
The Effects of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase and Erythropoietin, and Their Interactions in Angiogenesis: Implications in Retinopathy of Prematurity

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
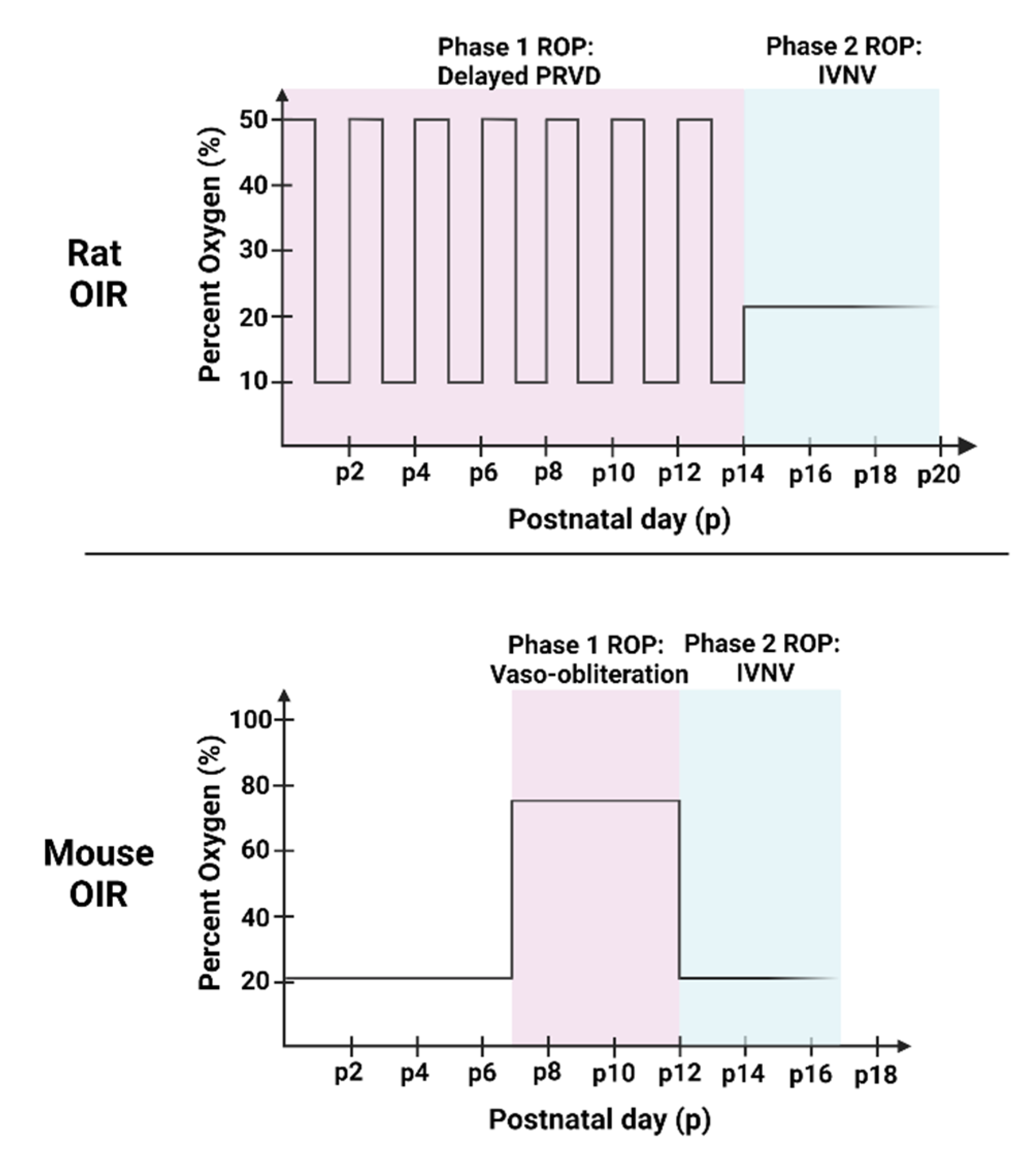
2. Animal Models of OIR
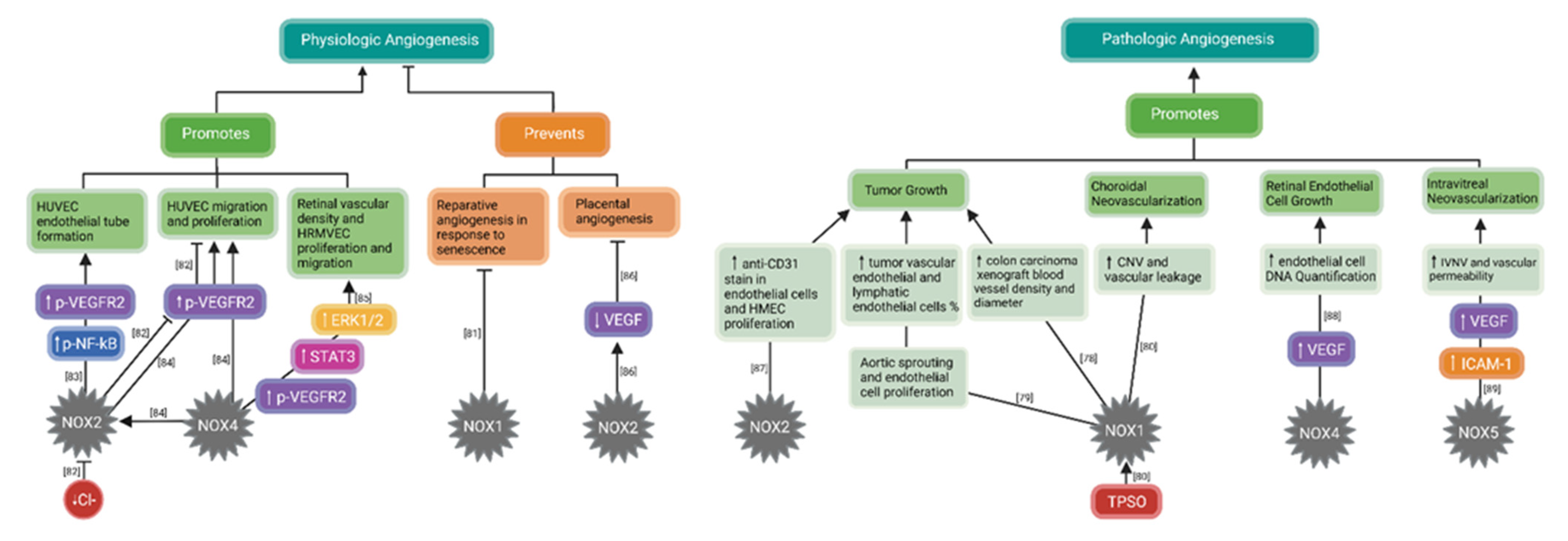
3. NADPH Oxidase (NOX) Involvement in Angiogenesis
3.1. ROS Generation from NOX
3.2. NADPH Oxidases (NOXs) in Angiogenesis

3.3. Intersection between NADPH Oxidase (NOX) and Erythropoietin
4. Erythropoietin Involvement in Angiogenesis
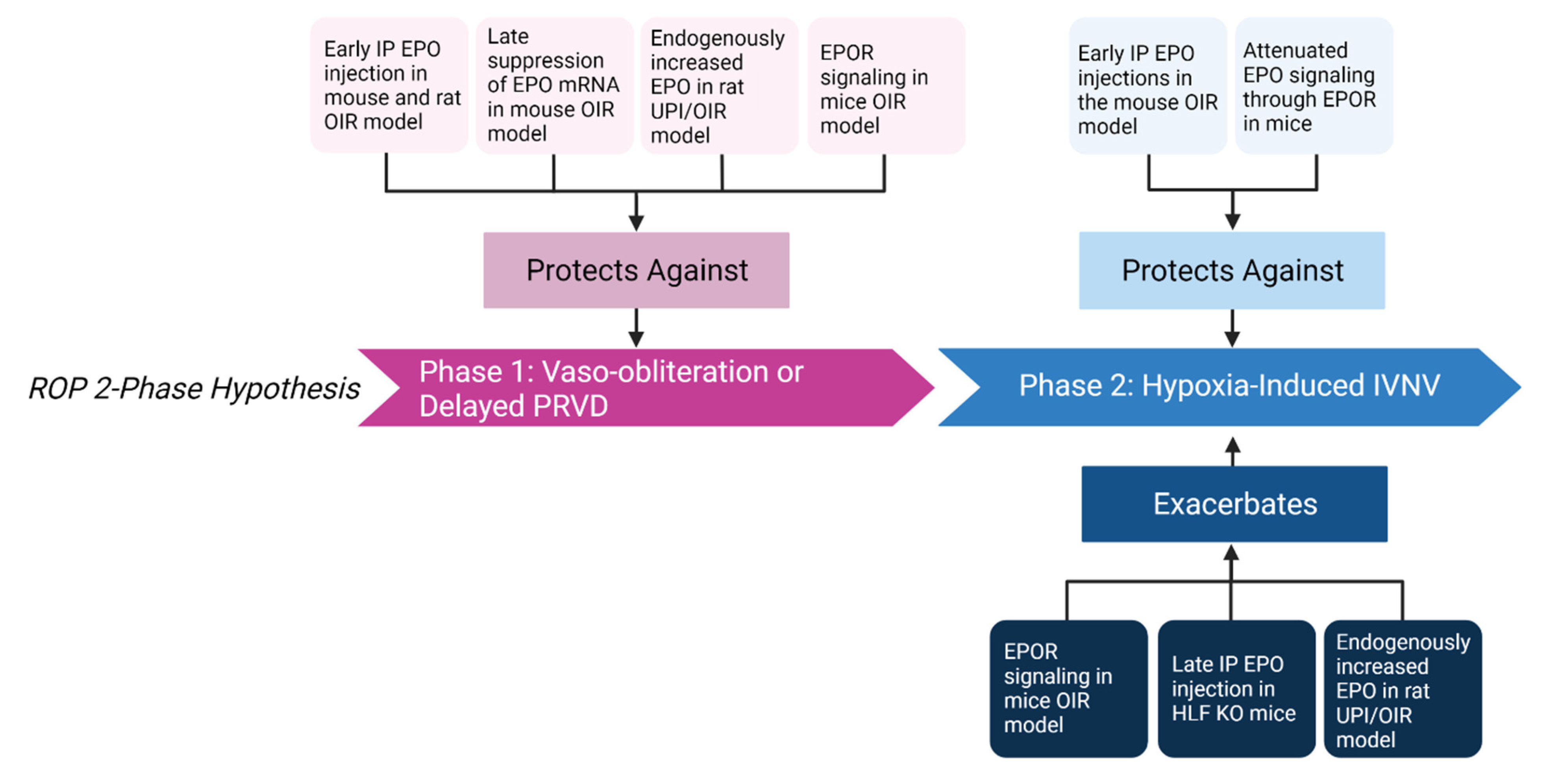
4.1. EPO-Mediated Protection against Vaso-Obliteration and Delayed Physiologic Retinal Vascular Development
4.2. EPO-Mediated Increase in Hypoxia-Induced Pathologic Neovascularization
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hartnett, M.E.; Penn, J.S. Mechanisms and Management of Retinopathy of Prematurity. N. Engl. J. Med. 2012, 367, 2515–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartnett, M.E. Pathophysiology and Mechanisms of Severe Retinopathy of Prematurity. Ophthalmology 2015, 122, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Ashton, N.; Ward, B.; Serpell, G. Effect of Oxygen on Developing Retinal Vessels with Particular Reference to the Problem of Retrolental Fibroplasia. Br. J. Ophthalmol. 1954, 38, 397–432. [Google Scholar] [CrossRef] [Green Version]
- Katz, M.L.; Robison, W.G., Jr. Autoxidative damage to the retina: Potential role in retinopathy of prematurity. Birth Defects 1988, 24, 237–248. [Google Scholar] [PubMed]
- Penn, J.S. Oxygen-induced retinopathy in the rat: Possible contribution of peroxidation reactions. Doc. Ophthalmol. 1990, 74, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Geisen, P.; Uppal, A.; Hartnett, M.E. Inhibition of NAD(P)H oxidase reduces apoptosis and avascular retina in an animal model of retinopathy of prematurity. Mol. Vis. 2007, 13, 840–853. [Google Scholar] [PubMed]
- Robinson, W.G.J.; Kuwabara, T.; Bieri, J.G. The roles of vitamin E and unsaturated fatty acids in the visual process. Retina 1982, 2, 263–281. [Google Scholar] [CrossRef]
- Stone, W.; Farnsworth, C.; Dratz, E. A reinvestigation of the fatty acid content of bovine, rat and frog retinal rod outer segments. Exp. Eye Res. 1979, 28, 387–397. [Google Scholar] [CrossRef]
- Du, Y.; Veenstra, A.; Palczewski, K.; Kern, T.S. Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc. Natl. Acad. Sci. USA 2013, 110, 16586–16591. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhang, S.X.; Hartnett, M.E. Signaling Pathways Triggered by Oxidative Stress That Mediate Features of Severe Retinopathy of Prematurity. JAMA Ophthalmol. 2013, 131, 80–85. [Google Scholar] [CrossRef] [Green Version]
- Frey, R.S.; Ushio–Fukai, M.; Malik, A.B. NADPH Oxidase-Dependent Signaling in Endothelial Cells: Role in Physiology and Pathophysiology. Antioxidants Redox Signal. 2009, 11, 791–810. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hartnett, M.E. Roles of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase in Angiogenesis: Isoform-Specific Effects. Antioxidants 2017, 6, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLeo, F.; Quinn, M.T. Assembly of the phagocyte NADPH oxidase: Molecular interaction of oxidase proteins. J. Leukoc. Biol. 1996, 60, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, C.; Giannantonio, C.; Cota, F.; Papacci, P.; Vento, G.; Valente, E.; Purcaro, V.; Costa, S. A prospective, randomized, double blind study comparing lutein to placebo for reducing occurrence and severity of retinopathy of prematurity. J. Matern. Neonatal Med. 2011, 24, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.H.; Quinn, G.E.; Abbasi, S.; Delivoria-Papadopoulos, M.; Peckham, G.; Bowen, F.W., Jr. Bilateral Stage 3-Plus Retinopathy of Prematurity (ROP) Effect of Treatment (Rx) with High-Dose Vitamin E. Ann. N. Y. Acad. Sci. 1989, 570, 464–466. [Google Scholar] [CrossRef]
- Parad, R.B.; Allred, E.N.; Rosenfeld, W.N.; Davis, J.M. Reduction of Retinopathy of Prematurity in Extremely Low Gestational Age Newborns Treated with Recombinant Human Cu/Zn Superoxide Dismutase. Neonatology 2012, 102, 139–144. [Google Scholar] [CrossRef]
- Brines, M.; Grasso, G.; Fiordaliso, F.; Sfacteria, A.; Ghezzi, P.; Fratelli, M.; Latini, R.; Xie, Q.W.; Smart, J.; Su-Rick, C.J.; et al. Erythropoietin mediates tissue protection through an erythropoietin and common beta-subunit heteroreceptor. Proc. Natl. Acad. Sci. USA 2004, 101, 14907–14912. [Google Scholar] [CrossRef] [Green Version]
- Jaquet, K.; Krause, K.; Tawakol-Khodai, M.; Geidel, S.; Kuck, K.H. Erythropoietin and VEGF exhibit equal angiogenic potential. Microvasc. Res. 2002, 64, 326–333. [Google Scholar] [CrossRef]
- Ozturk, E.; Demirbilek, S.; But, A.K.; Saricicek, V.; Gulec, M.; Akyol, O.; Ersoy, M.O. Antioxidant properties of propofol and erythropoietin after closed head injury in rats. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2005, 29, 922–927. [Google Scholar] [CrossRef]
- Öztürk, E.; Demirbilek, S.; Köroğlu, A.; But, A.; Begeç, Z.; Gülec, M.; Akyol, O.; Ersoy, M. Propofol and erythropoietin antioxidant properties in rat brain injured tissue. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 81–86. [Google Scholar] [CrossRef]
- Juul, S.E.; Comstock, B.A.; Wadhawan, R.; Mayock, D.E.; Courtney, S.E.; Robinson, T.; Ahmad, K.A.; Bendel-Stenzel, E.; Baserga, M.; LaGamma, E.F.; et al. A Randomized Trial of Erythropoietin for Neuroprotection in Preterm Infants. N. Engl. J. Med. 2020, 382, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.-H.; Chung, M.-Y.; Zhou, X.-G.; Lin, H.-C. Early Erythropoietin Administration does not Increase the Risk of Retinopathy in Preterm Infants. Pediatr. Neonatol. 2016, 58, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Song, J.; Kang, W.; Wang, Y.; Sun, X.; Zhou, C.; Xiong, H.; Xu, F.; Li, M.; Zhang, X.; et al. Effect of early prophylactic low-dose recombinant human erythropoietin on retinopathy of prematurity in very preterm infants. J. Transl. Med. 2020, 18, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ohls, R.K.; Kamath-Rayne, B.D.; Christensen, R.D.; Wiedmeier, S.E.; Rosenberg, A.; Fuller, J.; Lacy, C.B.; Roohi, M.; Lambert, D.K.; Burnett, J.J.; et al. Cognitive Outcomes of Preterm Infants Randomized to Darbepoetin, Erythropoietin, or Placebo. Pediatrics 2014, 133, 1023–1030. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Connor, K.M.; Aderman, C.M.; Smith, L.E. Erythropoietin deficiency decreases vascular stability in mice. J. Clin. Investig. 2008, 118, 526–533. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Connor, K.M.; Aderman, C.M.; Willett, K.L.; Aspegren, O.P.; Smith, L.E.H. Suppression of Retinal Neovascularization by Erythropoietin siRNA in a Mouse Model of Proliferative Retinopathy. Investig. Opthalmology Vis. Sci. 2009, 50, 1329–1335. [Google Scholar] [CrossRef]
- Wang, H.; Byfield, G.; Jiang, Y.; Smith, G.W.; McCloskey, M.; Hartnett, M.E. VEGF-Mediated STAT3 Activation Inhibits Retinal Vascularization by Down-Regulating Local Erythropoietin Expression. Am. J. Pathol. 2012, 180, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Bretz, C.A.; Simmons, A.B.; Kunz, E.; Ramshekar, A.; Kennedy, C.; Cardenas, I.; Hartnett, M.E. Erythropoietin Receptor Signaling Supports Retinal Function after Vascular Injury. Am. J. Pathol. 2020, 190, 630–641. [Google Scholar] [CrossRef]
- Bretz, C.A.; Ramshekar, A.; Kunz, E.; Wang, H.; Hartnett, M.E. Signaling Through the Erythropoietin Receptor Affects Angiogenesis in Retinovascular Disease. Investig. Opthalmology Vis. Sci. 2020, 61, 23. [Google Scholar] [CrossRef]
- Lutty, G.A.; McLeod, D.S.; Bhutto, I.; Wiegand, S.J. Effect of VEGF Trap on Normal Retinal Vascular Development and Oxygen-Induced Retinopathy in the Dog. Investig. Opthalmology Vis. Sci. 2011, 52, 4039–4047. [Google Scholar] [CrossRef] [Green Version]
- Penn, J.S.; Henry, M.M.; Tolman, B.L. Exposure to Alternating Hypoxia and Hyperoxia Causes Severe Proliferative Retinopathy in the Newborn Rat. Pediatr. Res. 1994, 36, 724–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.E.; Wesolowski, E.; McLellan, A.; Kostyk, S.K.; D’Amato, R.; Sullivan, R.; D’Amore, P.A. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol. Vis. Sci. 1994, 35, 101–111. [Google Scholar] [PubMed]
- Cunningham, S.; McColm, J.R.; Wade, J.; Sedowofia, K.; McIntosh, N.; Fleck, B. A novel model of retinopathy of prematurity simulating preterm oxygen variability in the rat. Investig. Ophthalmol. Vis. Sci. 2000, 41, 4275–4280. [Google Scholar]
- Cunningham, S.; Mclntosh, N.; Fleck, B.; Elton, R. Transcutaneous oxygen levels in retinopathy of prematurity. Lancet 1995, 346, 1464–1465. [Google Scholar] [CrossRef]
- York, J.R.; Landers, S.; Kirby, R.S.; Arbogast, P.G.; Penn, J.S. Arterial Oxygen Fluctuation and Retinopathy of Prematurity in Very-Low-Birth-Weight Infants. J. Perinatol. 2004, 24, 82–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McColm, J.R.; Cunningham, S.; Wade, J.; Sedowofia, K.; Gellén, B.; Sharma, T.; McIntosh, N.; Fleck, B.W. Hypoxic Oxygen Fluctuations Produce Less Severe Retinopathy than Hyperoxic Fluctuations in a Rat Model of Retinopathy of Prematurity. Pediatr. Res. 2004, 55, 107–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Lin, F.-L.; Leung, J.Y.K.; Tu, L.; Wang, J.-H.; Chuang, Y.-F.; Li, F.; Shen, H.-H.; Dusting, G.J.; Wong, V.H.Y.; et al. A drug-tunable Flt23k gene therapy for controlled intervention in retinal neovascularization. Angiogenesis 2020, 24, 97–110. [Google Scholar] [CrossRef]
- Lamartina, S.; Cimino, M.; Roscilli, G.; Dammassa, E.; Lazzaro, D.; Rota, R.; Ciliberto, G.; Toniatti, C. Helper-dependent adenovirus for the gene therapy of proliferative retinopathies: Stable gene transfer, regulated gene expression and therapeutic efficacy. J. Gene Med. 2007, 9, 862–874. [Google Scholar] [CrossRef]
- Wang, H.; Smith, G.W.; Yang, Z.; Jiang, Y.; McCloskey, M.; Greenberg, K.; Geisen, P.; Culp, W.D.; Flannery, J.; Kafri, T.; et al. Short Hairpin RNA-Mediated Knockdown of VEGFA in Müller Cells Reduces Intravitreal Neovascularization in a Rat Model of Retinopathy of Prematurity. Am. J. Pathol. 2013, 183, 964–974. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Wang, H.; Culp, D.; Yang, Z.; Fotheringham, L.; Flannery, J.; Hammond, S.; Kafri, T.; Hartnett, M.E. Targeting Müller Cell–Derived VEGF164 to Reduce Intravitreal Neovascularization in the Rat Model of Retinopathy of Prematurity. Investig. Opthalmology Vis. Sci. 2014, 55, 824–831. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.-Y.; Cringle, S.J. Oxygen Distribution and Consumption within the Retina in Vascularised and Avascular Retinas and in Animal Models of Retinal Disease. Prog. Retin. Eye Res. 2001, 20, 175–208. [Google Scholar] [CrossRef]
- Chan, T.C.; Berka, J.L.W.; Deliyanti, D.; Hunter, D.; Fung, A.; Liew, G.; White, A. The role of reactive oxygen species in the pathogenesis and treatment of retinal diseases. Exp. Eye Res. 2020, 201, 108255. [Google Scholar] [CrossRef]
- Auten, R.L.; Davis, J.M. Oxygen Toxicity and Reactive Oxygen Species: The Devil Is in the Details. Pediatr. Res. 2009, 66, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starkov, A.A. The Role of Mitochondria in Reactive Oxygen Species Metabolism and Signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef]
- Kang, Q.; Yang, C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol. 2020, 37, 101799. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Kowluru, V.; Xiong, Y.; Ho, Y.-S. Overexpression of mitochondrial superoxide dismutase in mice protects the retina from diabetes-induced oxidative stress. Free Radic. Biol. Med. 2006, 41, 1191–1196. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Atasi, L.; Ho, Y.-S. Role of Mitochondrial Superoxide Dismutase in the Development of Diabetic Retinopathy. Investig. Opthalmology Vis. Sci. 2006, 47, 1594–1599. [Google Scholar] [CrossRef] [Green Version]
- Kanwar, M.; Chan, P.-S.; Kern, T.S.; Kowluru, R.A. Oxidative Damage in the Retinal Mitochondria of Diabetic Mice: Possible Protection by Superoxide Dismutase. Investig. Opthalmology Vis. Sci. 2007, 48, 3805–3811. [Google Scholar] [CrossRef] [Green Version]
- Schütte, M.; Werner, P. Redistribution of glutathione in the ischemic rat retina. Neurosci. Lett. 1998, 246, 53–56. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH Oxidases in Vascular Pathology. Antioxidants Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, K.-H. Tissue distribution and putative physiological function of NOX family NADPH oxidases. Jpn. J. Infect. Dis. 2004, 57. [Google Scholar]
- Lassègue, B.; San Martín, A.; Griendling, K.K. Biochemistry, Physiology, and Pathophysiology of NADPH Oxidases in the Cardiovascular System. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef]
- Cheng, G.; Diebold, B.A.; Hughes, Y.; Lambeth, J.D. Nox1-dependent Reactive Oxygen Generation Is Regulated by Rac1. J. Biol. Chem. 2006, 281, 17718–17726. [Google Scholar] [CrossRef] [Green Version]
- Raad, H.; Paclet, M.H.; Boussetta, T.; Kroviarski, Y.; Morel, F.; Quinn, M.T.; Gougerot-Pocidalo, M.A.; Dang, P.M.; El-Benna, J. Regulation of the phagocyte NADPH oxidase activity: Phosphorylation of gp91phox/NOX2 by protein kinase C enhances its diaphorase activity and binding to Rac2, p67phox, and p47phox. FASEB J. 2009, 23, 1011–1022. [Google Scholar] [CrossRef] [Green Version]
- Dang, P.M.-C.; Stensballe, A.; Boussetta, T.; Raad, H.; Dewas, C.; Kroviarski, Y.; Hayem, G.; Jensen, O.N.; Gougerot-Pocidalo, M.-A.; El-Benna, J. A specific p47phox -serine phosphorylated by convergent MAPKs mediates neutrophil NADPH oxidase priming at inflammatory sites. J. Clin. Investig. 2006, 116, 2033–2043. [Google Scholar] [CrossRef] [Green Version]
- Banfi, B.; Tirone, F.; Durussel, I.; Knisz, J.; Moskwa, P.; Molnár, G.Z.; Krause, K.-H.; Cox, J.A. Mechanism of Ca2+ Activation of the NADPH Oxidase 5 (NOX5). J. Biol. Chem. 2004, 279, 18583–18591. [Google Scholar] [CrossRef] [Green Version]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Martyn, K.D.; Frederick, L.M.; von Loehneysen, K.; Dinauer, M.C.; Knaus, U.G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell. Signal. 2006, 18, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Brown, O.I.; Bridge, K.I.; Kearney, M.T. Nicotinamide Adenine Dinucleotide Phosphate Oxidases in Glucose Homeostasis and Diabetes-Related Endothelial Cell Dysfunction. Cells 2021, 10, 2315. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Petry, A.; Hess, J.; Görlach, A. The NADPH Oxidase Subunit NOX4 Is a New Target Gene of the Hypoxia-inducible Factor-1. Mol. Biol. Cell 2010, 21, 2087–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, G.; Khan, S.A.; Luo, W.; Nanduri, J.; Semenza, G.L.; Prabhakar, N.R. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J. Cell. Physiol. 2011, 226, 2925–2933. [Google Scholar] [CrossRef] [Green Version]
- Manea, A.; Manea, S.A.; Gafencu, A.V.; Raicu, M. Regulation of NADPH oxidase subunit p22phoxby NF-kB in human aortic smooth muscle cells. Arch. Physiol. Biochem. 2007, 113, 163–172. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2015, 1850, 794–801. [Google Scholar] [CrossRef] [Green Version]
- Manea, A.; Tanase, L.I.; Raicu, M.; Simionescu, M. JAK/STAT Signaling Pathway Regulates Nox1 and Nox4-Based NADPH Oxidase in Human Aortic Smooth Muscle Cells. Arter. Thromb. Vasc. Biol. 2010, 30, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Sanders, Y.Y.; Liu, H.; Liu, G.; Thannickal, V.J. Epigenetic mechanisms regulate NADPH oxidase-4 expression in cellular senescence. Free Radic. Biol. Med. 2015, 79, 197–205. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, Y.; Wang, Z.; Wang, L.; Wei, X.; Zhang, B.; Wen, Z.; Fang, H.; Pang, Q.; Yi, F. Regulation of NADPH Oxidase Activity Is Associated with miRNA-25-Mediated NOX4 Expression in Experimental Diabetic Nephropathy. Am. J. Nephrol. 2010, 32, 581–589. [Google Scholar] [CrossRef]
- Varga, Z.V.; Kupai, K.; Szűcs, G.; Gáspár, R.; Pálóczi, J.; Faragó, N.; Zvara, A.; Puskás, L.G.; Rázga, Z.; Tiszlavicz, L.; et al. MicroRNA-25-dependent up-regulation of NADPH oxidase 4 (NOX4) mediates hypercholesterolemia-induced oxidative/nitrative stress and subsequent dysfunction in the heart. J. Mol. Cell. Cardiol. 2013, 62, 111–121. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Cross-Talk between NADPH Oxidase and Mitochondria: Role in ROS Signaling and Angiogenesis. Cells 2020, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Alexander, R.W. Reactive oxygen species as mediators of angiogenesis signaling. Role of NAD(P)H oxidase. Mol. Cell. Biochem. 2004, 264, 85–97. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paravicini, T.M.; Touyz, R.M. NADPH oxidases, reactive oxygen species, and hypertension: Clinical implications and therapeutic possibilities. Diabetes Care 2008, 31 (Suppl. 2), S170–S180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Uppal, A.; Byfield, G.; Budd, S.; Hartnett, M.E. Activated NAD(P)H Oxidase from Supplemental Oxygen Induces Neovascularization Independent of VEGF in Retinopathy of Prematurity Model. Investig. Opthalmology Vis. Sci. 2008, 49, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Byfield, G.; Budd, S.; Hartnett, M.E. The Role of Supplemental Oxygen and JAK/STAT Signaling in Intravitreous Neovascularization in a ROP Rat Model. Investig. Opthalmology Vis. Sci. 2009, 50, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Garrido-Urbani, S.; Jemelin, S.; Deffert, C.; Carnesecchi, S.; Basset, O.; Szyndralewiez, C.; Heitz, F.; Page, P.; Montet, X.; Michalik, L.; et al. Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARalpha mediated mechanism. PLoS ONE 2011, 6, e14665. [Google Scholar] [CrossRef]
- Juhasz, A.; Markel, S.; Gaur, S.; Liu, H.; Lu, J.; Jiang, G.; Wu, X.; Antony, S.; Wu, Y.; Melillo, G.; et al. NADPH oxidase 1 supports proliferation of colon cancer cells by modulating reactive oxygen species-dependent signal transduction. J. Biol. Chem. 2017, 292, 7866–7887. [Google Scholar] [CrossRef] [Green Version]
- Stalin, J.; Garrido-Urbani, S.; Heitz, F.; Szyndralewiez, C.; Jemelin, S.; Coquoz, O.; Ruegg, C.; Imhof, B.A. Inhibition of host NOX1 blocks tumor growth and enhances checkpoint inhibitor–based immunotherapy. Life Sci. Alliance 2019, 2, e201800265. [Google Scholar] [CrossRef] [Green Version]
- Wolf, A.; Herb, M.; Schramm, M.; Langmann, T. The TSPO-NOX1 axis controls phagocyte-triggered pathological angiogenesis in the eye. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Li, Y.; Kračun, D.; Dustin, C.M.; El Massry, M.; Yuan, S.; Goossen, C.J.; DeVallance, E.R.; Sahoo, S.; Hilaire, C.S.; Gurkar, A.U.; et al. Forestalling age-impaired angiogenesis and blood flow by targeting NOX: Interplay of NOX1, IL-6, and SASP in propagating cell senescence. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Liu, Y.-Y.; Lv, X.-F.; Lin, Z.-M.; Zhang, T.-T.; Zhang, F.-R.; Guo, J.-W.; Hong, Y.; Liu, X.; Lin, X.-C.; et al. Reduced intracellular chloride concentration impairs angiogenesis by inhibiting oxidative stress-mediated VEGFR2 activation. Acta Pharmacol. Sin. 2020, 42, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Watt, J.M.; Fortunato, T.M.; Mellor, H.; Burgess, M.; Wicks, K.; Mace, K.; Reeksting, S.B.; Lubben, A.; Wheeler-Jones, C.P.D.; et al. Direct Activation of NADPH Oxidase 2 by 2-Deoxyribose-1-Phosphate Triggers Nuclear Factor Kappa B-Dependent Angiogenesis. Antioxid. Redox Signal. 2018, 28, 110–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.-M.; Kim, S.-J.; Tatsunami, R.; Yamamura, H.; Fukai, T.; Ushio-Fukai, M. ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. Am. J. Physiol. Physiol. 2017, 312, C749–C764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Wang, J.J.; Wang, J.; Abboud, H.E.; Chen, Y.; Zhang, S.X. Endothelium-specific deletion of Nox4 delays retinal vascular development and mitigates pathological angiogenesis. Angiogenesis 2020, 24, 363–377. [Google Scholar] [CrossRef]
- Hu, C.; Wu, Z.; Huang, Z.; Hao, X.; Wang, S.; Deng, J.; Yin, Y.; Tan, C. Nox2 impairs VEGF-A-induced angiogenesis in placenta via mitochondrial ROS-STAT3 pathway. Redox Biol. 2021, 45, 102051. [Google Scholar] [CrossRef]
- Harrison, I.P.; Vinh, A.; Johnson, I.; Luong, R.; Drummond, G.R.; Sobey, C.G.; Tiganis, T.; Williams, E.; O’Leary, J.; Brooks, D.; et al. NOX2 oxidase expressed in endosomes promotes cell proliferation and prostate tumour development. Oncotarget 2018, 9, 35378–35393. [Google Scholar] [CrossRef] [Green Version]
- Appukuttan, B.; Ma, Y.; Stempel, A.; Ashander, L.M.; Deliyanti, D.; Wilkinson-Berka, J.L.; Smith, J.R. Effect of NADPH oxidase 1 and 4 blockade in activated human retinal endothelial cells. Clin. Exp. Ophthalmol. 2018, 46, 652–660. [Google Scholar] [CrossRef] [Green Version]
- Deliyanti, D.; AlRashdi, S.F.; Touyz, R.M.; Kennedy, C.R.; Jha, J.C.; Cooper, M.E.; Jandeleit-Dahm, K.A.; Wilkinson-Berka, J.L. Nox (NADPH Oxidase) 1, Nox4, and Nox5 Promote Vascular Permeability and Neovascularization in Retinopathy. Hypertension 2020, 75, 1091–1101. [Google Scholar] [CrossRef]
- Wang, H.; Yang, Z.; Jiang, Y.; Hartnett, M.E. Endothelial NADPH oxidase 4 mediates vascular endothelial growth factor receptor 2–induced intravitreal neovascularization in a rat model of retinopathy of prematurity. Mol. Vis. 2014, 20, 231–241. [Google Scholar]
- Ushio-Fukai, M. Redox signaling in angiogenesis: Role of NADPH oxidase. Cardiovasc. Res. 2006, 71, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urao, N.; Inomata, H.; Razvi, M.; Kim, H.W.; Wary, K.; McKinney, R.; Fukai, T.; Ushio-Fukai, M. Role of Nox2-Based NADPH Oxidase in Bone Marrow and Progenitor Cell Function Involved in Neovascularization Induced by Hindlimb Ischemia. Circ. Res. 2008, 103, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Galougahi, K.K.; Liu, C.C.; Gentile, C.; Kok, C.; Nunez, A.; Garcia, A.; Fry, N.A.; Davies, M.J.; Hawkins, C.L.; Rasmussen, H.H.; et al. Glutathionylation mediates angiotensin II-induced eNOS uncoupling, amplifying NADPH oxidase-dependent endothelial dysfunction. J. Am. Heart Assoc. 2014, 3, e000731. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Gong, J.; Xu, Z.; Duh, E.J. Nrf2 promotes reparative angiogenesis through regulation of NADPH oxidase-2 in oxygen-induced retinopathy. Free Radic. Biol. Med. 2016, 99, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Johnson, I.R.; Parkinson-Lawrence, E.J.; Keegan, H.; Spillane, C.D.; Barry-O’Crowley, J.; Watson, W.R.; Selemidis, S.; Butler, L.M.; O’Leary, J.J.; Brooks, D.A. Endosomal gene expression: A new indicator for prostate cancer patient prognosis? Oncotarget 2015, 6, 37919–37929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCann, S.K.; Dusting, G.J.; Roulston, C.L. Nox2 Knockout Delays Infarct Progression and Increases Vascular Recovery through Angiogenesis in Mice following Ischaemic Stroke with Reperfusion. PLoS ONE 2014, 9, e110602. [Google Scholar] [CrossRef]
- Oshikawa, J.; Kim, S.-J.; Furuta, E.; Caliceti, C.; Chen, G.-F.; McKinney, R.D.; Kuhr, F.; Levitan, I.; Fukai, T.; Ushio-Fukai, M. Novel role of p66Shc in ROS-dependent VEGF signaling and angiogenesis in endothelial cells. Am. J. Physiol. Circ. Physiol. 2012, 302, H724–H732. [Google Scholar] [CrossRef] [Green Version]
- Pi, X.; Xie, L.; Portbury, A.L.; Kumar, S.; Lockyer, P.; Li, X.; Patterson, C. NADPH Oxidase–Generated Reactive Oxygen Species Are Required for Stromal Cell–Derived Factor-1α–Stimulated Angiogenesis. Arter. Thromb. Vasc. Biol. 2014, 34, 2023–2032. [Google Scholar] [CrossRef] [Green Version]
- Fulton, D.J. Nox5 and the Regulation of Cellular Function. Antioxidants Redox Signal. 2009, 11, 2443–2452. [Google Scholar] [CrossRef] [Green Version]
- Ebie, A.Z.; Fleming, K.G. Dimerization of the Erythropoietin Receptor Transmembrane Domain in Micelles. J. Mol. Biol. 2007, 366, 517–524. [Google Scholar] [CrossRef]
- Witthuhn, B.A.; Quelle, F.; Silvennoinen, O.; Yi, T.; Tang, B.; Miura, O.; Ihle, J.N. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell 1993, 74, 227–236. [Google Scholar] [CrossRef]
- Ostrowski, D.; Heinrich, R. Alternative Erythropoietin Receptors in the Nervous System. J. Clin. Med. 2018, 7, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suenobu, S.; Takakura, N.; Inada, T.; Yamada, Y.; Yuasa, H.; Zhang, X.-Q.; Sakano, S.; Oike, Y.; Suda, T. A role of EphB4 receptor and its ligand, ephrin-B2, in erythropoiesis. Biochem. Biophys. Res. Commun. 2002, 293, 1124–1131. [Google Scholar] [CrossRef]
- Wang, G.L.; Semenza, G.L. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc. Natl. Acad. Sci. USA 1993, 90, 4304–4308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acker, H.; Dufau, E.; Huber, J.; Sylvester, D. Indications to an NADPH oxidase as a possible pO2 sensor in the rat carotid body. FEBS Lett. 1989, 256, 75–78. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Youngson, C.; Wong, V.; Yeger, H.; Dinauer, M.C.; de Miera, E.V.-S.; Rudy, B.; Cutz, E. NADPH-oxidase and a hydrogen peroxide-sensitive K+ channel may function as an oxygen sensor complex in airway chemoreceptors and small cell lung carcinoma cell lines. Proc. Natl. Acad. Sci. USA 1996, 93, 13182–13187. [Google Scholar] [CrossRef] [Green Version]
- Bunn, H.F.; Gu, J.; E Huang, L.; Park, J.W.; Zhu, H. Erythropoietin: A model system for studying oxygen-dependent gene regulation. J. Exp. Biol. 1998, 201, 1197–1201. [Google Scholar] [CrossRef]
- Fandrey, J.; Frede, S.; Jelkmann, W. Role of hydrogen peroxide in hypoxia-induced erythropoietin production. Biochem. J. 1994, 303, 507–510. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.P.; Yang, X.H.; Wang, X.J.; Li, S.M.; Sun, N.; Zhang, T. Erythropoietin Decreases the Occurrence of Myocardial Fibrosis by Inhibiting the NADPH-ERK-NFκB Pathway. Cardiology 2016, 133, 97–108. [Google Scholar] [CrossRef]
- Toba, H.; Sawai, N.; Morishita, M.; Murata, S.; Yoshida, M.; Nakashima, K.; Morita, Y.; Kobara, M.; Nakata, T. Chronic treatment with recombinant human erythropoietin exerts renoprotective effects beyond hematopoiesis in streptozotocin-induced diabetic rat. Eur. J. Pharmacol. 2009, 612, 106–114. [Google Scholar] [CrossRef]
- Dayyat, E.A.; Zhang, S.X.; Wang, Y.; Cheng, Z.J.; Gozal, D. Exogenous erythropoietin administration attenuates intermittent hypoxia-induced cognitive deficits in a murine model of sleep apnea. BMC Neurosci. 2012, 13, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.-h.; Zhang, J.; Xiong, Q. Suppressive effect erythropoietin on oxidative stress by targeting AMPK/Nox4/ROS pathway in renal ischemia reperfusion injury. Transpl. Immunol. 2022, 72, 101537. [Google Scholar] [CrossRef] [PubMed]
- Toba, H.; Kojima, Y.; Wang, J.; Noda, K.; Tian, W.; Kobara, M.; Nakata, T. Erythropoietin attenuated vascular dysfunction and inflammation by inhibiting NADPH oxidase-derived superoxide production in nitric oxide synthase-inhibited hypertensive rat aorta. Eur. J. Pharmacol. 2012, 691, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Toba, H.; Morita, Y.; Nakashima, K.; Noda, K.; Tian, W.; Kobara, M.; Nakata, T. Endothelial Dysfunction, Macrophage Infiltration and NADPH Oxidase-Dependent Superoxide Production Were Attenuated by Erythropoietin in Streptozotocin-Induced Diabetic Rat Aorta. Pharmacology 2013, 91, 48–58. [Google Scholar] [CrossRef]
- Murasawa, S.; Asahara, T. Endothelial progenitor cells for vasculogenesis. Physiology 2005, 20, 36–42. [Google Scholar] [CrossRef]
- Heeschen, C.; Aicher, A.; Lehmann, R.; Fichtlscherer, S.; Vasa, M.; Urbich, C.; Mildner-Rihm, C.; Martin, H.; Zeiher, A.M.; Dimmeler, S. Erythropoietin is a potent physiologic stimulus for endothelial progenitor cell mobilization. Blood 2003, 102, 1340–1346. [Google Scholar] [CrossRef] [Green Version]
- Schröder, K.; Kohnen, A.; Aicher, A.; Liehn, E.A.; Büchse, T.; Stein, S.; Weber, C.; Dimmeler, S.; Brandes, R.P. NADPH Oxidase Nox2 Is Required for Hypoxia-Induced Mobilization of Endothelial Progenitor Cells. Circ. Res. 2009, 105, 537–544. [Google Scholar] [CrossRef]
- Sato, T.; Kusaka, S.; Shimojo, H.; Fujikado, T. Vitreous Levels of Erythropoietin and Vascular Endothelial Growth Factor in Eyes with Retinopathy of Prematurity. Ophthalmology 2009, 116, 1599–1603. [Google Scholar] [CrossRef]
- Becker, S.; Wang, H.; Yu, B.; Brown, R.; Han, X.; Lane, R.H.; Hartnett, M.E. Protective effect of maternal uteroplacental insufficiency on oxygen-induced retinopathy in offspring: Removing bias of premature birth. Sci. Rep. 2017, 7, 42301. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Ohneda, O.; Takahashi, S.; Higuchi, M.; Mukai, H.Y.; Nakahata, T.; Imagawa, S.; Yamamoto, M. Erythroid-specific expression of the erythropoietin receptor rescued its null mutant mice from lethality. Blood 2002, 100, 2279–2288. [Google Scholar] [CrossRef] [Green Version]
- Nakano, M.; Satoh, K.; Fukumoto, Y.; Ito, Y.; Kagaya, Y.; Ishii, N.; Sugamura, K.; Shimokawa, H. Important Role of Erythropoietin Receptor to Promote VEGF Expression and Angiogenesis in Peripheral Ischemia in Mice. Circ. Res. 2007, 100, 662–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCloskey, M.; Wang, H.; Jiang, Y.; Smith, G.W.; Strange, J.; Hartnett, M.E. Anti-VEGF Antibody Leads to Later Atypical Intravitreous Neovascularization and Activation of Angiogenic Pathways in a Rat Model of Retinopathy of Prematurity. Investig. Opthalmology Vis. Sci. 2013, 54, 2020–2026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, O.E.; Canning, P.; Reid, E.; Bertelli, P.M.; McKeown, S.; Brines, M.; Cerami, A.; Du, X.; Xu, H.; Chen, M.; et al. The vasoreparative potential of endothelial colony-forming cells in the ischemic retina is enhanced by cibinetide, a non-hematopoietic erythropoietin mimetic. Exp. Eye Res. 2019, 182, 144–155. [Google Scholar] [CrossRef]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1α regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278. [Google Scholar] [CrossRef] [Green Version]
- Morita, M.; Ohneda, O.; Yamashita, T.; Takahashi, S.; Suzuki, N.; Nakajima, O.; Kawauchi, S.; Ema, M.; Shibahara, S.; Udono, T.; et al. HLF/HIF-2α is a key factor in retinopathy of prematurity in association with erythropoietin. EMBO J. 2003, 22, 1134–1146. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Wang, H.; Jiang, Y.; Hartnett, M.E. VEGFA Activates Erythropoietin Receptor and Enhances VEGFR2-Mediated Pathological Angiogenesis. Am. J. Pathol. 2014, 184, 1230–1239. [Google Scholar] [CrossRef] [Green Version]
- Nagao, M.; Masuda, S.; Abe, S.; Ueda, M.; Sasaki, R. Production and ligand-binding characteristics of the soluble form of murine erythropoietin receptor. Biochem. Biophys. Res. Commun. 1992, 188, 888–897. [Google Scholar] [CrossRef]
- Watanabe, D.; Suzuma, K.; Matsui, S.; Kurimoto, M.; Kiryu, J.; Kita, M.; Suzuma, I.; Ohashi, H.; Ojima, T.; Murakami, T.; et al. Erythropoietin as a Retinal Angiogenic Factor in Proliferative Diabetic Retinopathy. New Engl. J. Med. 2005, 353, 782–792. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cung, T.; Wang, H.; Hartnett, M.E. The Effects of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase and Erythropoietin, and Their Interactions in Angiogenesis: Implications in Retinopathy of Prematurity. Cells 2022, 11, 1951. https://doi.org/10.3390/cells11121951
Cung T, Wang H, Hartnett ME. The Effects of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase and Erythropoietin, and Their Interactions in Angiogenesis: Implications in Retinopathy of Prematurity. Cells. 2022; 11(12):1951. https://doi.org/10.3390/cells11121951
Chicago/Turabian StyleCung, Thaonhi, Haibo Wang, and M. Elizabeth Hartnett. 2022. "The Effects of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase and Erythropoietin, and Their Interactions in Angiogenesis: Implications in Retinopathy of Prematurity" Cells 11, no. 12: 1951. https://doi.org/10.3390/cells11121951