
New Insights into Neuroinflammation Involved in Pathogenic Mechanism of Alzheimer’s Disease and Its Potential for Therapeutic Intervention


{kind=link}
Abstract
:1. Introduction
2. NF-κB
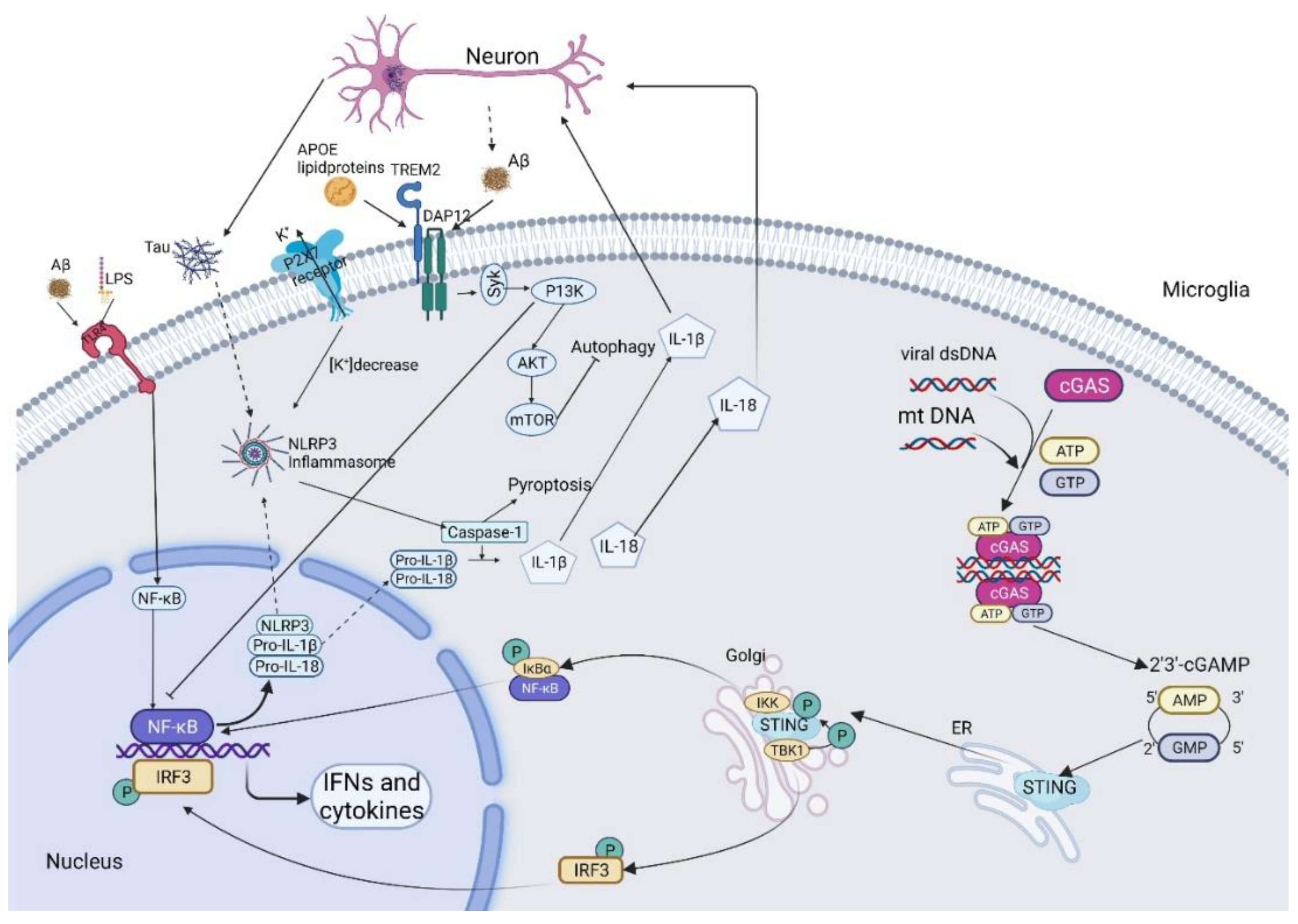
2.1. The NF-κB Pathway
2.2. Receptors for Advanced Glycation End Products (RAGE)
2.3. NF-κB-Targeting Drugs as Potential AD Therapeutics
3. NLRP3
3.1. The NLRP3 Inflammasome
3.2. The NLRP3 Activation Pathway
3.3. The Role of NLRP3 Inflammasome in AD
3.4. Therapeutic Targeting NLRP3 in the Treatment of AD
4. TREM2
4.1. TREM2 Structure and Variants Related to AD
4.2. TREM2 Functions in AD Pathology
4.3. Targeting TREM2 as a Novel Strategy for AD
5. cGAS-STING Pathway
5.1. The cGAMP-STING-IRF3 Signaling Pathway
5.2. Effects of cGAS-STING Pathway on Neuroinflammation
5.3. The Role of cGAS-STING Signaling in AD
5.4. Therapeutic Targeting of cGAS-STING Pathway in Treatment of AD
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Heath Organisation. Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 24 January 2022).
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knopman, D.S.; Jack, C.R., Jr.; Wiste, H.J.; Weigand, S.D.; Vemuri, P.; Lowe, V.J.; Kantarci, K.; Gunter, J.L.; Senjem, M.L.; Mielke, M.M.; et al. Brain injury biomarkers are not dependent on β-amyloid in normal elderly. Ann. Neurol. 2013, 73, 472–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knopman, D.S.; Jones, D.T.; Greicius, M.D. Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimer’s Dement. 2021, 17, 696–701. [Google Scholar] [CrossRef]
- Musiek, E.S.; Gomez-Isla, T.; Holtzman, D.M. Aducanumab for Alzheimer disease: The amyloid hypothesis moves from bench to bedside. J. Clin. Investig. 2021, 131, e154889. [Google Scholar] [CrossRef]
- Ferreira, S.T.; Lourenco, M.V.; Oliveira, M.M.; De Felice, F.G. Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front. Cell Neurosci. 2015, 9, 191. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.; Dickson, D.W.; Lin, W.L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef] [Green Version]
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimer’s Dementia. 2021, 17, 115–124. [Google Scholar] [CrossRef]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Folch, J.; Busquets, O.; Ettcheto, M.; Sánchez-López, E.; Castro-Torres, R.D.; Verdaguer, E.; Garcia, M.L.; Olloquequi, J.; Casadesús, G.; Beas-Zarate, C.; et al. Memantine for the Treatment of Dementia: A Review on its Current and Future Applications. J. Alzheimer’s Dis. 2018, 62, 1223–1240. [Google Scholar] [CrossRef] [Green Version]
- Lovestone, S.; Boada, M.; Dubois, B.; Hüll, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andrés, M.V.; Gómez-Carrillo, B.; León, T.; et al. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef]
- del Ser, T.; Steinwachs, K.C.; Gertz, H.J.; Andrés, M.V.; Gómez-Carrillo, B.; Medina, M.; Vericat, J.A.; Redondo, P.; Fleet, D.; León, T. Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: A pilot study. J. Alzheimer’s Dis. 2013, 33, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg Spine 2016, 1, 1003. [Google Scholar]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Gaiteri, C.; Bodea, L.G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Dong, Z.; Song, W. NLRP3 inflammasome as a novel therapeutic target for Alzheimer’s disease. Signal Transduct. Target. Ther. 2020, 5, 37. [Google Scholar] [CrossRef]
- Wang, C.; Fan, L.; Khawaja, R.R.; Liu, B.; Zhan, L.; Kodama, L.; Chin, M.; Li, Y.; Le, D.; Zhou, Y.; et al. Microglial NF-κB drives tau spreading and toxicity in a mouse model of tauopathy. Nat. Commun. 2022, 13, 1969. [Google Scholar] [CrossRef]
- Hopfner, K.-P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef]
- Seo, E.J.; Fischer, N.; Efferth, T. Phytochemicals as inhibitors of NF-κB for treatment of Alzheimer’s disease. Pharmacol Res. 2018, 129, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liang, H.; Zen, K. Molecular Mechanisms That Influence the Macrophage M1–M2 Polarization Balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Hu, L.; Zhao, J.; Hong, H.; Feng, F.; Qu, W.; Liu, W. Chotosan improves Aβ1-42-induced cognitive impairment and neuroinflammatory and apoptotic responses through the inhibition of TLR-4/NF-κB signaling in mice. J. Ethnopharmacol. 2016, 191, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; de Oliveira, A.C.P. Role of Microglia TLRs in Neurodegeneration. Front. Cell Neurosci. 2018, 12, 329. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol 2005, 6, 1087–1095. [Google Scholar] [CrossRef]
- Israël, A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Signal. Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef]
- Ullah, M.O.; Sweet, M.J.; Mansell, A.; Kellie, S.; Kobe, B. TRIF-dependent TLR signaling, its functions in host defense and inflammation, and its potential as a therapeutic target. J. Leukoc. Biol. 2016, 100, 27–45. [Google Scholar] [CrossRef]
- Gritsenko, A.; Yu, S.; Martin-Sanchez, F.; Diaz-del-Olmo, I.; Nichols, E.-M.; Davis, D.M.; Brough, D.; Lopez-Castejon, G. Priming Is Dispensable for NLRP3 Inflammasome Activation in Human Monocytes In Vitro. Front. Immunol. 2020, 11, 2573. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, J.A.; Jarosz-Griffiths, H.H.; Caseley, E.; Lara-Reyna, S.; Poulter, J.A.; Williams-Gray, C.H.; Peckham, D.; McDermott, M.F. Neurodegenerative Disease and the NLRP3 Inflammasome. Front. Pharmacol. 2021, 12, 193. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.R.; Cho, W.H.; Kim, J.; Lee, H.J.; Chung, C.; Jeon, W.K.; Han, J.S. Increased expression of the receptor for advanced glycation end products in neurons and astrocytes in a triple transgenic mouse model of Alzheimer’s disease. Exp. Mol. Med. 2014, 46, e75. [Google Scholar] [CrossRef] [Green Version]
- Li, X.H.; Lv, B.L.; Xie, J.Z.; Liu, J.; Zhou, X.W.; Wang, J.Z. AGEs induce Alzheimer-like tau pathology and memory deficit via RAGE-mediated GSK-3 activation. Neurobiol. Aging 2012, 33, 1400–1410. [Google Scholar] [CrossRef]
- Tóbon-Velasco, J.C.; Cuevas, E.; Torres-Ramos, M.A. Receptor for AGEs (RAGE) as mediator of NF-kB pathway activation in neuroinflammation and oxidative stress. CNS Neurol. Disord. Drug Targets 2014, 13, 1615–1626. [Google Scholar] [CrossRef]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef]
- Granic, I.; Dolga, A.M.; Nijholt, I.M.; van Dijk, G.; Eisel, U.L. Inflammation and NF-kappaB in Alzheimer’s disease and diabetes. J. Alzheimer’s Dis. 2009, 16, 809–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birch, A.M.; Katsouri, L.; Sastre, M. Modulation of inflammation in transgenic models of Alzheimer’s disease. J. Neuroinflammation 2014, 11, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillot-Sestier, M.V.; Doty, K.R.; Gate, D.; Rodriguez, J., Jr.; Leung, B.P.; Rezai-Zadeh, K.; Town, T. Il10 Deficiency Rebalances Innate Immunity to Mitigate Alzheimer-Like Pathology. Neuron 2015, 85, 534–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagisawa, D.; Ibrahim, N.F.; Taguchi, H.; Morikawa, S.; Hirao, K.; Shirai, N.; Sogabe, T.; Tooyama, I. Curcumin derivative with the substitution at C-4 position, but not curcumin, is effective against amyloid pathology in APP/PS1 mice. Neurobiol. Aging 2015, 36, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Hackler, L., Jr.; Ózsvári, B.; Gyuris, M.; Sipos, P.; Fábián, G.; Molnár, E.; Marton, A.; Faragó, N.; Mihály, J.; Nagy, L.I.; et al. The Curcumin Analog C-150, Influencing NF-κB, UPR and Akt/Notch Pathways Has Potent Anticancer Activity In Vitro and In Vivo. PLoS ONE 2016, 11, e0149832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libro, R.; Giacoppo, S.; Soundara Rajan, T.; Bramanti, P.; Mazzon, E. Natural Phytochemicals in the Treatment and Prevention of Dementia: An Overview. Molecules 2016, 21, 518. [Google Scholar] [CrossRef] [Green Version]
- Calder, P.C. Omega-3 polyunsaturated fatty acids and inflammatory processes: Nutrition or pharmacology? Br. J. Clin. Pharmacol. 2013, 75, 645–662. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Gao, L.; Liu, X.; Lu, T.; Xie, C.; Jia, J. Resveratrol Attenuates Microglial Activation via SIRT1-SOCS1 Pathway. Evid. Based Complementary Altern. Med. 2017, 2017, 8791832. [Google Scholar] [CrossRef]
- Capiralla, H.; Vingtdeux, V.; Zhao, H.; Sankowski, R.; Al-Abed, Y.; Davies, P.; Marambaud, P. Resveratrol mitigates lipopolysaccharide- and Aβ-mediated microglial inflammation by inhibiting the TLR4/NF-κB/STAT signaling cascade. J. Neurochem. 2012, 120, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Cianciulli, A.; Dragone, T.; Calvello, R.; Porro, C.; Trotta, T.; Lofrumento, D.D.; Panaro, M.A. IL-10 plays a pivotal role in anti-inflammatory effects of resveratrol in activated microglia cells. Int. Immunopharmacol. 2015, 24, 369–376. [Google Scholar] [CrossRef]
- Munoz, L.; Ralay Ranaivo, H.; Roy, S.M.; Hu, W.; Craft, J.M.; McNamara, L.K.; Chico, L.W.; Van Eldik, L.J.; Watterson, D.M. A novel p38 alpha MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer’s disease mouse model. J. Neuroinflammation 2007, 4, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ketelut-Carneiro, N.; Fitzgerald, K.A. Inflammasomes. Curr. Biol. 2020, 30, R689–R694. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P.Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Shaftel, S.S.; Griffin, W.S.T.; O’Banion, M.K. The role of interleukin-1 in neuroinflammation and Alzheimer disease: An evolving perspective. J. Neuroinflammation 2008, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Felderhoff-Mueser, U.; Schmidt, O.I.; Oberholzer, A.; Buhrer, C.; Stahel, P.F. IL-18: A key player in neuroinflammation and neurodegeneration? Trends Neurosci. 2005, 28, 487–493. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Boise, L.H.; Collins, C.M. Salmonella-induced cell death: Apoptosis, necrosis or programmed cell death? Trends Microbiol. 2001, 9, 64–67. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Xu, W.; Zhou, R. NLRP3 inflammasome activation and cell death. Cell. Mol. Immunol. 2021, 18, 2114–2127. [Google Scholar] [CrossRef] [PubMed]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, H.; Zhang, Q. Activation of NLRP3 Inflammasome and Onset of Alzheimer’s Disease. Front. Immunol. 2021, 12, 2998. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Gustin, A.; Kirchmeyer, M.; Koncina, E.; Felten, P.; Losciuto, S.; Heurtaux, T.; Tardivel, A.; Heuschling, P.; Dostert, C. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PLoS ONE 2015, 10, e0130624. [Google Scholar] [CrossRef] [Green Version]
- Yap, J.K.Y.; Pickard, B.S.; Chan, E.W.L.; Gan, S.Y. The Role of Neuronal NLRP1 Inflammasome in Alzheimer’s Disease: Bringing Neurons into the Neuroinflammation Game. Mol. Neurobiol. 2019, 56, 7741–7753. [Google Scholar] [CrossRef]
- Minkiewicz, J.; de Rivero Vaccari, J.P.; Keane, R.W. Human astrocytes express a novel NLRP2 inflammasome. Glia 2013, 61, 1113–1121. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M.; Hanan, M.; Greenberg, D.; Soreq, H.; Latz, E.; Golenbock, D.; Heneka, M.T. Systemic inflammation impairs microglial Aβ clearance through NLRP3 inflammasome. EMBO J. 2019, 38, e101064. [Google Scholar] [CrossRef] [PubMed]
- Krabbe, G.; Halle, A.; Matyash, V.; Rinnenthal, J.L.; Eom, G.D.; Bernhardt, U.; Miller, K.R.; Prokop, S.; Kettenmann, H.; Heppner, F.L. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE 2013, 8, e60921. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Bond, M.; Rogers, G.; Peters, J.; Anderson, R.; Hoyle, M.; Miners, A.; Moxham, T.; Davis, S.; Thokala, P.; Wailoo, A.; et al. The effectiveness and cost-effectiveness of donepezil, galantamine, rivastigmine and memantine for the treatment of Alzheimer’s disease (review of Technology Appraisal No. 111): A systematic review and economic model. Health Technol. Assess. 2012, 16, 1–470. [Google Scholar] [CrossRef]
- Kim, J.; Lee, H.-J.; Park, S.K.; Park, J.-H.; Jeong, H.-R.; Lee, S.; Lee, H.; Seol, E.; Hoe, H.-S. Donepezil Regulates LPS and Aβ-Stimulated Neuroinflammation through MAPK/NLRP3 Inflammasome/STAT3 Signaling. Int. J. Mol. Sci. 2021, 22, 10637. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [Green Version]
- Dempsey, C.; Rubio Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.A.B.; Cooper, M.A.; O’Neill, L.A.J.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef] [Green Version]
- Stancu, I.-C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brône, B.; Huaux, F.; Octave, J.-N.; et al. Aggregated Tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A.; et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonnemann, N.; Hosseini, S.; Marchetti, C.; Skouras, D.B.; Stefanoni, D.; D’Alessandro, A.; Dinarello, C.A.; Korte, M. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 32145–32154. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yang, H.; Yu, L.; Jin, J.; Qian, L.; Zhao, H.; Xu, Y.; Zhu, X. Oridonin attenuates Aβ1-42-induced neuroinflammation and inhibits NF-κB pathway. PLoS ONE 2014, 9, e104745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W.; et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 2018, 9, 2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniels, M.J.D.; Rivers-Auty, J.; Schilling, T.; Spencer, N.G.; Watremez, W.; Fasolino, V.; Booth, S.J.; White, C.S.; Baldwin, A.G.; Freeman, S.; et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun. 2016, 7, 12504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuve, O.; Weideman, R.A.; McMahan, D.M.; Jacob, D.A.; Little, B.B. Diclofenac reduces the risk of Alzheimer’s disease: A pilot analysis of NSAIDs in two US veteran populations. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420935676. [Google Scholar] [CrossRef]
- Indramohan, M.; Stehlik, C.; Dorfleutner, A. COPs and POPs Patrol Inflammasome Activation. J. Mol. Biol. 2018, 430, 153–173. [Google Scholar] [CrossRef]
- de Almeida, L.; Khare, S.; Misharin, A.V.; Patel, R.; Ratsimandresy, R.A.; Wallin, M.C.; Perlman, H.; Greaves, D.R.; Hoffman, H.M.; Dorfleutner, A.; et al. The PYRIN Domain-only Protein POP1 Inhibits Inflammasome Assembly and Ameliorates Inflammatory Disease. Immunity 2015, 43, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Khare, S.; Ratsimandresy, R.A.; de Almeida, L.; Cuda, C.M.; Rellick, S.L.; Misharin, A.V.; Wallin, M.C.; Gangopadhyay, A.; Forte, E.; Gottwein, E.; et al. The PYRIN domain–only protein POP3 inhibits ALR inflammasomes and regulates responses to infection with DNA viruses. Nat. Immunol. 2014, 15, 343–353. [Google Scholar] [CrossRef]
- Ratsimandresy, R.A.; Chu, L.H.; Khare, S.; de Almeida, L.; Gangopadhyay, A.; Indramohan, M.; Misharin, A.V.; Greaves, D.R.; Perlman, H.; Dorfleutner, A.; et al. The PYRIN domain-only protein POP2 inhibits inflammasome priming and activation. Nat. Commun. 2017, 8, 15556. [Google Scholar] [CrossRef] [PubMed]
- Devi, S.; Stehlik, C.; Dorfleutner, A. An Update on CARD Only Proteins (COPs) and PYD Only Proteins (POPs) as Inflammasome Regulators. Int. J. Mol. Sci. 2020, 21, 6901. [Google Scholar] [CrossRef] [PubMed]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQuade, A.; Kang, Y.J.; Hasselmann, J.; Jairaman, A.; Sotelo, A.; Coburn, M.; Shabestari, S.K.; Chadarevian, J.P.; Fote, G.; Tu, C.H.; et al. Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer’s disease. Nat. Commun. 2020, 11, 5370. [Google Scholar] [CrossRef]
- Nizami, S.; Hall-Roberts, H.; Warrier, S.; Cowley, S.A.; Di Daniel, E. Microglial inflammation and phagocytosis in Alzheimer’s disease: Potential therapeutic targets. Br. J. Pharmacol. 2019, 176, 3515–3532. [Google Scholar] [CrossRef] [Green Version]
- Forabosco, P.; Ramasamy, A.; Trabzuni, D.; Walker, R.; Smith, C.; Bras, J.; Levine, A.P.; Hardy, J.; Pocock, J.M.; Guerreiro, R.; et al. Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol. Aging 2013, 34, 2699–2714. [Google Scholar] [CrossRef] [Green Version]
- Schoch, K.M.; Ezerskiy, L.A.; Morhaus, M.M.; Bannon, R.N.; Sauerbeck, A.D.; Shabsovich, M.; Jafar-nejad, P.; Rigo, F.; Miller, T.M. Acute Trem2 reduction triggers increased microglial phagocytosis, slowing amyloid deposition in mice. Proc. Natl. Acad. Sci. USA 2021, 118, e2100356118. [Google Scholar] [CrossRef]
- Ellwanger, D.C.; Wang, S.; Brioschi, S.; Shao, Z.; Green, L.; Case, R.; Yoo, D.; Weishuhn, D.; Rathanaswami, P.; Bradley, J.; et al. Prior activation state shapes the microglia response to antihuman TREM2 in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2017742118. [Google Scholar] [CrossRef]
- Cui, X.; Qiao, J.; Liu, S.; Wu, M.; Gu, W. Mechanism of TREM2/DAP12 complex affecting β-amyloid plaque deposition in Alzheimer’s disease modeled mice through mediating inflammatory response. Brain Res. Bull. 2021, 166, 21–28. [Google Scholar] [CrossRef]
- Daws, M.R.; Sullam, P.M.; Niemi, E.C.; Chen, T.T.; Tchao, N.K.; Seaman, W.E. Pattern recognition by TREM-2: Binding of anionic ligands. J. Immunol. 2003, 171, 594–599. [Google Scholar] [CrossRef] [Green Version]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Morenas-Rodríguez, E.; Li, Y.; Nuscher, B.; Franzmeier, N.; Xiong, C.; Suárez-Calvet, M.; Fagan, A.M.; Schultz, S.; Gordon, B.A.; Benzinger, T.L.S.; et al. Soluble TREM2 in CSF and its association with other biomarkers and cognition in autosomal-dominant Alzheimer’s disease: A longitudinal observational study. Lancet Neurol. 2022, 21, 329–341. [Google Scholar] [CrossRef]
- Lichtenthaler, S.F.; Lemberg, M.K.; Fluhrer, R. Proteolytic ectodomain shedding of membrane proteins in mammals-hardware, concepts, and recent developments. EMBO J. 2018, 37, 99456. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Calvet, M.; Kleinberger, G.; Araque Caballero, M.Á.; Brendel, M.; Rominger, A.; Alcolea, D.; Fortea, J.; Lleó, A.; Blesa, R.; Gispert, J.D.; et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol. Med. 2016, 8, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, J.D.; Ulland, T.K.; Colonna, M.; Holtzman, D.M. Elucidating the Role of TREM2 in Alzheimer’s Disease. Neuron 2017, 94, 237–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korvatska, O.; Leverenz, J.B.; Jayadev, S.; McMillan, P.; Kurtz, I.; Guo, X.; Rumbaugh, M.; Matsushita, M.; Girirajan, S.; Dorschner, M.O.; et al. R47H Variant of TREM2 Associated With Alzheimer Disease in a Large Late-Onset Family: Clinical, Genetic, and Neuropathological Study. JAMA Neurol. 2015, 72, 920–927. [Google Scholar] [CrossRef] [Green Version]
- Hall-Roberts, H.; Agarwal, D.; Obst, J.; Smith, T.B.; Monzón-Sandoval, J.; Di Daniel, E.; Webber, C.; James, W.S.; Mead, E.; Davis, J.B.; et al. TREM2 Alzheimer’s variant R47H causes similar transcriptional dysregulation to knockout, yet only subtle functional phenotypes in human iPSC-derived macrophages. Alzheimer’s Res. Ther. 2020, 12, 151. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.-L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031. [Google Scholar] [CrossRef] [Green Version]
- Gratuze, M.; Chen, Y.; Parhizkar, S.; Jain, N.; Strickland, M.R.; Serrano, J.R.; Colonna, M.; Ulrich, J.D.; Holtzman, D.M. Activated microglia mitigate Aβ-associated tau seeding and spreading. J. Exp. Med. 2021, 218, e20210542. [Google Scholar] [CrossRef]
- Gratuze, M.; Leyns, C.E.G.; Sauerbeck, A.D.; St-Pierre, M.-K.; Xiong, M.; Kim, N.; Serrano, J.R.; Tremblay, M.-È.; Kummer, T.T.; Colonna, M.; et al. Impact of TREM2R47H variant on tau pathology–induced gliosis and neurodegeneration. J. Clin. Investig. 2020, 130, 4954–4968. [Google Scholar] [CrossRef]
- Bemiller, S.M.; McCray, T.J.; Allan, K.; Formica, S.V.; Xu, G.; Wilson, G.; Kokiko-Cochran, O.N.; Crish, S.D.; Lasagna-Reeves, C.A.; Ransohoff, R.M.; et al. TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol. Neurodegener. 2017, 12, 74. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, I.R.; Gilfillan, S.; Cella, M.; Aoshi, T.; Miller, M.; Piccio, L.; Hernandez, M.; Colonna, M. Cutting edge: TREM-2 attenuates macrophage activation. J. Immunol. 2006, 177, 3520–3524. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yu, W.; Zhang, M.; Tian, X.; Li, Y.; Lü, Y. Imbalance of Microglial TLR4/TREM2 in LPS-Treated APP/PS1 Transgenic Mice: A Potential Link Between Alzheimer’s Disease and Systemic Inflammation. Neurochem. Res. 2019, 44, 1138–1151. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Taso, O.; Wang, R.; Bayram, S.; Graham, A.C.; Garcia-Reitboeck, P.; Mallach, A.; Andrews, W.D.; Piers, T.M.; Botia, J.A.; et al. Trem2 promotes anti-inflammatory responses in microglia and is suppressed under pro-inflammatory conditions. Hum. Mol. Genet. 2020, 29, 3224–3248. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, Y.; Wang, L.; Zhan, H.; Luo, X.; Zeng, Y.; Wu, W.; Zhang, X.; Wang, F. TREM2 ameliorates neuroinflammatory response and cognitive impairment via PI3K/AKT/FoxO3a signaling pathway in Alzheimer’s disease mice. Aging 2020, 12, 20862–20879. [Google Scholar] [CrossRef]
- Chen, S.; Peng, J.; Sherchan, P.; Ma, Y.; Xiang, S.; Yan, F.; Zhao, H.; Jiang, Y.; Wang, N.; Zhang, J.H.; et al. TREM2 activation attenuates neuroinflammation and neuronal apoptosis via PI3K/Akt pathway after intracerebral hemorrhage in mice. J. Neuroinflammation 2020, 17, 168. [Google Scholar] [CrossRef]
- Cosker, K.; Mallach, A.; Limaye, J.; Piers, T.M.; Staddon, J.; Neame, S.J.; Hardy, J.; Pocock, J.M. Microglial signalling pathway deficits associated with the patient derived R47H TREM2 variants linked to AD indicate inability to activate inflammasome. Sci. Rep. 2021, 11, 13316. [Google Scholar] [CrossRef]
- Lin, Y.C.; Huang, D.Y.; Wang, J.S.; Lin, Y.L.; Hsieh, S.L.; Huang, K.C.; Lin, W.W. Syk is involved in NLRP3 inflammasome-mediated caspase-1 activation through adaptor ASC phosphorylation and enhanced oligomerization. J. Leukoc. Biol. 2015, 97, 825–835. [Google Scholar] [CrossRef]
- Deming, Y.; Li, Z.; Benitez, B.A.; Cruchaga, C. Triggering receptor expressed on myeloid cells 2 (TREM2): A potential therapeutic target for Alzheimer disease? Expert Opin. Ther. Targets 2018, 22, 587–598. [Google Scholar] [CrossRef]
- Zhong, L.; Xu, Y.; Zhuo, R.; Wang, T.; Wang, K.; Huang, R.; Wang, D.; Gao, Y.; Zhu, Y.; Sheng, X.; et al. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 1365. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Chen, X.-F.; Wang, T.; Wang, Z.; Liao, C.; Wang, Z.; Huang, R.; Wang, D.; Li, X.; Wu, L.; et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 2017, 214, 597–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suárez-Calvet, M.; Morenas-Rodríguez, E.; Kleinberger, G.; Schlepckow, K.; Araque Caballero, M.Á.; Franzmeier, N.; Capell, A.; Fellerer, K.; Nuscher, B.; Eren, E.; et al. Early increase of CSF sTREM2 in Alzheimer’s disease is associated with tau related-neurodegeneration but not with amyloid-β pathology. Mol. Neurodegener. 2019, 14, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlepckow, K.; Kleinberger, G.; Fukumori, A.; Feederle, R.; Lichtenthaler, S.F.; Steiner, H.; Haass, C. An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol. Med. 2017, 9, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Schlepckow, K.; Monroe, K.M.; Kleinberger, G.; Cantuti-Castelvetri, L.; Parhizkar, S.; Xia, D.; Willem, M.; Werner, G.; Pettkus, N.; Brunner, B.; et al. Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region. EMBO Mol. Med. 2020, 12, e11227. [Google Scholar] [CrossRef]
- Price, B.R.; Sudduth, T.L.; Weekman, E.M.; Johnson, S.; Hawthorne, D.; Woolums, A.; Wilcock, D.M. Therapeutic Trem2 activation ameliorates amyloid-beta deposition and improves cognition in the 5XFAD model of amyloid deposition. J. Neuroinflammation 2020, 17, 1–13. [Google Scholar] [CrossRef]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020, 217, e20200785. [Google Scholar] [CrossRef]
- Ward, M.; Long, H.; Schwabe, T.; Rhinn, H.; Tassi, I.; Salazar, S.V.; Rychkova, A.; Tu, G.; Barner, C.; Yeh, F.L.; et al. A phase 1 study of AL002 in healthy volunteers. Alzheimer’s Dementia 2021, 17, e054669. [Google Scholar] [CrossRef]
- Zhao, Y.; Bhattacharjee, S.; Jones, B.M.; Dua, P.; Alexandrov, P.N.; Hill, J.M.; Lukiw, W.J. Regulation of TREM2 expression by an NF-κB-sensitive miRNA-34a. Neuroreport 2013, 24, 318–323. [Google Scholar] [CrossRef] [Green Version]
- Stefano, L.; Racchetti, G.; Bianco, F.; Passini, N.; Gupta, R.S.; Panina Bordignon, P.; Meldolesi, J. The surface-exposed chaperone, Hsp60, is an agonist of the microglial TREM2 receptor. J. Neurochem. 2009, 110, 284–294. [Google Scholar] [CrossRef]
- Griciuc, A.; Patel, S.; Federico, A.N.; Choi, S.H.; Innes, B.J.; Oram, M.K.; Cereghetti, G.; McGinty, D.; Anselmo, A.; Sadreyev, R.I.; et al. TREM2 Acts Downstream of CD33 in Modulating Microglial Pathology in Alzheimer’s Disease. Neuron 2019, 103, 820–835.e7. [Google Scholar] [CrossRef]
- Chin, A.C. Neuroinflammation and the cGAS-STING pathway. J. Neurophysiol. 2019, 121, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- de Weerd, N.A.; Nguyen, T. The interferons and their receptors--distribution and regulation. Immunol. Cell Biol. 2012, 90, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Mathur, V.; Burai, R.; Vest, R.T.; Bonanno, L.N.; Lehallier, B.; Zardeneta, M.E.; Mistry, K.N.; Do, D.; Marsh, S.E.; Abud, E.M.; et al. Activation of the STING-Dependent Type I Interferon Response Reduces Microglial Reactivity and Neuroinflammation. Neuron 2017, 96, 1290–1302.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, C.W.; Zhang, Y.; Herrup, K. Non-Neuronal Cells Are Required to Mediate the Effects of Neuroinflammation: Results from a Neuron-Enriched Culture System. PLoS ONE 2016, 11, e0147134. [Google Scholar] [CrossRef] [PubMed]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 2019, 567, 262–266. [Google Scholar] [CrossRef]
- Su, M.; Mei, Y.; Sinha, S. Role of the Crosstalk between Autophagy and Apoptosis in Cancer. J. Oncol. 2013, 2013, 102735. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Meng, Q.; Qin, Y.; Liang, P.; Tan, P.; He, L.; Zhou, Y.; Chen, Y.; Huang, J.; Wang, R.F.; et al. TRIM14 Inhibits cGAS Degradation Mediated by Selective Autophagy Receptor p62 to Promote Innate Immune Responses. Mol. Cell 2016, 64, 105–119. [Google Scholar] [CrossRef] [Green Version]
- Konno, H.; Konno, K.; Barber, G.N. Cyclic Dinucleotides Trigger ULK1 (ATG1) Phosphorylation of STING to Prevent Sustained Innate Immune Signaling. Cell 2013, 155, 688–698. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V. The Trinity of cGAS, TLR9, and ALRs Guardians of the Cellular Galaxy Against Host-Derived Self-DNA. Front. Immunol. 2021, 11, 3902. [Google Scholar] [CrossRef]
- Wu, P.J.; Hung, Y.F.; Liu, H.Y.; Hsueh, Y.P. Deletion of the Inflammasome Sensor Aim2 Mitigates Aβ Deposition and Microglial Activation but Increases Inflammatory Cytokine Expression in an Alzheimer Disease Mouse Model. Neuroimmunomodulation 2017, 24, 29–39. [Google Scholar] [CrossRef]
- Nazmi, A.; Field, R.H.; Griffin, E.W.; Haugh, O.; Hennessy, E.; Cox, D.; Reis, R.; Tortorelli, L.; Murray, C.L.; Lopez-Rodriguez, A.B.; et al. Chronic neurodegeneration induces type I interferon synthesis via STING, shaping microglial phenotype and accelerating disease progression. Glia 2019, 67, 1254–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choubey, D. Type I interferon (IFN)-inducible Absent in Melanoma 2 proteins in neuroinflammation: Implications for Alzheimer’s disease. J. Neuroinflammation 2019, 16, 236. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, L.; Hiller, B.; Kostrewa, D.; Lässig, C.; de Oliveira Mann, C.C.; Jan Drexler, D.; Maiser, A.; Gaidt, M.; Leonhardt, H.; Hornung, V.; et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein–DNA ladders. Nature 2017, 549, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.J.; Field, R.H.; Williams, D.G.; Baran, M.; Bowie, A.G.; Cunningham, C.; Dunne, A. DNA sensors are expressed in astrocytes and microglia in vitro and are upregulated during gliosis in neurodegenerative disease. Glia 2015, 63, 812–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Xu, W.; Cheng, H.; Yuan, H.; Tan, X. Efficacy and mechanism of cGAMP to suppress Alzheimer’s disease by elevating TREM2. Brain Behav. Immun. 2019, 81, 495–508. [Google Scholar] [CrossRef]
- Wang, W.; Hu, D.; Wu, C.; Feng, Y.; Li, A.; Liu, W.; Wang, Y.; Chen, K.; Tian, M.; Xiao, F. STING promotes NLRP3 localization in ER and facilitates NLRP3 deubiquitination to activate the inflammasome upon HSV-1 infection. PLoS Pathog. 2020, 16, e1008335. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.; Shiwaku, H.; Tanaka, H.; Obita, T.; Ohuchi, S.; Yoshioka, Y.; Jin, X.; Kondo, K.; Fujita, K.; Homma, H.; et al. Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat. Commun. 2021, 12, 6565. [Google Scholar] [CrossRef]
- Fryer, A.L.; Abdullah, A.; Taylor, J.M.; Crack, P.J. The Complexity of the cGAS-STING Pathway in CNS Pathologies. Front. Neurosci. 2021, 15, 82. [Google Scholar] [CrossRef]
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD(+) supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Lu, L.; Pember, E.; Li, X.; Zhang, B.; Zhu, Z. New Insights into Neuroinflammation Involved in Pathogenic Mechanism of Alzheimer’s Disease and Its Potential for Therapeutic Intervention. Cells 2022, 11, 1925. https://doi.org/10.3390/cells11121925
Li T, Lu L, Pember E, Li X, Zhang B, Zhu Z. New Insights into Neuroinflammation Involved in Pathogenic Mechanism of Alzheimer’s Disease and Its Potential for Therapeutic Intervention. Cells. 2022; 11(12):1925. https://doi.org/10.3390/cells11121925
Chicago/Turabian StyleLi, Tiantian, Li Lu, Eloise Pember, Xinuo Li, Bocheng Zhang, and Zheying Zhu. 2022. "New Insights into Neuroinflammation Involved in Pathogenic Mechanism of Alzheimer’s Disease and Its Potential for Therapeutic Intervention" Cells 11, no. 12: 1925. https://doi.org/10.3390/cells11121925