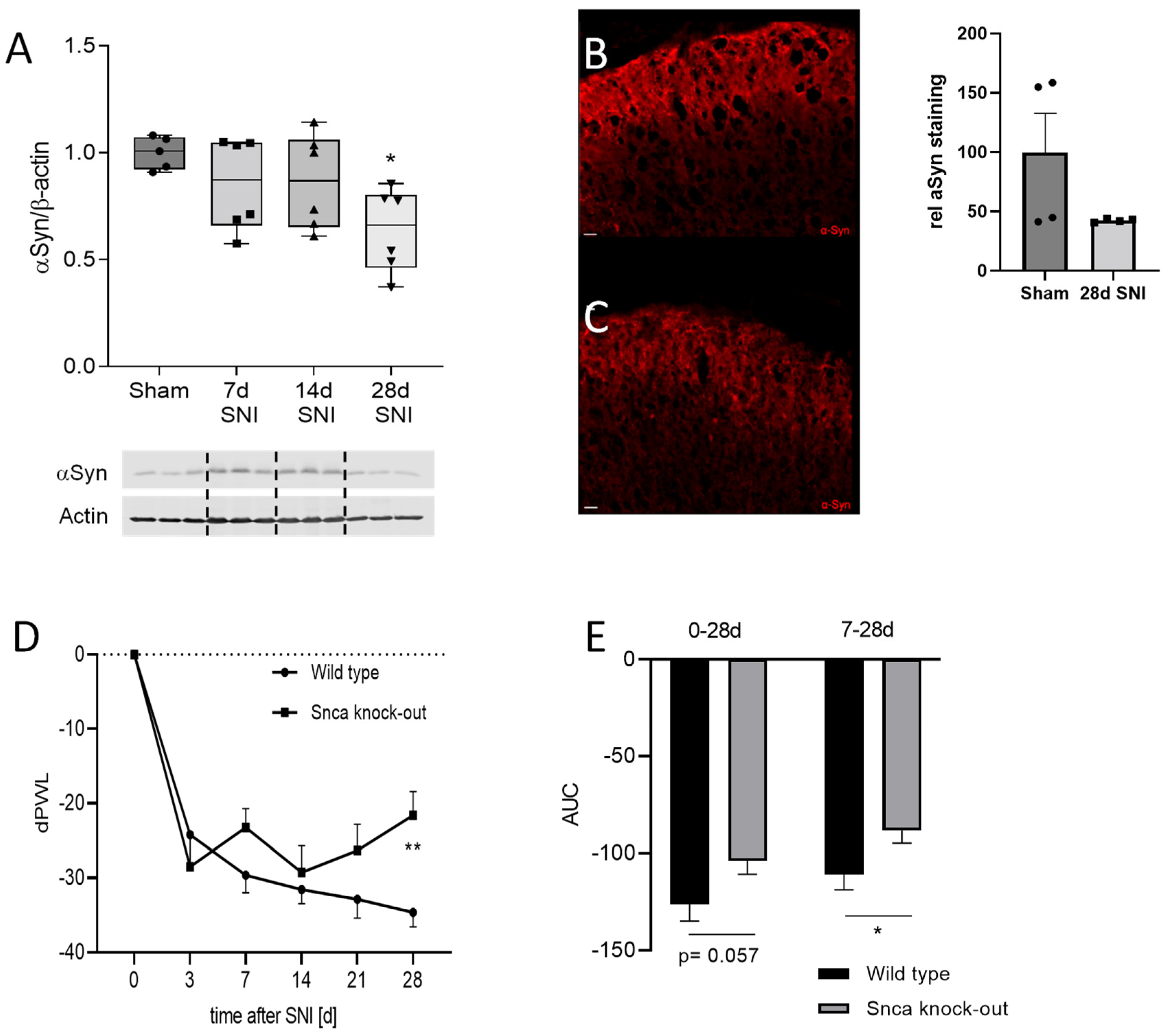
3.1. Expression and Localization of αSyn in the Spinal Cord
3.2. Effects of αSyn Knock-Out on Motor Function and Acute Nociception
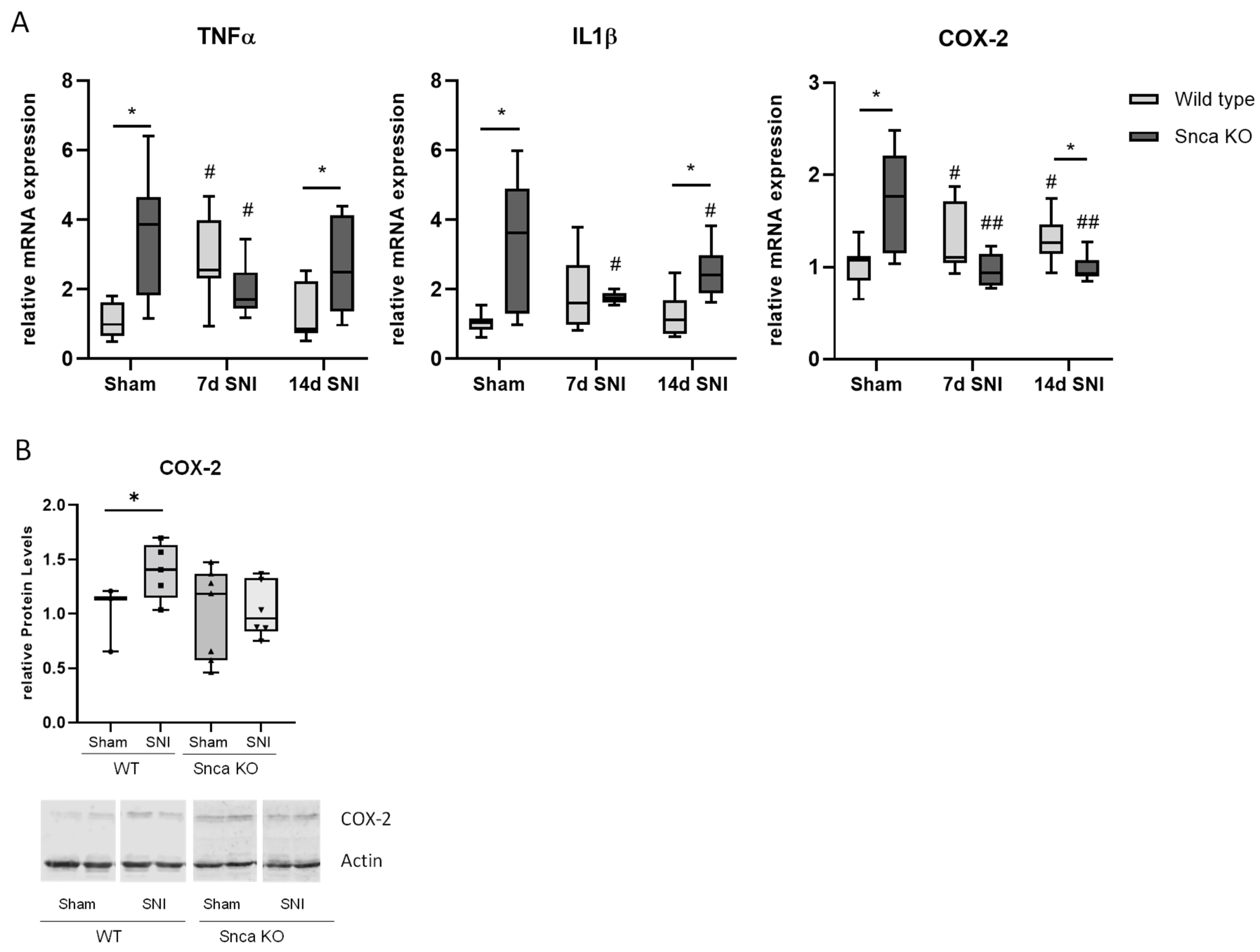
3.3. αSyn Does Not Affect Inflammatory Nociceptive Responses
3.4. Impact of αSyn on Neuropathic Pain in the SNI Model
3.5. Potential Mechanisms of Reduced Neuropathic Pain in Snca Knock-Out Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burre, J.; Sharma, M.; Sudhof, T.C. Cell Biology and Pathophysiology of alpha-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef] [PubMed]
- Withers, G.S.; George, J.M.; Banker, G.A.; Clayton, D.F. Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Brain Res. Dev. Brain Res. 1997, 99, 87–94. [Google Scholar] [CrossRef]
- Logan, T.; Bendor, J.; Toupin, C.; Thorn, K.; Edwards, R.H. alpha-Synuclein promotes dilation of the exocytotic fusion pore. Nat. Neurosci. 2017, 20, 681–689. [Google Scholar] [CrossRef]
- Scott, D.; Roy, S. alpha-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J. Neurosci. 2012, 32, 10129–10135. [Google Scholar] [CrossRef] [Green Version]
- George, J.M.; Jin, H.; Woods, W.S.; Clayton, D.F. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 1995, 15, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Abeliovich, A.; Schmitz, Y.; Farinas, I.; Choi-Lundberg, D.; Ho, W.H.; Castillo, P.E.; Shinsky, N.; Verdugo, J.M.; Armanini, M.; Ryan, A.; et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000, 25, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [Green Version]
- Maraganore, D.M.; de Andrade, M.; Elbaz, A.; Farrer, M.J.; Ioannidis, J.P.; Kruger, R.; Rocca, W.A.; Schneider, N.K.; Lesnick, T.G.; Lincoln, S.J.; et al. Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA 2006, 296, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Mallory, M.; Hashimoto, M.; Takeda, A.; Sagara, Y.; Sisk, A.; Mucke, L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: Implications for neurodegenerative disorders. Science 2000, 287, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Kirik, D.; Rosenblad, C.; Burger, C.; Lundberg, C.; Johansen, T.E.; Muzyczka, N.; Mandel, R.J.; Bjorklund, A. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J. Neurosci. 2002, 22, 2780–2791. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Reichmann, H.; Brandt, M.D.; Klingelhoefer, L. The nonmotor features of Parkinson’s disease: Pathophysiology and management advances. Curr. Opin. Neurol. 2016, 29, 467–473. [Google Scholar] [CrossRef]
- Beiske, A.G.; Loge, J.H.; Ronningen, A.; Svensson, E. Pain in Parkinson’s disease: Prevalence and characteristics. Pain 2009, 141, 173–177. [Google Scholar] [CrossRef]
- Hanagasi, H.A.; Akat, S.; Gurvit, H.; Yazici, J.; Emre, M. Pain is common in Parkinson’s disease. Clin. Neurol. Neurosurg. 2011, 113, 11–13. [Google Scholar] [CrossRef]
- Perez-Lloret, S.; Rey, M.V.; Dellapina, E.; Pellaprat, J.; Brefel-Courbon, C.; Rascol, O. Emerging analgesic drugs for Parkinson’s disease. Expert Opin. Emerg. Drugs 2012, 17, 157–171. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef]
- Djaldetti, R.; Shifrin, A.; Rogowski, Z.; Sprecher, E.; Melamed, E.; Yarnitsky, D. Quantitative measurement of pain sensation in patients with Parkinson disease. Neurology 2004, 62, 2171–2175. [Google Scholar] [CrossRef]
- Chudler, E.H.; Dong, W.K. The role of the basal ganglia in nociception and pain. Pain 1995, 60, 3–38. [Google Scholar] [CrossRef]
- Boecker, H.; Ceballos-Baumann, A.; Bartenstein, P.; Weindl, A.; Siebner, H.R.; Fassbender, T.; Munz, F.; Schwaiger, M.; Conrad, B. Sensory processing in Parkinson’s and Huntington’s disease: Investigations with 3D H(2)(15)O-PET. Brain 1999, 122, 1651–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lees, A.J. L-dopa treatment and Parkinson’s disease. Q. J. Med. 1986, 59, 535–547. [Google Scholar] [PubMed]
- Snider, S.R.; Fahn, S.; Isgreen, W.P.; Cote, L.J. Primary sensory symptoms in parkinsonism. Neurology 1976, 26, 423–429. [Google Scholar] [CrossRef]
- Ferreira, N.; Goncalves, N.P.; Jan, A.; Jensen, N.M.; van der Laan, A.; Mohseni, S.; Vaegter, C.B.; Jensen, P.H. Trans-synaptic spreading of alpha-synuclein pathology through sensory afferents leads to sensory nerve degeneration and neuropathic pain. Acta Neuropathol. Commun. 2021, 9, 31. [Google Scholar] [CrossRef]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef] [Green Version]
- Yekkirala, A.S.; Roberson, D.P.; Bean, B.P.; Woolf, C.J. Breaking barriers to novel analgesic drug development. Nat. Rev. Drug Discov. 2017, 16, 810. [Google Scholar] [CrossRef] [Green Version]
- Cabin, D.E.; Shimazu, K.; Murphy, D.; Cole, N.B.; Gottschalk, W.; McIlwain, K.L.; Orrison, B.; Chen, A.; Ellis, C.E.; Paylor, R.; et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci. 2002, 22, 8797–8807. [Google Scholar] [CrossRef] [Green Version]
- Ekstrand, M.I.; Terzioglu, M.; Galter, D.; Zhu, S.; Hofstetter, C.; Lindqvist, E.; Thams, S.; Bergstrand, A.; Hansson, F.S.; Trifunovic, A.; et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 1325–1330. [Google Scholar] [CrossRef] [Green Version]
- Decosterd, I.; Woolf, C.J. Spared nerve injury: An animal model of persistent peripheral neuropathic pain. Pain 2000, 87, 149–158. [Google Scholar] [CrossRef]
- Tegeder, I.; Costigan, M.; Griffin, R.S.; Abele, A.; Belfer, I.; Schmidt, H.; Ehnert, C.; Nejim, J.; Marian, C.; Scholz, J.; et al. GTP cyclohydrolase and tetrahydrobiopterin regulate pain sensitivity and persistence. Nat. Med. 2006, 12, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Mogil, J.S.; Wilson, S.G.; Bon, K.; Lee, S.E.; Chung, K.; Raber, P.; Pieper, J.O.; Hain, H.S.; Belknap, J.K.; Hubert, L.; et al. Heritability of nociception I: Responses of 11 inbred mouse strains on 12 measures of nociception. Pain 1999, 80, 67–82. [Google Scholar] [CrossRef]
- Dubuisson, D.; Dennis, S.G. The formalin test: A quantitative study of the analgesic effects of morphine, meperidine, and brain stem stimulation in rats and cats. Pain 1977, 4, 161–174. [Google Scholar] [CrossRef]
- Meller, S.T.; Gebhart, G.F. Intraplantar zymosan as a reliable, quantifiable model of thermal and mechanical hyperalgesia in the rat. Eur. J. Pain 1997, 1, 43–52. [Google Scholar] [CrossRef]
- Vivacqua, G.; Yin, J.J.; Casini, A.; Li, X.; Li, Y.H.; D’Este, L.; Chan, P.; Renda, T.G.; Yu, S. Immunolocalization of alpha-synuclein in the rat spinal cord by two novel monoclonal antibodies. Neuroscience 2009, 158, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Fornai, F.; Kwon, H.B.; Yazdani, U.; Atasoy, D.; Liu, X.; Hammer, R.E.; Battaglia, G.; German, D.C.; Castillo, P.E.; et al. Double-knockout mice for alpha- and beta-synucleins: Effect on synaptic functions. Proc. Natl. Acad. Sci. USA 2004, 101, 14966–14971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Wang, Q.; Chao, D.; Xia, T.C.; Sheng, S.; Li, Z.R.; Zhao, J.N.; Wen, G.Q.; Ding, G.; Xia, Y. δ-Opioid Receptor Activation Attenuates the Oligomer Formation Induced by Hypoxia and/or alpha-Synuclein Overexpression/Mutation Through Dual Signaling Pathways. Mol. Neurobiol. 2019, 56, 2463–3475. [Google Scholar] [CrossRef]
- Kurz, A.; Double, K.L.; Lastres-Becker, I.; Tozzi, A.; Tantucci, M.; Bockhart, V.; Bonin, M.; Garcia-Arencibia, M.; Nuber, S.; Schlaudraff, F.; et al. A53T-alpha-synuclein overexpression impairs dopamine signaling and striatal synaptic plasticity in old mice. PLoS ONE 2010, 5, e11464. [Google Scholar] [CrossRef]
- Valek, L.; Auburger, G.; Tegeder, I. Sensory neuropathy and nociception in rodent models of Parkinson’s disease. Dis. Models Mech. 2019, 12, dmm039396. [Google Scholar] [CrossRef] [Green Version]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of alpha-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Jimenez, A.; Walter, N.A.; Gine, E.; Santos, A.; Echeverry-Alzate, V.; Buhler, K.M.; Olmos, P.; Giezendanner, S.; Moratalla, R.; Montoliu, L.; et al. A spontaneous deletion of alpha-synuclein is associated with an increase in CB1 mRNA transcript and receptor expression in the hippocampus and amygdala: Effects on alcohol consumption. Synapse 2013, 67, 280–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.; Mehta, S.L.; Morris-Blanco, K.C.; Chokkalla, A.K.; Chelluboina, B.; Lopez, M.; Sullivan, R.; Kim, H.T.; Cook, T.D.; Kim, J.Y.; et al. The microRNA miR-7a-5p ameliorates ischemic brain damage by repressing alpha-synuclein. Sci. Signal. 2018, 11, eaat4285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, H.; Zhang, Q.; Yuan, H.; Li, Y.; Wang, D.; Wang, R.; He, X. Elevated neuronal alpha-synuclein promotes microglia activation after spinal cord ischemic/reperfused injury. Neuroreport 2015, 26, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Mehta, S.L.; Kaimal, B.; Lyons, K.; Dempsey, R.J.; Vemuganti, R. Poststroke Induction of alpha-Synuclein Mediates Ischemic Brain Damage. J. Neurosci. 2016, 36, 7055–7065. [Google Scholar] [CrossRef]
- Sauerbeck, A.D.; Goldstein, E.Z.; Alfredo, A.N.; Norenberg, M.; Marcillo, A.; McTigue, D.M. Alpha-synuclein increases in rodent and human spinal cord injury and promotes inflammation and tissue loss. Sci. Rep. 2021, 11, 11720. [Google Scholar] [CrossRef]
- Lee, H.J.; Suk, J.E.; Patrick, C.; Bae, E.J.; Cho, J.H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.J. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.L.; Long, C.X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Liu, N.; Yang, Y.Y.; Xing, H.Y.; Liu, X.X.; Li, F.; La, G.Y.; Huang, M.J.; Zhou, M.W. Lentivirus-mediated downregulation of alpha-synuclein reduces neuroinflammation and promotes functional recovery in rats with spinal cord injury. J. Neuroinflammation 2019, 16, 283. [Google Scholar] [CrossRef] [Green Version]
- Ji, R.R.; Gereau, R.W.t.; Malcangio, M.; Strichartz, G.R. MAP kinase and pain. Brain Res. Rev. 2009, 60, 135–148. [Google Scholar] [CrossRef]
- Rannikko, E.H.; Weber, S.S.; Kahle, P.J. Exogenous alpha-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 2015, 16, 57. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Mao, K.; Yu, H.; Wen, Y.; She, H.; Zhang, H.; Liu, L.; Li, M.; Li, W.; Zou, F. p38-TFEB pathways promote microglia activation through inhibiting CMA-mediated NLRP3 degradation in Parkinson’s disease. J. Neuroinflammation 2021, 18, 295. [Google Scholar] [CrossRef] [PubMed]
- Wilms, H.; Rosenstiel, P.; Romero-Ramos, M.; Arlt, A.; Schafer, H.; Seegert, D.; Kahle, P.J.; Odoy, S.; Claasen, J.H.; Holzknecht, C.; et al. Suppression of MAP kinases inhibits microglial activation and attenuates neuronal cell death induced by alpha-synuclein protofibrils. Int. J. Immunopathol. Pharmacol. 2009, 22, 897–909. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test System | Snca Wild Type (Latency Time, Mean ± SEM) | Snca Knock-Out (Latency Time, Mean ± SEM) | Significance/n |
|---|---|---|---|
| Hanging Wire | 60 s | 60 s | n.s. (n = 12) |
| Rotarod | 90 s | 90 s | n.s. (n = 12) |
| Cold Plate | 23.93 ± 1.59 s | 34.88 ± 2.11 s | *** p < 0.001 (n = 11) |
| Hot Plate | 13.58 ± 0.99 s | 16.32 ± 1.29 s | n.s. (n = 12) |
| Dynamic Plantar | 9.17 ± 0.44 s | 8.54 ± 0.42 s | n.s. (n = 12) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Möller, M.; Möser, C.V.; Weiß, U.; Niederberger, E. The Role of AlphαSynuclein in Mouse Models of Acute, Inflammatory and Neuropathic Pain. Cells 2022, 11, 1967. https://doi.org/10.3390/cells11121967
Möller M, Möser CV, Weiß U, Niederberger E. The Role of AlphαSynuclein in Mouse Models of Acute, Inflammatory and Neuropathic Pain. Cells. 2022; 11(12):1967. https://doi.org/10.3390/cells11121967
Chicago/Turabian StyleMöller, Moritz, Christine V. Möser, Ulrike Weiß, and Ellen Niederberger. 2022. "The Role of AlphαSynuclein in Mouse Models of Acute, Inflammatory and Neuropathic Pain" Cells 11, no. 12: 1967. https://doi.org/10.3390/cells11121967