
CAR-T Cells Shoot for New Targets: Novel Approaches to Boost Adoptive Cell Therapy for B Cell-Derived Malignancies


Abstract
:1. Introduction
2. Clinically Available CAR-T Therapies
3. Managing Major Pitfalls of CAR-T Cell Therapy
3.1. Improvement of CAR-T Cells Persistence
3.2. Modulation of CAR-T Cells Exhaustion
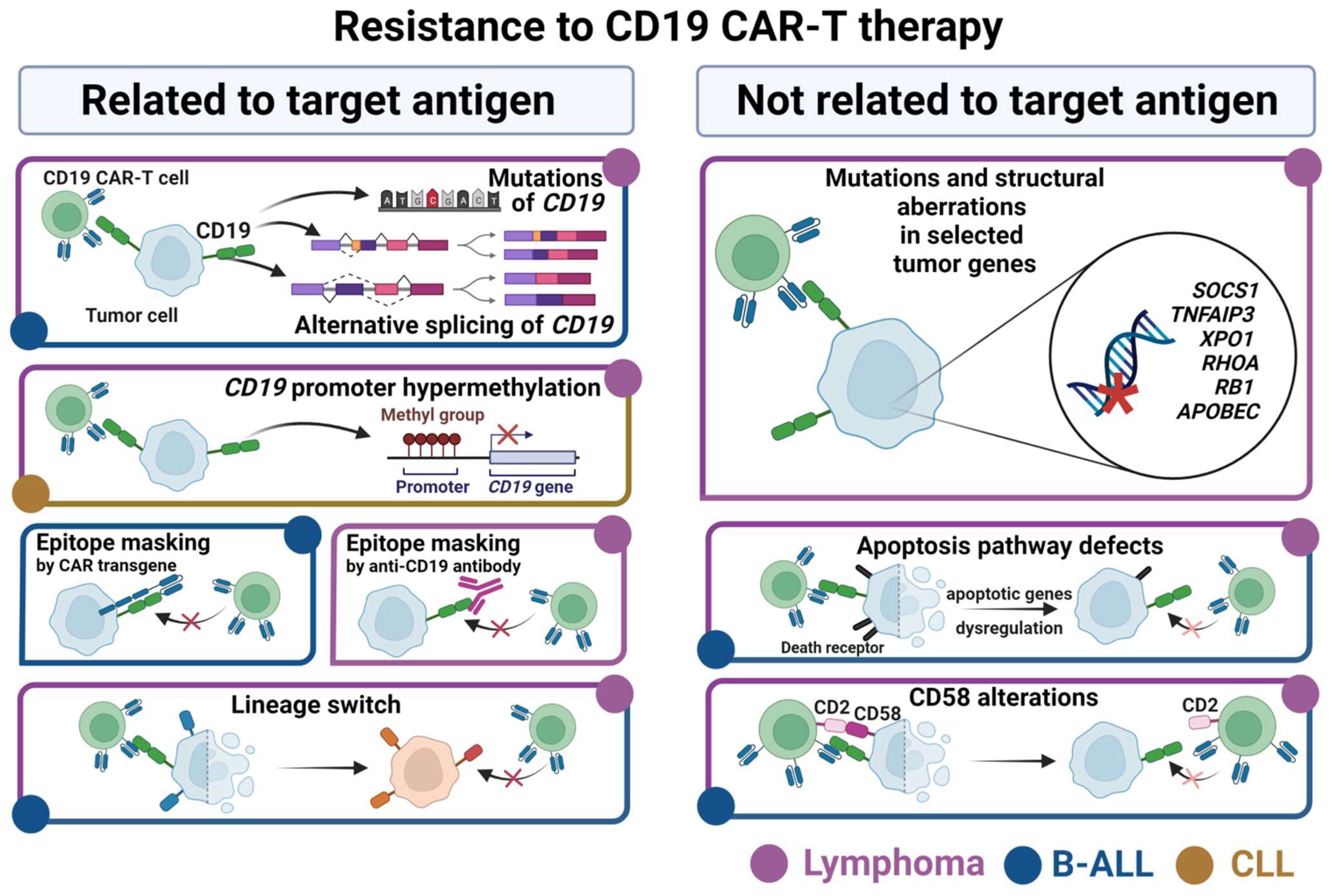
4. Mechanisms of Resistance to the CD19 CAR-T Therapy in B Cell Malignancies
4.1. Resistance Related to CD19 Antigen
4.2. Resistance Not Related to CD19 Antigen
5. New CAR-T Targets Explored in Hematological Malignancies
5.1. Novel Antigens under Investigation in Pre-Clinical Studies
5.2. Promising Antigens Tested in Clinical Trials
5.3. New Directions for Alternative CAR-T Targets Development
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gökbuget, N.; Dombret, H.; Ribera, J.M.; Fielding, A.K.; Advani, A.; Bassan, R.; Chia, V.; Doubek, M.; Giebel, S.; Hoelzer, D.; et al. International Reference Analysis of Outcomes in Adults with B-Precursor Ph-Negative Relapsed/Refractory Acute Lymphoblastic Leukemia. Haematologica 2016, 101, 1524–1533. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Malvar, J.; Sposto, R.; Verma, A.; Wilkes, J.J.; Dennis, R.; Heym, K.; Laetsch, T.W.; Widener, M.; Rheingold, S.R.; et al. Outcome of Children with Multiply Relapsed B-Cell Acute Lymphoblastic Leukemia: A Therapeutic Advances in Childhood Leukemia & Lymphoma Study. Leukemia 2018, 32, 2316–2325. [Google Scholar] [CrossRef]
- Freyer, D.R.; Devidas, M.; La, M.; Carroll, W.L.; Gaynon, P.S.; Hunger, S.P.; Seibel, N.L. Postrelapse Survival in Childhood Acute Lymphoblastic Leukemia Is Independent of Initial Treatment Intensity: A Report from the Children’s Oncology Group. Blood 2011, 117, 3010–3015. [Google Scholar] [CrossRef] [Green Version]
- Modi, D.; Potugari, B.; Uberti, J. Immunotherapy for Diffuse Large B-Cell Lymphoma: Current Landscape and Future Directions. Cancers 2021, 13, 5827. [Google Scholar] [CrossRef]
- Bazarbachi, A.H.; Al Hamed, R.; Malard, F.; Harousseau, J.-L.; Mohty, M. Relapsed Refractory Multiple Myeloma: A Comprehensive Overview. Leukemia 2019, 33, 2343–2357. [Google Scholar] [CrossRef]
- Durer, C.; Durer, S.; Lee, S.; Chakraborty, R.; Malik, M.N.; Rafae, A.; Zar, M.A.; Kamal, A.; Rosko, N.; Samaras, C.; et al. Treatment of Relapsed Multiple Myeloma: Evidence-Based Recommendations. Blood Rev. 2020, 39, 100616. [Google Scholar] [CrossRef]
- Susanibar, A.S.; Stadtmauer, E.A.; Cohen, A.D. CAR T Cell Therapy for Multiple Myeloma: What Have We Learned? Leukemia 2022, 22, 39–47. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Santos, E.; Nikhamin, Y.; Yeh, R.; Matsushita, M.; La Perle, K.; Quintás-Cardama, A.; Larson, S.M.; Sadelain, M. Genetically Targeted T Cells Eradicate Systemic Acute Lymphoblastic Leukemia Xenografts. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 5426–5435. [Google Scholar] [CrossRef] [Green Version]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric Receptors Containing CD137 Signal Transduction Domains Mediate Enhanced Survival of T Cells and Increased Antileukemic Efficacy in vivo. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef]
- Zhong, X.S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric Antigen Receptors Combining 4–1BB and CD28 Signaling Domains Augment PI3kinase/AKT/Bcl-XL Activation and CD8+ T Cell-Mediated Tumor Eradication. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef]
- Tokarew, N.; Ogonek, J.; Endres, S.; von Bergwelt-Baildon, M.; Kobold, S. Teaching an Old Dog New Tricks: Next-Generation CAR T Cells. Br. J. Cancer 2019, 120, 26–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Shah, N.N.; Lee, D.W.; Yates, B.; Yuan, C.M.; Shalabi, H.; Martin, S.; Wolters, P.L.; Steinberg, S.M.; Baker, E.H.; Delbrook, C.P.; et al. Long-Term Follow-Up of CD19-CAR T-Cell Therapy in Children and Young Adults With B-ALL. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Schuster, S.J.; Tam, C.S.; Borchmann, P.; Worel, N.; McGuirk, J.P.; Holte, H.; Waller, E.K.; Jaglowski, S.; Bishop, M.R.; Damon, L.E.; et al. Long-Term Clinical Outcomes of Tisagenlecleucel in Patients with Relapsed or Refractory Aggressive B-Cell Lymphomas (JULIET): A Multicentre, Open-Label, Single-Arm, Phase 2 Study. Lancet. Oncol. 2021, 22, 1403–1415. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Jacobson, C.; Locke, F.L.; Ghobadi, A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Hill, B.T.; Timmerman, J.M.; Deol, A.; et al. Long-Term (≥4 Year and ≥5 Year) Overall Survival (OS) By 12- and 24-Month Event-Free Survival (EFS): An Updated Analysis of ZUMA-1, the Pivotal Study of Axicabtagene Ciloleucel (Axi-Cel) in Patients (Pts) with Refractory Large B-Cell Lymphoma (LBCL). Blood 2021, 138, 1764. [Google Scholar] [CrossRef]
- Jacobson, C.A.; Chavez, J.C.; Sehgal, A.R.; William, B.M.; Munoz, J.; Salles, G.; Munshi, P.N.; Casulo, C.; Maloney, D.G.; de Vos, S.; et al. Axicabtagene Ciloleucel in Relapsed or Refractory Indolent Non-Hodgkin Lymphoma (ZUMA-5): A Single-Arm, Multicentre, Phase 2 Trial. Lancet Oncol. 2022, 23, 91–103. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Shah, B.D.; Ghobadi, A.; Oluwole, O.O.; Logan, A.C.; Boissel, N.; Cassaday, R.D.; Leguay, T.; Bishop, M.R.; Topp, M.S.; Tzachanis, D.; et al. KTE-X19 for Relapsed or Refractory Adult B-Cell Acute Lymphoblastic Leukaemia: Phase 2 Results of the Single-Arm, Open-Label, Multicentre ZUMA-3 Study. Lancet 2021, 398, 491–502. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene Maraleucel for Patients with Relapsed or Refractory Large B-Cell Lymphomas (TRANSCEND NHL 001): A Multicentre Seamless Design Study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.E.; Purev, E.; Maloney, D.G.; Andreadis, C.; Sehgal, A.R.; et al. Two-Year Follow-up of Transcend NHL 001, a Multicenter Phase 1 Study of Lisocabtagene Maraleucel (Liso-Cel) in Relapsed or Refractory (R/R) Large B-Cell Lymphomas (LBCL). Blood 2021, 138, 2840. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D.J.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Kanapuru, B.; George, B.; Lin, X.; Xu, Z.; Bryan, W.W.; Pazdur, R.; Theoret, M.R. FDA Approval Summary: Idecabtagene Vicleucel for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 21, 3803. [Google Scholar] [CrossRef]
- Shah, B.D.; Bishop, M.R.; Oluwole, O.O.; Logan, A.C.; Baer, M.R.; Donnellan, W.B.; O’Dwyer, K.M.; Holmes, H.; Arellano, M.L.; Ghobadi, A.; et al. KTE-X19 Anti-CD19 CAR T-Cell Therapy in Adult Relapsed/Refractory Acute Lymphoblastic Leukemia: ZUMA-3 Phase 1 Results. Blood 2021, 138, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Munoz, J.; Goy, A.H.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. One-Year Follow-up of ZUMA-2, the Multicenter, Registrational Study of KTE-X19 in Patients with Relapsed/Refractory Mantle Cell Lymphoma. Blood 2020, 136, 20–22. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of Resistance to CAR T Cell Therapy. Nature reviews. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Recent Advances in CAR T-Cell Toxicity: Mechanisms, Manifestations and Management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J.J.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, 1109. [Google Scholar] [CrossRef]
- Zhylko, A.; Winiarska, M.; Graczyk-Jarzynka, A. The Great War of Today: Modifications of CAR-T Cells to Effectively Combat Malignancies. Cancers 2020, 12, 2030. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Mueller, K.T.; Maude, S.L.; Porter, D.L.; Frey, N.; Wood, P.; Han, X.; Waldron, E.; Chakraborty, A.; Awasthi, R.; Levine, B.L.; et al. Cellular Kinetics of CTL019 in Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia and Chronic Lymphocytic Leukemia. Blood 2017, 130, 2317–2325. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Lu, W.; Chen, D.; Tu, H.; Guo, Z.; Zhou, X.; Li, M.; Tu, S.; Li, Y. Mechanisms Underlying CD19-Positive ALL Relapse after Anti-CD19 CAR T Cell Therapy and Associated Strategies. Biomark. Res. 2020, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Gill, S. CAR-T Cell Persistence in the Treatment of Leukemia and Lymphoma. Leuk. Lymphoma 2021, 62, 2587–2599. [Google Scholar] [CrossRef]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D.J.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Yang, J.; Zhang, X.; Lu, X.A.; Xiong, M.; Zhang, J.; Zhou, X.; Qi, F.; He, T.; Ding, Y.; et al. Efficacy and Safety of CD28- or 4-1BB-Based CD19 CAR-T Cells in B Cell Acute Lymphoblastic Leukemia. Mol. Ther. Oncol. 2020, 18, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Philipson, B.I.; O’Connor, R.S.; May, M.J.; June, C.H.; Albelda, S.M.; Milone, M.C. 4–1BB Costimulation Promotes CAR T Cell Survival through Noncanonical NF-ΚB Signaling. Sci. Signal. 2020, 13, 8248. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Condomines, M.; van der Stegen, S.J.C.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Boucher, J.C.; Li, G.; Kotani, H.; Cabral, M.L.; Morrissey, D.; Lee, S.B.; Spitler, K.; Beatty, N.J.; Cervantes, E.V.; Shrestha, B.; et al. CD28 Costimulatory Domain-Targeted Mutations Enhance Chimeric Antigen Receptor T-Cell Function. Cancer Immunol. Res. 2021, 9, 62–74. [Google Scholar] [CrossRef]
- Roselli, E.; Boucher, J.C.; Li, G.; Kotani, H.; Spitler, K.; Reid, K.; Cervantes, E.V.; Bulliard, Y.; Tu, N.; Lee, S.B.; et al. 4-1BB and Optimized CD28 Co-Stimulation Enhances Function of Human Mono-Specific and Bi-Specific Third-Generation CAR T Cells. J. Immunother. Cancer 2021, 9, 3354. [Google Scholar] [CrossRef]
- Wang, J.; Mou, N.; Yang, Z.; Li, Q.; Jiang, Y.; Meng, J.; Liu, X.; Deng, Q. Efficacy and Safety of Humanized Anti-CD19-CAR-T Therapy Following Intensive Lymphodepleting Chemotherapy for Refractory/Relapsed B Acute Lymphoblastic Leukaemia. Br. J. Haematol. 2020, 191, 212–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabrizio, V.A.; Boelens, J.J.; Mauguen, A.; Baggott, C.; Prabhu, S.; Egeler, E.; Mavroukakis, S.; Pacenta, H.; Phillips, C.L.; Rossoff, J.; et al. Optimal Fludarabine Lymphodepletion Is Associated with Improved Outcomes after CAR T-Cell Therapy. Blood Adv. 2022, 6, 1961–1968. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Hudecek, M.; Pender, B.; Robinson, E.; Hawkins, R.; Chaney, C.; Cherian, S.; Chen, X.; et al. Immunotherapy of Non-Hodgkin’s Lymphoma with a Defined Ratio of CD8+ and CD4+ CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Sci. Transl. Med. 2016, 8, 355ra116. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, A.V.; Gauthier, J.; Hay, K.A.; Voutsinas, J.M.; Wu, Q.; Gooley, T.; Li, D.; Cherian, S.; Chen, X.; Pender, B.S.; et al. The Response to Lymphodepletion Impacts PFS in Patients with Aggressive Non-Hodgkin Lymphoma Treated with CD19 CAR T cells. Blood 2019, 133, 1876–1887. [Google Scholar] [CrossRef] [Green Version]
- Lyu, C.; Cui, R.; Wang, J.; Mou, N.; Jiang, Y.; Li, W.; Deng, Q. Intensive Debulking Chemotherapy Improves the Short-Term and Long-Term Efficacy of Anti-CD19-CAR-T in Refractory/Relapsed DLBCL with High Tumor Bulk. Front. Oncol. 2021, 11, 706087. [Google Scholar] [CrossRef]
- Neelapu, S.S. CAR-T Efficacy: Is Conditioning the Key? Blood 2019, 133, 1799–1800. [Google Scholar] [CrossRef]
- Chen, M.L.; Pittet, M.J.; Gorelik, L.; Flavell, R.A.; Weissleder, R.; von Boehmer, H.; Khazaie, K. Regulatory T Cells Suppress Tumor-Specific CD8 T Cell Cytotoxicity through TGF-Beta Signals in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Arita, M.; Takahashi, M.; Saida, Y.; Koya, T.; Kikuchi, T. Effect of Lymphodepletion on Donor T Cells and the Role of Recipient Cells Persisting after Cytotoxic Treatments in Cancer Immunotherapies. Crit. Rev. Immunol. 2017, 37, 59–73. [Google Scholar] [CrossRef]
- Hay, K.A.; Hanafi, L.A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; López, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and Biomarkers of Severe Cytokine Release Syndrome after CD19 Chimeric Antigen Receptor-Modified T-Cell Therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef] [Green Version]
- Dawicki, W.; Allen, K.J.H.; Garg, R.; Geoghegan, E.M.; Berger, M.S.; Ludwig, D.L.; Dadachova, E. Targeted Lymphodepletion with a CD45-Directed Antibody Radioconjugate as a Novel Conditioning Regimen Prior to Adoptive Cell Therapy. Oncotarget 2020, 11, 3571–3581. [Google Scholar] [CrossRef]
- Williams, K.M.; Hakim, F.T.; Gress, R.E. T Cell Immune Reconstitution Following Lymphodepletion. Semin. Immunol. 2007, 19, 318–330. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Abken, H. TRUCKS, the Fourth-Generation CAR T Cells: Current Developments and Clinical Translation. Adv. Cell Gene Ther. 2020, 3, e84. [Google Scholar] [CrossRef]
- Jin, J.; Cheng, J.; Huang, M.; Luo, H.; Zhou, J. Fueling Chimeric Antigen Receptor T Cells with Cytokines. Am. J. Cancer Res. 2020, 10, 4038–4055. [Google Scholar]
- Pegram, H.J.; Lee, J.C.; Hayman, E.G.; Imperato, G.H.; Tedder, T.F.; Sadelain, M.; Brentjens, R.J. Tumor-Targeted T Cells Modified to Secrete IL-12 Eradicate Systemic Tumors without Need for Prior Conditioning. Blood 2012, 119, 4133–4141. [Google Scholar] [CrossRef]
- Kueberuwa, G.; Kalaitsidou, M.; Cheadle, E.; Hawkins, R.E.; Gilham, D.E. CD19 CAR T Cells Expressing IL-12 Eradicate Lymphoma in Fully Lymphoreplete Mice through Induction of Host Immunity. Mol. Ther. Oncol. 2018, 8, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhang, M.; Ramos, C.A.; Durett, A.; Liu, E.; Dakhova, O.; Liu, H.; Creighton, C.J.; Gee, A.P.; Heslop, H.E.; et al. Closely Related T-Memory Stem Cells Correlate with in Vivo Expansion of CAR.CD19-T Cells and Are Preserved by IL-7 and IL-15. Blood 2014, 123, 3750–3759. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lv, X.; Song, Y. Short-Term Culture with IL-2 Is Beneficial for Potent Memory Chimeric Antigen Receptor T Cell Production. Biochem. Biophys. Res. Commun. 2018, 495, 1833–1838. [Google Scholar] [CrossRef]
- Alizadeh, D.; Wong, R.A.; Yang, X.; Wang, D.; Pecoraro, J.R.; Kuo, C.F.; Aguilar, B.; Qi, Y.; Ann, D.K.; Starr, R.; et al. IL15 Enhances CAR-T Cell Antitumor Activity by Reducing MTORC1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol. Res. 2019, 7, 759–772. [Google Scholar] [CrossRef]
- Ataca Atilla, P.; McKenna, M.K.; Tashiro, H.; Srinivasan, M.; Mo, F.; Watanabe, N.; Simons, B.W.; McLean Stevens, A.; Redell, M.S.; Heslop, H.E.; et al. Modulating TNFα Activity Allows Transgenic IL15-Expressing CLL-1 CAR T Cells to Safely Eliminate Acute Myeloid Leukemia. J. Immunother. Cancer 2020, 8, 1229. [Google Scholar] [CrossRef]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 Release by Engineered T Cells Expressing Chimeric Antigen Receptors Can Effectively Muster an Antigen-Independent Macrophage Response on Tumor Cells That Have Shut down Tumor Antigen Expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [Green Version]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A Novel Chimeric Antigen Receptor Containing a JAK-STAT Signaling Domain Mediates Superior Antitumor Effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Štach, M.; Ptáčková, P.; Mucha, M.; Musil, J.; Klener, P.; Otáhal, P. Inducible Secretion of IL-21 Augments Anti-Tumor Activity of PiggyBac-Manufactured Chimeric Antigen Receptor T Cells. Cytotherapy 2020, 22, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Hoyos, V.; Savoldo, B.; Quintarelli, C.; Mahendravada, A.; Zhang, M.; Vera, J.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Dotti, G. Engineering CD19-Specific T Lymphocytes with Interleukin-15 and a Suicide Gene to Enhance Their Anti-Lymphoma/Leukemia Effects and Safety. Leukemia 2010, 24, 1160–1170. [Google Scholar] [CrossRef] [Green Version]
- Diaconu, I.; Ballard, B.; Zhang, M.; Chen, Y.; West, J.; Dotti, G.; Savoldo, B. Inducible Caspase-9 Selectively Modulates the Toxicities of CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 580–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guercio, M.; Manni, S.; Boffa, I.; Caruso, S.; Di Cecca, S.; Sinibaldi, M.; Abbaszadeh, Z.; Camera, A.; Ciccone, R.; Polito, V.A.; et al. Inclusion of the Inducible Caspase 9 Suicide Gene in CAR Construct Increases Safety of CAR.CD19 T Cell Therapy in B-Cell Malignancies. Front. Immunol. 2021, 12, 755639. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The Diverse Functions of the PD1 Inhibitory Pathway. Nature reviews. Immunology 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Roemer, M.G.M.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 2690–2697. [Google Scholar] [CrossRef] [Green Version]
- Cader, F.Z.; Schackmann, R.C.J.; Hu, X.; Wienand, K.; Redd, R.; Chapuy, B.; Ouyang, J.; Paul, N.; Gjini, E.; Lipschitz, M.; et al. Mass Cytometry of Hodgkin Lymphoma Reveals a CD4(+) Regulatory T-Cell-Rich and Exhausted T-Effector Microenvironment. Blood 2018, 132, 825–836. [Google Scholar] [CrossRef]
- Xie, W.; Medeiros, L.J.; Li, S.; Yin, C.C.; Khoury, J.D.; Xu, J. PD-1/PD-L1 Pathway and Its Blockade in Patients with Classic Hodgkin Lymphoma and Non-Hodgkin Large-Cell Lymphomas. Curr. Hematol. Malig. Rep. 2020, 15, 372–381. [Google Scholar] [CrossRef]
- Park, S.H.; You, E.; Park, C.-J.; Cho, Y.U.; Jang, S.; Im, H.J.; Seo, J.J.; Park, H.S.; Lee, J.H. Increased Expression of Immune Checkpoint Programmed Cell Death Protein-1 (PD-1) on T Cell Subsets of Bone Marrow Aspirates in Patients with B-Lymphoblastic Leukemia, Especially in Relapse and at Diagnosis. Cytometry. Part B Clin. Cytom. 2020, 98, 336–347. [Google Scholar] [CrossRef]
- Brudno, J.N.; Somerville, R.P.T.; Shi, V.; Rose, J.J.; Halverson, D.C.; Fowler, D.H.; Gea-Banacloche, J.C.; Pavletic, S.Z.; Hickstein, D.D.; Lu, T.L.; et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J. Clin. Oncol. J. Am. Soc. Clin. Oncol. 2016, 34, 1112–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.M.; Hucks, G.E.; Dinofia, A.M.; Seif, A.E.; Teachey, D.T.; Baniewicz, D.; Callahan, C.; Fasano, C.; McBride, B.; Gonzalez, V.; et al. Checkpoint Inhibitors Augment CD19-Directed Chimeric Antigen Receptor (CAR) T Cell Therapy in Relapsed B-Cell Acute Lymphoblastic Leukemia. Blood 2018, 132, 556. [Google Scholar] [CrossRef]
- Cao, Y.; Lu, W.; Sun, R.; Jin, X.; Cheng, L.; He, X.; Wang, L.; Yuan, T.; Lyu, C.; Zhao, M. Anti-CD19 Chimeric Antigen Receptor T Cells in Combination with Nivolumab Are Safe and Effective Against Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma. Front. Oncol. 2019, 9, 767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, E.A.; Alanio, C.; Svoboda, J.; Nasta, S.D.; Landsburg, D.J.; Lacey, S.F.; Ruella, M.; Bhattacharyya, S.; Wherry, E.J.; Schuster, S.J. Pembrolizumab for B-Cell Lymphomas Relapsing after or Refractory to CD19-Directed CAR T-Cell Therapy. Blood 2022, 139, 1026–1038. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted Delivery of a PD-1-Blocking ScFv by CAR-T Cells Enhances Anti-Tumor Efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Liu, H.; Lei, W.; Zhang, C.; Yang, C.; Wei, J.; Guo, Q.; Guo, X.; Chen, Z.; Lu, Y.; Young, K.H.; et al. CD19-Specific CAR T Cells That Express a PD-1/CD28 Chimeric Switch-Receptor Are Effective in Patients with PD-L1-Positive B-Cell Lymphoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 473–484. [Google Scholar] [CrossRef]
- Prinzing, B.; Zebley, C.C.; Petersen, C.T.; Fan, Y.; Anido, A.A.; Yi, Z.; Nguyen, P.; Houke, H.; Bell, M.; Haydar, D.; et al. Deleting DNMT3A in CAR T Cells Prevents Exhaustion and Enhances Antitumor Activity. Sci. Transl. Med. 2021, 13, eabh0272. [Google Scholar] [CrossRef]
- Gennert, D.G.; Lynn, R.C.; Granja, J.M.; Weber, E.W.; Mumbach, M.R.; Zhao, Y.; Duren, Z.; Sotillo, E.; Greenleaf, W.J.; Wong, W.H.; et al. Dynamic Chromatin Regulatory Landscape of Human CAR T Cell Exhaustion. Proc. Natl. Acad. Sci. USA 2021, 118, 8118. [Google Scholar] [CrossRef]
- Zebley, C.C.; Brown, C.; Mi, T.; Fan, Y.; Alli, S.; Boi, S.; Galletti, G.; Lugli, E.; Langfitt, D.; Metais, J.-Y.; et al. CD19-CAR T Cells Undergo Exhaustion DNA Methylation Programming in Patients with Acute Lymphoblastic Leukemia. Cell Rep. 2021, 37, 110079. [Google Scholar] [CrossRef]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V.; et al. C-Jun Overexpression in CAR T Cells Induces Exhaustion Resistance. Nature 2019, 576, 293–300. [Google Scholar] [CrossRef]
- Ajina, A.; Maher, J. Strategies to Address Chimeric Antigen Receptor Tonic Signaling. Mol. Cancer Ther. 2018, 17, 1795–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.H.; Suresh, M. Role of PI3K/Akt Signaling in Memory CD8 T Cell Differentiation. Front. Immunol. 2013, 4, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caforio, M.; de Billy, E.; De Angelis, B.; Iacovelli, S.; Quintarelli, C.; Paganelli, V.; Folgiero, V. PI3K/Akt Pathway: The Indestructible Role of a Vintage Target as a Support to the Most Recent Immunotherapeutic Approaches. Cancers 2021, 13, 4040. [Google Scholar] [CrossRef] [PubMed]
- Stock, S.; Übelhart, R.; Schubert, M.L.; Fan, F.; He, B.; Hoffmann, J.M.; Wang, L.; Wang, S.; Gong, W.; Neuber, B.; et al. Idelalisib for Optimized CD19-Specific Chimeric Antigen Receptor T Cells in Chronic Lymphocytic Leukemia Patients. Int. J. Cancer 2019, 145, 1312–1324. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Yoo, H.J.; Stock, S.; Wang, L.; Liu, Y.; Schubert, M.L.; Wang, S.; Neuber, B.; Hückelhoven-Krauss, A.; Gern, U.; et al. Ibrutinib for Improved Chimeric Antigen Receptor T-Cell Production for Chronic Lymphocytic Leukemia Patients. Int. J. Cancer 2021, 148, 419–428. [Google Scholar] [CrossRef]
- Funk, C.R.; Wang, S.; Chen, K.Z.; Waller, A.; Sharma, A.; Edgar, C.L.; Gupta, V.A.; Chandrakasan, S.; Zoine, J.T.; Fedanov, A.; et al. PI3Kδ/γ Inhibition Promotes Human CART Cell Epigenetic and Metabolic Reprogramming to Enhance Antitumor Cytotoxicity. Blood 2022, 139, 523–537. [Google Scholar] [CrossRef]
- Richman, S.A.; Wang, L.C.; Moon, E.K.; Khire, U.R.; Albelda, S.M.; Milone, M.C. Ligand-Induced Degradation of a CAR Permits Reversible Remote Control of CAR T Cell Activity In Vitro and In Vivo. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 1600–1613. [Google Scholar] [CrossRef]
- Weber, E.W.; Parker, K.R.; Sotillo, E.; Lynn, R.C.; Anbunathan, H.; Lattin, J.; Good, Z.; Belk, J.A.; Daniel, B.; Klysz, D.; et al. Transient Rest Restores Functionality in Exhausted CAR-T Cells through Epigenetic Remodeling. Science 2021, 372, 1786. [Google Scholar] [CrossRef]
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The Tyrosine Kinase Inhibitor Dasatinib Acts as a Pharmacologic on/off Switch for CAR T Cells. Sci. Transl. Med. 2019, 11, 5907. [Google Scholar] [CrossRef]
- Aamir, S.; Anwar, M.Y.; Khalid, F.; Khan, S.I.; Ali, M.A.; Khattak, Z.E. Systematic Review and Meta-Analysis of CD19-Specific CAR-T Cell Therapy in Relapsed/Refractory Acute Lymphoblastic Leukemia in the Pediatric and Young Adult Population: Safety and Efficacy Outcomes. Clin. Lymphoma Myeloma Leuk. 2021, 21, e334–e347. [Google Scholar] [CrossRef]
- Lemoine, J.; Ruella, M.; Houot, R. Born to Survive: How Cancer Cells Resist CAR T Cell Therapy. J. Hematol. Oncol. 2021, 14, 199. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-Treat Leukemia Remission by CD19 CAR T Cells of Defined Formulation and Dose in Children and Young Adults. Blood 2017, 129, 3322–3331. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T Cells of Defined CD4+:CD8+ Composition in Adult B Cell ALL Patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Spiegel, J.Y.; Dahiya, S.; Jain, M.D.; Tamaresis, J.; Nastoupil, L.J.; Jacobs, M.T.; Ghobadi, A.; Lin, Y.; Lunning, M.; Lekakis, L.; et al. Outcomes of Patients with Large B-Cell Lymphoma Progressing after Axicabtagene Ciloleucel Therapy. Blood 2021, 137, 1832–1835. [Google Scholar]
- Fousek, K.; Watanabe, J.; Joseph, S.K.; George, A.; An, X.; Byrd, T.T.; Morris, J.S.; Luong, A.; Martínez-Paniagua, M.A.; Sanber, K.; et al. CAR T-Cells That Target Acute B-Lineage Leukemia Irrespective of CD19 Expression. Leukemia 2021, 35, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Shalabi, H.; Kraft, I.L.; Wang, H.-W.; Yuan, C.M.; Yates, B.; Delbrook, C.; Zimbelman, J.D.; Giller, R.; Stetler-Stevenson, M.; Jaffe, E.S.; et al. Sequential Loss of Tumor Surface Antigens Following Chimeric Antigen Receptor T-Cell Therapies in Diffuse Large B-Cell Lymphoma. Haematologica 2018, 103, e215–e218. [Google Scholar] [CrossRef]
- Plaks, V.; Rossi, J.M.; Chou, J.; Wang, L.; Poddar, S.; Han, G.; Wang, Z.; Kuang, S.Q.; Chu, F.; Davis, R.E.; et al. CD19 Target Evasion as a Mechanism of Relapse in Large B-Cell Lymphoma Treated with Axicabtagene Ciloleucel. Blood 2021, 138, 1081–1085. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic Mechanisms of Target Antigen Loss in CAR19 Therapy of Acute Lymphoblastic Leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Paret, C.; El Malki, K.; Alt, F.; Wingerter, A.; Neu, M.A.; Kron, B.; Russo, A.; Lehmann, N.; Roth, L.; et al. CD19 Isoforms Enabling Resistance to CART-19 Immunotherapy Are Expressed in B-ALL Patients at Initial Diagnosis. J. Immunother. 2017, 40, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Asnani, M.; Hayer, K.E.; Naqvi, A.S.; Zheng, S.; Yang, S.Y.; Oldridge, D.; Ibrahim, F.; Maragkakis, M.; Gazzara, M.R.; Black, K.L.; et al. Retention of CD19 Intron 2 Contributes to CART-19 Resistance in Leukemias with Subclonal Frameshift Mutations in CD19. Leukemia 2020, 34, 1202–1207. [Google Scholar] [CrossRef] [PubMed]
- Ledererova, A.; Dostalova, L.; Kozlova, V.; Peschelova, H.; Ladungova, A.; Culen, M.; Loja, T.; Verner, J.; Pospisilova, S.; Smida, M.; et al. Hypermethylation of CD19 Promoter Enables Antigen-Negative Escape to CART-19 in Vivo and in Vitro. J. Immunother. Cancer 2021, 9, 2352. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Xu, J.; Barrett, D.M.; Fraietta, J.A.; Reich, T.J.; Ambrose, D.E.; Klichinsky, M.; Shestova, O.; Patel, P.R.; Kulikovskaya, I.; et al. Induction of Resistance to Chimeric Antigen Receptor T Cell Therapy by Transduction of a Single Leukemic B Cell. Nat. Med. 2018, 24, 1499–1503. [Google Scholar] [CrossRef]
- Ruella, M.; Barrett, D.M.; Shestova, O.; Perazzelli, J.; Posey, A.D.; Hong, S.J.; Kozlowski, M.; Lacey, S.F.; Melenhorst, J.J.; June, C.H.; et al. A Cellular Antidote to Specifically Deplete Anti-CD19 Chimeric Antigen Receptor-Positive Cells. Blood 2020, 135, 505–509. [Google Scholar] [CrossRef]
- Quintarelli, C.; Guercio, M.; Manni, S.; Boffa, I.; Sinibaldi, M.; Di Cecca, S.; Caruso, S.; Abbaszadeh, Z.; Camera, A.; Cembrola, B.; et al. Strategy to Prevent Epitope Masking in CAR.CD19+ B-Cell Leukemia Blasts. J. Immunother. Cancer 2021, 9, 1514. [Google Scholar] [CrossRef]
- Fitzgerald, K.N.; Quesada, A.E.; von Keudell, G.; Raj, S.; Lewis, N.E.; Dogan, A.; Salles, G.; Palomba, M.L. CD19 Epitope Masking by Tafasitamab Leads to Delays in Subsequent Use of CD19 CAR T-Cell Therapy in Two Patients with Aggressive Mature B-Cell Lymphomas. Leuk. Lymphoma 2022, 63, 751–754. [Google Scholar] [CrossRef]
- Liao, W.; Kohler, M.E.; Fry, T.; Ernst, P. Does Lineage Plasticity Enable Escape from CAR-T Cell Therapy? Lessons from MLL-r Leukemia. Exp. Hematol. 2021, 100, 1–11. [Google Scholar] [CrossRef]
- Jacoby, E.; Nguyen, S.M.; Fountaine, T.J.; Welp, K.; Gryder, B.; Qin, H.; Yang, Y.; Chien, C.D.; Seif, A.E.; Lei, H.; et al. CD19 CAR Immune Pressure Induces B-Precursor Acute Lymphoblastic Leukaemia Lineage Switch Exposing Inherent Leukaemic Plasticity. Nat. Commun. 2016, 7, 12320. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.-A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-Negative Myeloid Phenotype Allows Immune Escape of MLL-Rearranged B-ALL from CD19 CAR-T-Cell Therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [Green Version]
- Lamble, A.J.; Myers, R.M.; Taraseviciute, A.; John, S.; Yates, B.; Steinberg, S.M.; Sheppard, J.; Kovach, A.E.; Wood, B.L.; Borowitz, M.; et al. KMT2A Rearrangements Are Associated with Lineage Switch Following CD19 Targeting CAR T-Cell Therapy. Blood 2021, 138, 256. [Google Scholar] [CrossRef]
- Li, L.Z.; Sun, Q.; Fang, Y.; Yang, L.J.; Xu, Z.Y.; Hu, J.H.; Cao, L.; Huang, J.Y.; Hong, M.; Li, J.Y.; et al. A Report on Lineage Switch at Relapse of CD19 CAR-T Therapy for Philadelphia Chromosome-Positive B-Precursor Acute Lymphoblastic Leukemia. Chin. Med. J. 2020, 133, 2001–2003. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Orlando, E.J.; Wang, H.Y.; Bogusz, A.M.; Liu, X.; Lacey, S.F.; Strauser, H.T.; Nunez-Cruz, S.; Nejati, R.; Zhang, P.; et al. Transdifferentiation of Lymphoma into Sarcoma Associated with Profound Reprogramming of the Epigenome. Blood 2020, 136, 1980–1983. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Syrykh, C.; Hamon, M.; Adélaïde, J.; Guille, A.; Escudié, F.; Jalowicki, G.; Fina, F.; Bardet, A.; Mescam, L.; et al. Resistance of B-Cell Lymphomas to CAR T-Cell Therapy Is Associated with Genomic Tumor Changes Which Can Result in Transdifferentiation. Am. J. Surg. Pathol. 2021, 97, 1834. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhao, H.; Shao, Y.; Chen, X.; Hong, R.; Wang, L.; Ni, F.; Nagler, A.; Hu, Y.; Huang, H. Serial Surveillance by Circulating Tumor DNA Profiling after Chimeric Antigen Receptor T Therapy for the Guidance of r/r Diffuse Large B Cell Lymphoma Precise Treatment. J. Cancer 2021, 12, 5423–5431. [Google Scholar] [CrossRef]
- Jain, M.D.; Ziccheddu, B.; Coughlin, C.A.; Faramand, R.; Griswold, A.J.; Reid, K.M.; Landgren, O.; Locke, F.L.; Maura, F.; Davila, M.L.; et al. Genomic Drivers of Large B-Cell Lymphoma Resistance to CD19 CAR-T Therapy. Blood 2021, 138, 42. [Google Scholar] [CrossRef]
- Jain, M.D.; Ziccheddu, B.; Coughlin, C.A.; Faramand, R.; Griswold, A.J.; Reid, K.M.; Landgren, O.; Locke, F.L.; Maura, F.; Davila, M.L.; et al. Genomic Drivers of Large B-Cell Lymphoma Resistance to CD19 CAR-T Therapy. bioRxiv 2021, 25, 457649. [Google Scholar] [CrossRef]
- Lemoine, J.; Ruella, M.; Houot, R. Overcoming Intrinsic Resistance of Cancer Cells to CAR T-Cell Killing. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 6298–6306. [Google Scholar] [CrossRef]
- Dufva, O.; Koski, J.; Maliniemi, P.; Ianevski, A.; Klievink, J.; Leitner, J.; Pölönen, P.; Hohtari, H.; Saeed, K.; Hannunen, T.; et al. Integrated Drug Profiling and CRISPR Screening Identify Essential Pathways for CAR T-Cell Cytotoxicity. Blood 2020, 135, 597–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.; Lee, Y.G.; Shestova, O.; Ravikumar, P.; Hayer, K.E.; Hong, S.J.; Lu, X.M.; Pajarillo, R.; Agarwal, S.; Kuramitsu, S.; et al. Impaired Death Receptor Signaling in Leukemia Causes Antigen-Independent Resistance by Inducing CAR T-Cell Dysfunction. Cancer Discov. 2020, 10, 552–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyay, R.; Boiarsky, J.A.; Pantsulaia, G.; Svensson-Arvelund, J.; Lin, M.J.; Wroblewska, A.; Bhalla, S.; Scholler, N.; Bot, A.; Rossi, J.M.; et al. A Critical Role for Fas-Mediated Off-Target Tumor Killing in T-Cell Immunotherapy. Cancer Discov. 2021, 11, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Challa-Malladi, M.; Lieu, Y.K.; Califano, O.; Holmes, A.B.; Bhagat, G.; Murty, V.V.; Dominguez-Sola, D.; Pasqualucci, L.; Dalla-Favera, R. Combined Genetic Inactivation of Β2-Microglobulin and CD58 Reveals Frequent Escape from Immune Recognition in Diffuse Large B Cell Lymphoma. Cancer Cell 2011, 20, 728–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majzner, R.G.; Frank, M.J.; Mount, C.; Tousley, A.; Kurtz, D.M.; Sworder, B.; Murphy, K.A.; Manousopoulou, A.; Kohler, K.; Rotiroti, M.C.; et al. CD58 Aberrations Limit Durable Responses to CD19 CAR in Large B Cell Lymphoma Patients Treated with Axicabtagene Ciloleucel but Can Be Overcome through Novel CAR Engineering. Blood 2020, 136, 53–54. [Google Scholar] [CrossRef]
- Jiang, H.; Hu, Y.; Mei, H. Consolidative Allogeneic Hematopoietic Stem Cell Transplantation after Chimeric Antigen Receptor T-Cell Therapy for Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia: Who? When? Why? Biomark. Res. 2020, 8, 66. [Google Scholar] [CrossRef]
- Henig, I.; Zuckerman, T. Hematopoietic Stem Cell Transplantation-50 Years of Evolution and Future Perspectives. Rambam Maimonides Med. J. 2014, 5, e0028. [Google Scholar] [CrossRef]
- Savani, M.; Oluwole, O.; Dholaria, B. New Targets for CAR T Therapy in Hematologic Malignancies. Best practice & research. Clin. Haematol. 2021, 34, 101277. [Google Scholar] [CrossRef]
- Vercelli, D.; Jabara, H.H.; Lee, B.W.; Woodland, N.; Geha, R.S.; Leung, D.Y. Human Recombinant Interleukin 4 Induces Fc Epsilon R2/CD23 on Normal Human Monocytes. J. Exp. Med. 1988, 167, 1406–1416. [Google Scholar] [CrossRef]
- Kumagai, S.; Ishida, H.; Iwai, K.; Tsubata, T.; Umehara, H.; Ozaki, S.; Suginoshita, T.; Araya, S.; Imura, H. Possible Different Mechanisms of B Cell Activation in Systemic Lupus Erythematosus and Rheumatoid Arthritis: Opposite Expression of Low-Affinity Receptors for IgE (CD23) on Their Peripheral B Cells. Clin. Exp. Immunol. 1989, 78, 348–353. [Google Scholar]
- Armitage, R.J.; Goff, L.K.; Beverley, P.C.L. Expression and Functional Role of CD23 on T Cells. Eur. J. Immunol. 1989, 19, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, C.; Arock, M.; Ktorza, S.; Ouaaz, F.; Merle-Béral, H.; Mentz, F.; Kilchherr, E.; Debré, P.; Mossalayi, M.D. Expression of CD23 by Human Bone Marrow Stromal Cells. Eur. Cytokine Netw. 1992, 3, 539–543. [Google Scholar]
- Capron, M.; Truong, M.J.; Aldebert, D.; Gruart, V.; Suemura, M.; Delespesse, G.; Tourvieille, B.; Capron, A. Eosinophil IgE Receptor and CD23. Immunol. Res. 1992, 11, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Krauss, S.; Mayer, E.; Rank, G.; Rieber, E.P. Induction of the Low Affinity Receptor for IgE (Fc Epsilon RII/CD23) on Human Blood Dendritic Cells by Interleukin-4. Adv. Exp. Med. Biol. 1993, 329, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, K.A.; Arock, M.; Issaly, F.; Dugas, N.; Le Goff, L.; Kolb, J.P. Granulocyte Macrophage Colony Stimulating Factor Induces Fc Epsilon RII/CD23 Expression on Normal Human Polymorphonuclear Neutrophils. Int. Immunol. 1996, 8, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Giordano Attianese, G.M.P.; Marin, V.; Hoyos, V.; Savoldo, B.; Pizzitola, I.; Tettamanti, S.; Agostoni, V.; Parma, M.; Ponzoni, M.; Bertilaccio, M.T.S.; et al. In Vitro and in Vivo Model of a Novel Immunotherapy Approach for Chronic Lymphocytic Leukemia by Anti-CD23 Chimeric Antigen Receptor. Blood 2011, 117, 4736–4745. [Google Scholar] [CrossRef]
- Tettamanti, S.; Rotiroti, M.C.; Giordano Attianese, G.M.P.; Arcangeli, S.; Zhang, R.; Banerjee, P.; Galletti, G.; McManus, S.; Mazza, M.; Nicolini, F.; et al. Lenalidomide Enhances CD23.CAR T Cell Therapy in Chronic Lymphocytic Leukemia. Leuk. Lymphoma 2022, 94, 1–14. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J. V Fcgamma Receptors: Old Friends and New Family Members. Immunity 2006, 24, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.C.; Schuliga, M.; Shepherd, M.; Powell, M.; Harris, T.; Langenbach, S.Y.; Tan, P.S.; Gerthoffer, W.T.; Hogarth, P.M.; Stewart, A.G.; et al. Functional Expression of IgG-Fc Receptors in Human Airway Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 2011, 44, 665–672. [Google Scholar] [CrossRef] [Green Version]
- Bruggeman, C.W.; Houtzager, J.; Dierdorp, B.; Kers, J.; Pals, S.T.; Lutter, R.; van Gulik, T.; den Haan, J.M.M.; van den Berg, T.K.; van Bruggen, R.; et al. Tissue-Specific Expression of IgG Receptors by Human Macrophages Ex Vivo. PLoS ONE 2019, 14, e0223264. [Google Scholar] [CrossRef]
- Wang, G.; Sun, X.; Zuo, S.; Li, C.; Niu, Q.; Xia, Y.; Meng, Y.; Liu, M.; Fang, Z.; Yang, X.; et al. Homogeneously High Expression of CD32b Makes It a Potential Target for CAR-T Therapy for Chronic Lymphocytic Leukemia. J. Hematol. Oncol. 2021, 14, 149. [Google Scholar] [CrossRef] [PubMed]
- Hintzen, R.Q.; Lens, S.M.; Koopman, G.; Pals, S.T.; Spits, H.; van Lier, R.A. CD70 Represents the Human Ligand for CD27. Int. Immunol. 1994, 6, 477–480. [Google Scholar] [CrossRef]
- Wajant, H. Therapeutic Targeting of CD70 and CD27. Expert Opin. Ther. Targets 2016, 20, 959–973. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, D.R.; Savoldo, B.; Yi, Z.; Chow, K.K.H.; Kakarla, S.; Spencer, D.M.; Dotti, G.; Wu, M.F.; Liu, H.; Kenney, S.; et al. T Cells Redirected against CD70 for the Immunotherapy of CD70-Positive Malignancies. Blood 2011, 117, 4304–4314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.; Chen, P.; Lei, W.; Xu, Y.; Xu, N.; Pu, J.J.; Liang, A.; Qian, W. CD70-Targeting CAR-T Cells Have Potential Activity against CD19-Negative B-Cell Lymphoma. Cancer Commun. 2021, 41, 925–929. [Google Scholar] [CrossRef] [PubMed]
- Schwarting, R.; Castello, R.; Moldenhauer, G.; Pezzutto, A.; von Hoegen, I.; Ludwig, W.D.; Parnes, J.R.; Dörken, B. Human Lyb-2 Homolog CD72 Is a Marker for Progenitor B-Cell Leukemias. Am. J. Hematol. 1992, 41, 151–158. [Google Scholar] [CrossRef]
- Nix, M.A.; Mandal, K.; Geng, H.; Paranjape, N.; Lin, Y.H.T.; Rivera, J.M.; Marcoulis, M.; White, K.L.; Whitman, J.D.; Bapat, S.P.; et al. Surface Proteomics Reveals CD72 as a Target for In Vitro-Evolved Nanobody-Based CAR-T Cells in KMT2A/MLL1-Rearranged B-ALL. Cancer Discov. 2021, 11, 2032–2049. [Google Scholar] [CrossRef]
- Yin, A.H.; Miraglia, S.; Zanjani, E.D.; Almeida-Porada, G.; Ogawa, M.; Leary, A.G.; Olweus, J.; Kearney, J.; Buck, D.W. AC133, a Novel Marker for Human Hematopoietic Stem and Progenitor Cells. Blood 1997, 90, 5002–5012. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Hu, Y.; Jin, Z.; Zhai, Y.; Tan, Y.; Sun, Y.; Zhu, S.; Zhao, C.; Chen, B.; Zhu, J.; et al. TanCAR T Cells Targeting CD19 and CD133 Efficiently Eliminate MLL Leukemic Cells. Leukemia 2018, 32, 2012–2016. [Google Scholar] [CrossRef]
- Bueno, C.; Velasco-Hernandez, T.; Gutiérrez-Agüera, F.; Zanetti, S.R.; Baroni, M.L.; Sánchez-Martínez, D.; Molina, O.; Closa, A.; Agraz-Doblás, A.; Marín, P.; et al. CD133-Directed CAR T-Cells for MLL Leukemia: On-Target, off-Tumor Myeloablative Toxicity. Leukemia 2019, 33, 2090–2125. [Google Scholar] [CrossRef]
- Kubagawa, H.; Oka, S.; Kubagawa, Y.; Torii, I.; Takayama, E.; Kang, D.-W.; Gartland, G.L.; Bertoli, L.F.; Mori, H.; Takatsu, H.; et al. Identity of the Elusive IgM Fc Receptor (FcmuR) in Humans. J. Exp. Med. 2009, 206, 2779–2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faitschuk, E.; Hombach, A.A.; Frenzel, L.P.; Wendtner, C.-M.; Abken, H. Chimeric Antigen Receptor T Cells Targeting Fc μ Receptor Selectively Eliminate CLL Cells While Sparing Healthy B Cells. Blood 2016, 128, 1711–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kardava, L.; Moir, S.; Wang, W.; Ho, J.; Buckner, C.M.; Posada, J.G.; O’Shea, M.A.; Roby, G.; Chen, J.; Sohn, H.W.; et al. Attenuation of HIV-Associated Human B Cell Exhaustion by SiRNA Downregulation of Inhibitory Receptors. J. Clin. Investig. 2011, 121, 2614–2624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumer, K.K.; Uyenishi, J.; Hoffman, M.C.; Fisher, B.M.; Winn, V.D. Siglec-6 Expression Is Increased in Placentas from Pregnancies Complicated by Preterm Preeclampsia. Reprod. Sci. 2013, 20, 646–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Blokhuis, B.R.J.; Diks, M.A.P.; Keshavarzian, A.; Garssen, J.; Redegeld, F.A. Functional Inhibitory Siglec-6 Is Upregulated in Human Colorectal Cancer-Associated Mast Cells. Front. Immunol. 2018, 9, 2138. [Google Scholar] [CrossRef]
- Kovalovsky, D.; Yoon, J.H.; Cyr, M.G.; Simon, S.; Voynova, E.; Rader, C.; Wiestner, A.; Alejo, J.; Pittaluga, S.; Gress, R.E. Siglec-6 Is a Target for Chimeric Antigen Receptor T-Cell Treatment of Chronic Lymphocytic Leukemia. Leukemia 2021, 35, 2581–2591. [Google Scholar] [CrossRef]
- Reche, P.A.; Soumelis, V.; Gorman, D.M.; Clifford, T.; Mr, L.; Travis, M.; Zurawski, S.M.; Johnston, J.; Liu, Y.J.; Spits, H.; et al. Human Thymic Stromal Lymphopoietin Preferentially Stimulates Myeloid Cells. J. Immunol. 2001, 167, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Cho, M.; Haso, W.; Zhang, L.; Tasian, S.K.; Oo, H.Z.; Negri, G.L.; Lin, Y.; Zou, J.; Mallon, B.S.; et al. Eradication of B-ALL Using Chimeric Antigen Receptor-Expressing T Cells Targeting the TSLPR Oncoprotein. Blood 2015, 126, 629–639. [Google Scholar] [CrossRef]
- Rodig, S.J.; Shahsafaei, A.; Li, B.; Mackay, C.R.; Dorfman, D.M. BAFF-R, the Major B Cell-Activating Factor Receptor, Is Expressed on Most Mature B Cells and B-Cell Lymphoproliferative Disorders. Hum. Pathol. 2005, 36, 1113–1119. [Google Scholar] [CrossRef]
- Saeland, S.; Duvert, V.; Pandrau, D.; Caux, C.; Durand, I.; Wrighton, N.; Wideman, J.; Lee, F.; Banchereau, J. Interleukin-7 Induces the Proliferation of Normal Human B-Cell Precursors. Blood 1991, 78, 2229–2238. [Google Scholar] [CrossRef] [Green Version]
- Wentink, M.W.J.; Kalina, T.; Perez-Andres, M.; Del Pino Molina, L.; IJspeert, H.; Kavelaars, F.G.; Lankester, A.C.; Lecrevisse, Q.; van Dongen, J.J.M.; Orfao, A.; et al. Delineating Human B Cell Precursor Development with Genetically Identified PID Cases as a Model. Front. Immunol. 2019, 10, 2680. [Google Scholar] [CrossRef] [PubMed]
- Otipoby, K.L.; Andersson, K.B.; Draves, K.E.; Klaus, S.J.; Farr, A.G.; Kerner, J.D.; Perlmutter, R.M.; Law, C.L.; Clark, E.A. CD22 Regulates Thymus-Independent Responses and the Lifespan of B Cells. Nature 1996, 384, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Viemann, D.; Schlenke, P.; Hammers, H.J.; Kirchner, H.; Kruse, A. Differential Expression of the B Cell-Restricted Molecule CD22 on Neonatal B Lymphocytes Depending upon Antigen Stimulation. Eur. J. Immunol. 2000, 30, 550–559. [Google Scholar] [CrossRef]
- Gilfillan, M.C.; Noel, P.J.; Podack, E.R.; Reiner, S.L.; Thompson, C.B. Expression of the Costimulatory Receptor CD30 Is Regulated by Both CD28 and Cytokines. J. Immunol. 1998, 160, 2180–2187. [Google Scholar] [PubMed]
- Weniger, M.A.; Tiacci, E.; Schneider, S.; Arnolds, J.; Rüschenbaum, S.; Duppach, J.; Seifert, M.; Döring, C.; Hansmann, M.L.; Küppers, R. Human CD30+ B Cells Represent a Unique Subset Related to Hodgkin Lymphoma Cells. J. Clin. Investig. 2018, 128, 2996–3007. [Google Scholar] [CrossRef] [Green Version]
- De Winde, C.M.; Zuidscherwoude, M.; Vasaturo, A.; van der Schaaf, A.; Figdor, C.G.; van Spriel, A.B. Multispectral Imaging Reveals the Tissue Distribution of Tetraspanins in Human Lymphoid Organs. Histochem. Cell Biol. 2015, 144, 133–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.C.J.; Weiss, B.M.; et al. Daratumumab Depletes CD38+ Immune Regulatory Cells, Promotes T-Cell Expansion, and Skews T-Cell Repertoire in Multiple Myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [Green Version]
- Garcia Vela, J.; Delgado, I.; Benito, L.; Monteserin, M.; Garcia Alonso, L.; Somolinos, N.; Andreu, M.; Oña, F. CD79b Expression in B Cell Chronic Lymphocytic Leukemia: Its Implication for Minimal Residual Disease Detection. Leukemia 1999, 13, 1501–1505. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Ikeda, K.; Maeda, Y.; Shinagawa, K.; Ohtsuka, A.; Yamamura, H.; Tanimoto, M. Identification of CD123+ Myeloid Dendritic Cells as an Early-Stage Immature Subset with Strong Tumoristatic Potential. Cancer Lett. 2008, 270, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.F.; Bécares, N.; Stephens, A.; Turcanu, V.; Lack, G. The Expression of CD123 Can Decrease with Basophil Activation: Implications for the Gating Strategy of the Basophil Activation Test. Clin. Transl. Allergy 2016, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Chilosi, M.; Adami, F.; Lestani, M.; Montagna, L.; Cimarosto, L.; Semenzato, G.; Pizzolo, G.; Menestrina, F. CD138/Syndecan-1: A Useful Immunohistochemical Marker of Normal and Neoplastic Plasma Cells on Routine Trephine Bone Marrow Biopsies. Mod. Pathol. Off. J. USA Can. Acad. Pathol. 1999, 12, 1101–1106. [Google Scholar]
- Kind, S.; Merenkow, C.; Büscheck, F.; Möller, K.; Dum, D.; Chirico, V.; Luebke, A.M.; Höflmayer, D.; Hinsch, A.; Jacobsen, F.; et al. Prevalence of Syndecan-1 (CD138) Expression in Different Kinds of Human Tumors and Normal Tissues. Dis. Mark. 2019, 2019, 4928315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D Is a Target for the Immunotherapy of Multiple Myeloma with Rationally Designed CAR T Cells. Sci. Transl. Med. 2019, 11, 7746. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Kochi, Y.; Nakai, W.; Mizuno, H.; Baba, T.; Habu, K.; Sawada, N.; Tsunoda, H.; Shima, T.; Miyawaki, K.; et al. Anti-GPRC5D/CD3 Bispecific T-Cell-Redirecting Antibody for the Treatment of Multiple Myeloma. Mol. Cancer Ther. 2019, 18, 1555–1564. [Google Scholar] [CrossRef]
- Verkleij, C.P.M.; Broekmans, M.E.C.; van Duin, M.; Frerichs, K.A.; Kuiper, R.; de Jonge, A.V.; Kaiser, M.; Morgan, G.; Axel, A.; Boominathan, R.; et al. Preclinical Activity and Determinants of Response of the GPRC5DxCD3 Bispecific Antibody Talquetamab in Multiple Myeloma. Blood Adv. 2021, 5, 2196–2215. [Google Scholar] [CrossRef] [PubMed]
- Salahuddin, S.; Fath, E.K.; Biel, N.; Ray, A.; Moss, C.R.; Patel, A.; Patel, S.; Hilding, L.; Varn, M.; Ross, T.; et al. Epstein-Barr Virus Latent Membrane Protein-1 Induces the Expression of SUMO-1 and SUMO-2/3 in LMP1-Positive Lymphomas and Cells. Sci. Rep. 2019, 9, 208. [Google Scholar] [CrossRef]
- Al-Shawi, R.; Ashton, S.V.; Underwood, C.; Simons, J.P. Expression of the Ror1 and Ror2 Receptor Tyrosine Kinase Genes during Mouse Development. Dev. Genes Evol. 2001, 211, 161–171. [Google Scholar] [CrossRef]
- Boles, K.S.; Mathew, P.A. Molecular Cloning of CS1, a Novel Human Natural Killer Cell Receptor Belonging to the CD2 Subset of the Immunoglobulin Superfamily. Immunogenetics 2001, 52, 302–307. [Google Scholar] [CrossRef]
- Bouchon, A.; Cella, M.; Grierson, H.L.; Cohen, J.I.; Colonna, M. Activation of NK Cell-Mediated Cytotoxicity by a SAP-Independent Receptor of the CD2 Family. J. Immunol. 2001, 167, 5517–5521. [Google Scholar] [CrossRef] [Green Version]
- Muta, T.; Kurosaki, T.; Misulovin, Z.; Sanchez, M.; Nussenzweig, M.C.; Ravetch, J. V A 13-Amino-Acid Motif in the Cytoplasmic Domain of Fc Gamma RIIB Modulates B-Cell Receptor Signalling. Nature 1994, 369, 340. [Google Scholar] [CrossRef]
- Espéli, M.; Bashford-Rogers, R.; Sowerby, J.M.; Alouche, N.; Wong, L.; Denton, A.E.; Linterman, M.A.; Smith, K.G.C. FcγRIIb Differentially Regulates Pre-Immune and Germinal Center B Cell Tolerance in Mouse and Human. Nat. Commun. 2019, 10, 1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Comenzo, R.L.; Olshen, A.B.; Bonvini, E.; Koenig, S.; Maslak, P.G.; Fleisher, M.; Hoffman, J.; Jhanwar, S.; Young, J.W.; et al. CD32B Is Highly Expressed on Clonal Plasma Cells from Patients with Systemic Light-Chain Amyloidosis and Provides a Target for Monoclonal Antibody-Based Therapy. Blood 2008, 111, 3403–3406. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Ashton-Key, M.; Cogliatti, S.; Rondeau, S.; Schmitz, S.F.H.; Ghielmini, M.; Cragg, M.S.; Johnson, P. Expression of the Inhibitory Fc Gamma Receptor IIB (FCGR2B, CD32B) on Follicular Lymphoma Cells Lowers the Response Rate to Rituximab Monotherapy (SAKK 35/98). Br. J. Haematol. 2015, 168, 145–148. [Google Scholar] [CrossRef] [Green Version]
- Nowicka, M.; Hilton, L.K.; Ashton-Key, M.; Hargreaves, C.E.; Lee, C.; Foxall, R.; Carter, M.J.; Beers, S.A.; Potter, K.N.; Bolen, C.R.; et al. Prognostic Significance of FCGR2B Expression for the Response of DLBCL Patients to Rituximab or Obinutuzumab Treatment. Blood Adv. 2021, 5, 2945–2957. [Google Scholar] [CrossRef] [PubMed]
- Roghanian, A.; Teige, I.; Mårtensson, L.; Cox, K.L.; Kovacek, M.; Ljungars, A.; Mattson, J.; Sundberg, A.; Vaughan, A.T.; Shah, V.; et al. Antagonistic Human FcγRIIB (CD32B) Antibodies Have Anti-Tumor Activity and Overcome Resistance to Antibody Therapy in Vivo. Cancer Cell 2015, 27, 473–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, M.R.; Crimmins, M.A.; Yetz-Aldape, J.; Kriz, R.; Kelleher, K.; Herrmann, S. The Cloning of CD70 and Its Identification as the Ligand for CD27. J. Immunol. 1994, 152, 1756–1761. [Google Scholar]
- Hintzen, R.Q.; Lens, S.M.; Beckmann, M.P.; Goodwin, R.G.; Lynch, D.; van Lier, R.A. Characterization of the Human CD27 Ligand, a Novel Member of the TNF Gene Family. J. Immunol. 1994, 152, 1762–1773. [Google Scholar]
- Jacquot, S. CD27/CD70 Interactions Regulate T Dependent B Cell Differentiation. Immunol. Res. 2000, 21, 23–30. [Google Scholar] [CrossRef]
- Borst, J.; Hendriks, J.; Xiao, Y. CD27 and CD70 in T Cell and B Cell Activation. Curr. Opin. Immunol. 2005, 17, 275–281. [Google Scholar] [CrossRef]
- Lens, S.M.; Drillenburg, P.; den Drijver, B.F.; van Schijndel, G.; Pals, S.T.; van Lier, R.A.; van Oers, M.H. Aberrant Expression and Reverse Signalling of CD70 on Malignant B Cells. Br. J. Haematol. 1999, 106, 491–503. [Google Scholar] [CrossRef]
- McEarchern, J.A.; Smith, L.M.; McDonagh, C.F.; Klussman, K.; Gordon, K.A.; Morris-Tilden, C.A.; Duniho, S.; Ryan, M.; Boursalian, T.E.; Carter, P.J.; et al. Preclinical Characterization of SGN-70, a Humanized Antibody Directed against CD70. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 7763–7772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balsas, P.; Veloza, L.; Clot, G.; Sureda-Gómez, M.; Rodríguez, M.L.; Masaoutis, C.; Frigola, G.; Navarro, A.; Beà, S.; Nadeu, F.; et al. SOX11, CD70, and Treg Cells Configure the Tumor-Immune Microenvironment of Aggressive Mantle Cell Lymphoma. Blood 2021, 138, 2202–2215. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Z.; Novak, A.J.; Ziesmer, S.C.; Witzig, T.E.; Ansell, S.M. CD70+ Non-Hodgkin Lymphoma B Cells Induce Foxp3 Expression and Regulatory Function in Intratumoral CD4+CD25 T Cells. Blood 2007, 110, 2537–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Israel, B.F.; Gulley, M.; Elmore, S.; Ferrini, S.; Feng, W.; Kenney, S.C. Anti-CD70 Antibodies: A Potential Treatment for EBV+ CD70-Expressing Lymphomas. Mol. Cancer Ther. 2005, 4, 2037–2044. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Nadler, M.J.; Brennan, L.A.; Gish, G.D.; Timms, J.F.; Fusaki, N.; Jongstra-Bilen, J.; Tada, N.; Pawson, T.; Wither, J.; et al. The B-Cell Transmembrane Protein CD72 Binds to and is an in Vivo Substrate of the Protein Tyrosine Phosphatase SHP-1. Curr. Biol. 1998, 8, 1009–1017. [Google Scholar] [CrossRef] [Green Version]
- Adachi, T.; Wakabayashi, C.; Nakayama, T.; Yakura, H.; Tsubata, T. CD72 Negatively Regulates Signaling through the Antigen Receptor of B Cells. J. Immunol. 2000, 164, 1223–1229. [Google Scholar] [CrossRef] [Green Version]
- Planken, E.V.; Van de Velde, H.; Falkenburg, J.H.; Thielemans, K.; Willemze, R.; Kluin-Nelemans, J.C. Selective Response of CD5+ B Cell Malignancies to Activation of the CD72 Antigen. Clin. Immunol. Immunopathol. 1998, 87, 42–49. [Google Scholar] [CrossRef]
- Kumanogoh, A.; Watanabe, C.; Lee, I.; Wang, X.; Shi, W.; Araki, H.; Hirata, H.; Iwahori, K.; Uchida, J.; Yasui, T.; et al. Identification of CD72 as a Lymphocyte Receptor for the Class IV Semaphorin CD100: A Novel Mechanism for Regulating B Cell Signaling. Immunity 2000, 13, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Akatsu, C.; Shinagawa, K.; Numoto, N.; Liu, Z.; Ucar, A.K.; Aslam, M.; Phoon, S.; Adachi, T.; Furukawa, K.; Ito, N.; et al. CD72 Negatively Regulates B Lymphocyte Responses to the Lupus-Related Endogenous Toll-like Receptor 7 Ligand Sm/RNP. J. Exp. Med. 2016, 213, 2691–2706. [Google Scholar] [CrossRef]
- Li, D.H.H.; Winslow, M.M.; Cao, T.M.; Chen, A.H.; Davis, C.R.; Mellins, E.D.; Utz, P.J.; Crabtree, G.R.; Parnes, J.R. Modulation of Peripheral B Cell Tolerance by CD72 in a Murine Model. Arthritis Rheum. 2008, 58, 3192–3204. [Google Scholar] [CrossRef]
- Asmiyou, A.; Bakr, A.M.; Shahin, D.A.; Wahba, Y. CD40 and CD72 Expression and Prognostic Values among Children with Systemic Lupus Erythematosus: A Case-Control Study. Lupus 2020, 29, 1270–1276. [Google Scholar] [CrossRef] [PubMed]
- Garand, R.; Robillard, N.; Bataille, R. CD72 Is Constantly Expressed in Chronic Lymphocytic Leukemia and Other B-Cell Lymphoproliferative Disorders. Leuk. Res. 1994, 18, 651–652. [Google Scholar] [CrossRef]
- Alcón, V.L.; Luther, C.; Balce, D.; Takei, F. B-Cell Co-Receptor CD72 Is Expressed on NK Cells and Inhibits IFN-Gamma Production but Not Cytotoxicity. Eur. J. Immunol. 2009, 39, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, T.R.; Kumanogoh, A.; Bandara, G.; Metcalfe, D.D.; Gilfillan, A.M. CD72 Negatively Regulates KIT-Mediated Responses in Human Mast Cells. J. Immunol. 2010, 184, 2468–2475. [Google Scholar] [CrossRef] [PubMed]
- Bossen, C.; Schneider, P. BAFF, APRIL and Their Receptors: Structure, Function and Signaling. Semin. Immunol. 2006, 18, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Shulga-Morskaya, S.; Dobles, M.; Walsh, M.E.; Ng, L.G.; MacKay, F.; Rao, S.P.; Kalled, S.L.; Scott, M.L. B Cell-Activating Factor Belonging to the TNF Family Acts through Separate Receptors to Support B Cell Survival and T Cell-Independent Antibody Formation. J. Immunol. 2004, 173, 2331–2341. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, Y.; Casola, S.; Kutok, J.L.; Rajewsky, K.; Schmidt-Supprian, M. TNF Family Member B Cell-Activating Factor (BAFF) Receptor-Dependent and-Independent Roles for BAFF in B Cell Physiology. J. Immunol. 2004, 173, 2245–2252. [Google Scholar] [CrossRef] [Green Version]
- Warnatz, K.; Salzer, U.; Rizzi, M.; Fischer, B.; Gutenberger, S.; Böhm, J.; Kienzler, A.-K.; Pan-Hammarström, Q.; Hammarström, L.; Rakhmanov, M.; et al. B-Cell Activating Factor Receptor Deficiency Is Associated with an Adult-Onset Antibody Deficiency Syndrome in Humans. Proc. Natl. Acad. Sci. USA 2009, 106, 13945–13950. [Google Scholar] [CrossRef] [Green Version]
- Wada, K.; Maeda, K.; Tajima, K.; Kato, T.; Kobata, T.; Yamakawa, M. Expression of BAFF-R and TACI in Reactive Lymphoid Tissues and B-Cell Lymphomas. Histopathology 2009, 54, 221–232. [Google Scholar] [CrossRef]
- Takahata, H.; Ohara, N.; Ichimura, K.; Tanaka, T.; Sato, Y.; Morito, T.; Takata, K.; Kojima, M.; Kobata, T.; Yoshino, T. BAFF-R Is Expressed on B-Cell Lymphomas Depending on Their Origin and Is Related to Proliferation Index of Nodal Diffuse Large B-Cell Lymphomas. J. Clin. Exp. Hematop. 2010, 50, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.J.; Jiang, W.Q.; Rao, H.L.; Huang, J.J.; Xia, Y.; Huang, H.Q.; Lin, T.Y.; Xia, Z.J.; Li, S.; Li, Z.-M. Expression of BAFF and BAFF-R in Follicular Lymphoma: Correlation with Clinicopathologic Characteristics and Survival Outcomes. PLoS ONE 2012, 7, e50936. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.J.; Jiang, W.Q.; Rao, H.L.; Huang, J.J.; Xia, Y.; Bi, X.; Sun, P.; Huang, H.Q.; Lin, T.Y.; et al. Expression of BAFF-R, but Not BAFF, Is an Independent Prognostic Factor in Diffuse Large B-Cell Lymphoma Patients Treated with R-CHOP. Annu. Hematol. 2015, 94, 1865–1873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Roy, N.K.; Vicioso, Y.; Woo, J.; Beck, R.; de Lima, M.; Caimi, P.; Feinberg, D.; Parameswaran, R. BAFF Receptor Antibody for Mantle Cell Lymphoma Therapy. Oncoimmunology 2021, 10, 1893501. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.; Nishio, M.; Enzler, T.; Cottam, H.B.; Fukuda, T.; James, D.F.; Karin, M.; Kipps, T.J. BAFF and APRIL Support Chronic Lymphocytic Leukemia B-Cell Survival through Activation of the Canonical NF-ΚB Pathway. Blood 2006, 109, 703–710. [Google Scholar] [CrossRef]
- Parameswaran, R.; Müschen, M.; Kim, Y.M.; Groffen, J.; Heisterkamp, N. A Functional Receptor for B-Cell-Activating Factor Is Expressed on Human Acute Lymphoblastic Leukemias. Cancer Res. 2010, 70, 4346–4356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maia, S.; Pelletier, M.; Ding, J.; Hsu, Y.M.; Sallan, S.E.; Rao, S.P.; Nadler, L.M.; Cardoso, A.A. Aberrant Expression of Functional BAFF-System Receptors by Malignant B-Cell Precursors Impacts Leukemia Cell Survival. PLoS ONE 2011, 6, e20787. [Google Scholar] [CrossRef] [Green Version]
- Fazio, G.; Turazzi, N.; Cazzaniga, V.; Kreuzaler, M.; Maglia, O.; Magnani, C.F.; Biagi, E.; Rolink, A.; Biondi, A.; Cazzaniga, G. TNFRSF13C (BAFFR) Positive Blasts Persist after Early Treatment and at Relapse in Childhood B-Cell Precursor Acute Lymphoblastic Leukaemia. Br. J. Haematol. 2018, 182, 434–436. [Google Scholar] [CrossRef] [Green Version]
- Parameswaran, R.; Lim, M.; Fei, F.; Abdel-Azim, H.; Arutyunyan, A.; Schiffer, I.; McLaughlin, M.E.; Gram, H.; Huet, H.; Groffen, J.; et al. Effector-Mediated Eradication of Precursor B Acute Lymphoblastic Leukemia with a Novel Fc-Engineered Monoclonal Antibody Targeting the BAFF-R. Mol. Cancer Ther. 2014, 13, 1567–1577. [Google Scholar] [CrossRef] [Green Version]
- McWilliams, E.M.; Lucas, C.R.; Chen, T.; Harrington, B.K.; Wasmuth, R.; Campbell, A.; Rogers, K.A.; Cheney, C.M.; Mo, X.; Andritsos, L.A.; et al. Anti-BAFF-R Antibody VAY-736 Demonstrates Promising Preclinical Activity in CLL and Enhances Effectiveness of Ibrutinib. Blood Adv. 2019, 3, 447–460. [Google Scholar] [CrossRef]
- Qin, H.; Dong, Z.; Wang, X.; Cheng, W.A.; Wen, F.; Xue, W.; Sun, H.; Walter, M.; Wei, G.; Smith, D.L.; et al. CAR T Cells Targeting BAFF-R Can Overcome CD19 Antigen Loss in B Cell Malignancies. Sci. Transl. Med. 2019, 11, 9414. [Google Scholar] [CrossRef]
- Schwartz-Albiez, R.; Dörken, B.; Hofmann, W.; Moldenhauer, G. The B Cell-Associated CD37 Antigen (Gp40–52). Structure and Subcellular Expression of an Extensively Glycosylated Glycoprotein. J. Immunol. 1988, 140, 905–914. [Google Scholar] [PubMed]
- Jones, E.L.; Demaria, M.C.; Wright, M.D. Tetraspanins in Cellular Immunity. Biochem. Soc. Trans. 2011, 39, 506–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Spriel, A.B. Tetraspanins in the Humoral Immune Response. Biochem. Soc. Trans. 2011, 39, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Barrena, S.; Almeida, J.; Yunta, M.; López, A.; Fernández-Mosteirín, N.; Giralt, M.; Romero, M.; Perdiguer, L.; Delgado, M.; Orfao, A.; et al. Aberrant Expression of Tetraspanin Molecules in B-Cell Chronic Lymphoproliferative Disorders and Its Correlation with Normal B-Cell Maturation. Leukemia 2005, 19, 1376–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobrowicz, M.; Kubacz, M.; Slusarczyk, A.; Winiarska, M. CD37 in B Cell Derived Tumors-More than Just a Docking Point for Monoclonal Antibodies. Int. J. Mol. Sci. 2020, 21, 9531. [Google Scholar] [CrossRef] [PubMed]
- Pagel, J.M.; Spurgeon, S.E.; Byrd, J.C.; Awan, F.T.; Flinn, I.W.; Lanasa, M.C.; Eisenfeld, A.J.; Stromatt, S.C.; Gopal, A.K. Otlertuzumab (TRU-016), an Anti-CD37 Monospecific ADAPTIR(TM) Therapeutic Protein for Relapsed or Refractory NHL Patients. Br. J. Haematol. 2015, 168, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Kroschinsky, F.; Middeke, J.M.; Janz, M.; Lenz, G.; Witzens-Harig, M.; Bouabdallah, R.; La Rosée, P.; Viardot, A.; Salles, G.; Kim, S.J.; et al. Phase I Dose Escalation Study of BI 836826 (CD37 Antibody) in Patients with Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma. Investig. New Drugs 2020, 38, 1472–1482. [Google Scholar] [CrossRef]
- Pereira, D.S.; Guevara, C.I.; Jin, L.; Mbong, N.; Verlinsky, A.; Hsu, S.J.; Aviña, H.; Karki, S.; Abad, J.D.; Yang, P.; et al. AGS67E, an Anti-CD37 Monomethyl Auristatin E Antibody-Drug Conjugate as a Potential Therapeutic for B/T-Cell Malignancies and AML: A New Role for CD37 in AML. Mol. Cancer Ther. 2015, 14, 1650–1660. [Google Scholar] [CrossRef] [Green Version]
- Stathis, A.; Flinn, I.W.; Madan, S.; Maddocks, K.; Freedman, A.; Weitman, S.; Zucca, E.; Munteanu, M.C.; Lia Palomba, M. Safety, Tolerability, and Preliminary Activity of IMGN529, a CD37-Targeted Antibody-Drug Conjugate, in Patients with Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma: A Dose-Escalation, Phase I Study. Investig. New Drugs 2018, 36, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Repetto-Llamazares, A.H.V.; Larsen, R.H.; Giusti, A.M.; Riccardi, E.; Bruland, Ø.S.; Selbo, P.K.; Dahle, J. 177Lu-DOTA-HH1, a Novel Anti-CD37 Radio-Immunoconjugate: A Study of Toxicity in Nude Mice. PLoS ONE 2014, 9, e103070. [Google Scholar] [CrossRef] [Green Version]
- Scarfò, I.; Ormhøj, M.; Frigault, M.J.; Castano, A.P.; Lorrey, S.; Bouffard, A.A.; van Scoyk, A.; Rodig, S.J.; Shay, A.J.; Aster, J.C.; et al. Anti-CD37 Chimeric Antigen Receptor T Cells Are Active against B- and T-Cell Lymphomas. Blood 2018, 132, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Köksal, H.; Dillard, P.; Josefsson, S.E.; Maggadottir, S.M.; Pollmann, S.; Fåne, A.; Blaker, Y.N.; Beiske, K.; Huse, K.; Kolstad, A.; et al. Preclinical Development of CD37CAR T-Cell Therapy for Treatment of B-Cell Lymphoma. Blood Adv. 2019, 3, 1230–1243. [Google Scholar] [CrossRef] [PubMed]
- Frigault, M.J.; Chen, Y.B.; Gallagher, K.M.E.; Horick, N.K.; El-Jawahri, A.; Scarfò, I.; Wehrli, M.; Huang, L.; Casey, K.; Cook, D.; et al. Phase 1 Study of CD37-Directed CAR T Cells in Patients with Relapsed or Refractory CD37+ Hematologic Malignancies. Blood 2021, 138, 653. [Google Scholar] [CrossRef]
- Bräuner-Osborne, H.; Jensen, A.A.; Sheppard, P.O.; Brodin, B.; Krogsgaard-Larsen, P.; O’Hara, P. Cloning and Characterization of a Human Orphan Family C G-Protein Coupled Receptor GPRC5D. Biochim. Biophys. Acta 2001, 1518, 237–248. [Google Scholar] [CrossRef]
- Inoue, S.; Nambu, T.; Shimomura, T. The RAIG Family Member, GPRC5D, Is Associated with Hard-Keratinized Structures. J. Investig. Dermatol. 2004, 122, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Pillarisetti, K.; Edavettal, S.; Mendonça, M.; Li, Y.; Tornetta, M.; Babich, A.; Majewski, N.; Husovsky, M.; Reeves, D.; Walsh, E.; et al. A T-Cell-Redirecting Bispecific G-Protein-Coupled Receptor Class 5 Member D x CD3 Antibody to Treat Multiple Myeloma. Blood 2020, 135, 1232–1243. [Google Scholar] [CrossRef]
- Atamaniuk, J.; Gleiss, A.; Porpaczy, E.; Kainz, B.; Grunt, T.W.; Raderer, M.; Hilgarth, B.; Drach, J.; Ludwig, H.; Gisslinger, H.; et al. Overexpression of G Protein-Coupled Receptor 5D in the Bone Marrow Is Associated with Poor Prognosis in Patients with Multiple Myeloma. Eur. J. Clin. Investig. 2012, 42, 953–960. [Google Scholar] [CrossRef]
- Chari, A.; Berdeja, J.G.; Oriol, A.; van de Donk, N.W.C.J.; Rodriguez, P.; Askari, E.; Mateos, M.-V.; Minnema, M.C.; Verona, R.; Girgis, S.; et al. A Phase 1, First-in-Human Study of Talquetamab, a G Protein-Coupled Receptor Family C Group 5 Member D (GPRC5D) x CD3 Bispecific Antibody, in Patients with Relapsed and/or Refractory Multiple Myeloma (RRMM). Blood 2020, 136, 40–41. [Google Scholar] [CrossRef]
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.; Ghoddusi, M.; Purdon, T.J.; Do, T.; Wang, X.; et al. CAR T Cell Therapy Targeting G Protein-Coupled Receptor Class C Group 5 Member D (GPRC5D), a Novel Target for the Immunotherapy of Multiple Myeloma. Blood 2018, 132, 589. [Google Scholar] [CrossRef]
- Mailankody, S.; Diamonte, C.; Fitzgerald, L.; Kane, P.; Wang, X.; Sikder, D.S.; Senechal, B.; Bermudez, V.P.; Frias, D.; Morgan, J.; et al. Phase I First-in-Class Trial of MCARH109, a G Protein Coupled Receptor Class C Group 5 Member D (GPRC5D) Targeted CAR T Cell Therapy in Patients with Relapsed or Refractory Multiple Myeloma. Blood 2021, 138, 827. [Google Scholar] [CrossRef]
- Shi, X.; Yan, L.; Shang, J.; Qu, S.; Kang, L.; Zhou, J.; Jin, S.; Yao, W.; Yao, Y.; Yan, S.; et al. Tandom Autologous Transplantation and Combined Infusion of CD19 and Bcma-Specific Chimeric Antigen Receptor T Cells for High Risk MM: Initial Safety and Efficacy Report from a Clinical Pilot Study. Blood 2018, 132, 1009. [Google Scholar] [CrossRef]
- Jiang, H.; Dong, B.; Gao, L.; Liu, L.; Ge, J.; He, A.; Du, J.; Li, L.; Lu, J.; Chen, X.; et al. Clinical Results of a Multicenter Study of the First-in-Human Dual BCMA and CD19 Targeted Novel Platform Fast CAR-T Cell Therapy for Patients with Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 25–26. [Google Scholar] [CrossRef]
- Tang, Y.; Yin, H.; Zhao, X.; Jin, D.; Liang, Y.; Xiong, T.; Li, L.; Tang, W.; Zhang, J.; Liu, M.; et al. High Efficacy and Safety of CD38 and BCMA Bispecific CAR-T in Relapsed or Refractory Multiple Myeloma. J. Exp. Clin. Cancer Res. 2021, 41, 2. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Johnson, B.D.; Schneider, D.; Zhu, F.; Szabo, A.; Keever-Taylor, C.A.; Krueger, W.; Worden, A.A.; Kadan, M.J.; Yim, S.; et al. Bispecific Anti-CD20, Anti-CD19 CAR T Cells for Relapsed B Cell Malignancies: A Phase 1 Dose Escalation and Expansion Trial. Nat. Med. 2020, 26, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Ramakrishna, S.; Nguyen, S.; Fountaine, T.J.; Ponduri, A.; Stetler-Stevenson, M.; Yuan, C.M.; Haso, W.; Shern, J.F.; Shah, N.N.; et al. Preclinical Development of Bivalent Chimeric Antigen Receptors Targeting Both CD19 and CD22. Mol. Ther. Oncol. 2018, 11, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, J.Y.; Patel, S.; Muffly, L.; Hossain, N.M.; Oak, J.; Baird, J.H.; Frank, M.J.; Shiraz, P.; Sahaf, B.; Craig, J.; et al. CAR T Cells with Dual Targeting of CD19 and CD22 in Adult Patients with Recurrent or Refractory B Cell Malignancies: A Phase 1 Trial. Nat. Med. 2021, 27, 1419–1431. [Google Scholar] [CrossRef]
- Cordoba, S.; Onuoha, S.; Thomas, S.; Pignataro, D.S.; Hough, R.; Ghorashian, S.; Vora, A.; Bonney, D.; Veys, P.; Rao, K.; et al. CAR T Cells with Dual Targeting of CD19 and CD22 in Pediatric and Young Adult Patients with Relapsed or Refractory B Cell Acute Lymphoblastic Leukemia: A Phase 1 Trial. Nat. Med. 2021, 27, 1797–1805. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Highfill, S.L.; Walsh, Z.; Nguyen, S.M.; Lei, H.; Shern, J.F.; Qin, H.; Kraft, I.L.; Stetler-Stevenson, M.; Yuan, C.M.; et al. Modulation of Target Antigen Density Improves CAR T-Cell Functionality and Persistence. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5329–5341. [Google Scholar] [CrossRef] [Green Version]
- Damm, J.K.; Gordon, S.; Ehinger, M.; Jerkeman, M.; Gullberg, U.; Hultquist, A.; Drott, K. Pharmacologically Relevant Doses of Valproate Upregulate CD20 Expression in Three Diffuse Large B-Cell Lymphoma Patients in Vivo. Exp. Hematol. Oncol. 2015, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Bobrowicz, M.; Dwojak, M.; Pyrzynska, B.; Stachura, J.; Muchowicz, A.; Berthel, E.; Dalla-Venezia, N.; Kozikowski, M.; Siernicka, M.; Miazek, N.; et al. HDAC6 Inhibition Upregulates CD20 Levels and Increases the Efficacy of Anti-CD20 Monoclonal Antibodies. Blood 2017, 130, 1628–1638. [Google Scholar] [CrossRef] [Green Version]
- Scialdone, A.; Hasni, M.S.; Damm, J.K.; Lennartsson, A.; Gullberg, U.; Drott, K. The HDAC Inhibitor Valproate Induces a Bivalent Status of the CD20 Promoter in CLL Patients Suggesting Distinct Epigenetic Regulation of CD20 Expression in CLL in Vivo. Oncotarget 2017, 8, 37409–37422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poorebrahim, M.; Mohammadkhani, N.; Mahmoudi, R.; Gholizadeh, M.; Fakhr, E.; Cid-Arregui, A. TCR-like CARs and TCR-CARs Targeting Neoepitopes: An Emerging Potential. Cancer Gene Ther. 2021, 28, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Raskin, S.; Van Pelt, S.; Toner, K.; Balakrishnan, P.B.; Dave, H.; Bollard, C.M.; Yvon, E. Novel TCR-like CAR-T Cells Targeting an HLA * 0201-Restricted SSX2 Epitope Display Strong Activity against Acute Myeloid Leukemia. Molecular therapy. Methods Clin. Dev. 2021, 23, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Dragon, A.C.; Zimmermann, K.; Nerreter, T.; Sandfort, D.; Lahrberg, J.; Klöß, S.; Kloth, C.; Mangare, C.; Bonifacius, A.; Tischer-Zimmermann, S.; et al. CAR-T Cells and TRUCKs That Recognize an EBNA-3C-Derived Epitope Presented on HLA-B*35 Control Epstein-Barr Virus-Associated Lymphoproliferation. J. Immunother. Cancer 2020, 8, 736. [Google Scholar] [CrossRef]


{kind=link}
| Generic Name and Brand Name | Indication | Target (Single Chain Variable Fragment, scFv) | Signaling Domain/ Hinge and TM | Gene Editing Vector | First FDA/EMA Registration Date | Approval-Based Clinical Trials, Number of Participants | CR or ORR Rate | Long-Term Follow-Up Studies, Number of Participants | Survival (Median Follow-Up in Years) |
|---|---|---|---|---|---|---|---|---|---|
| Tisagenlecleucel (KYMRIAH) | R/R pediatric and YA B-ALL | CD19 (FMC63) | 4-1BB-CD3ζ/CD8α | Lentivirus | August 2017/ August 2018 | ELIANA, n = 75 [NCT02435849] [12] | CR 60% | NCT01593696, n = 50 [13] | median OS 10.5 months (4.8 years) |
| R/R adult DLBCL | May 2018/ August 2018 | JULIET, n = 93 [NCT02445248] [14] | CR 40% | JULIET follow-up, n = 115 [15] | median OS 11.1 months (3.4 years) | ||||
| Axicabtagene Ciloleucel (YESCARTA) | R/R adult DLBCL (including DLBCL arising from FL) | CD19 (FMC63) | CD28-CD3ζ/CD28 | Retrovirus | October 2017/ August 2018 | ZUMA-1, n = 101 [NCT02348216] [16] | CR 54% | ZUMA-1 follow up, n = 101 [17] | median OS 25.8 months (4 years) |
| R/R adult PMBCL | |||||||||
| R/R adult FL | Mar 2021/ not yet registered | ZUMA-5, n = 84 [NCT03105336] [18] | CR 79% | not yet available | not yet available | ||||
| Brexucabtagene Autoleucel (TECARTUS) | R/R adult MCL | CD19 (FMC63) | CD28-CD3ζ/CD28 | Retrovirus | July 2020/ December 2020 | ZUMA-2, n = 60 (efficacy group) [NCT02601313] [19] | CR 67% | not yet available | not yet available |
| R/R adult B-ALL | October 2021/ not yet registered | ZUMA-3, n = 55 [NCT02614066] [20] | CR 56% | not yet available | not yet available | ||||
| Lisocabtagene Maraleucel (BREYANZI) | R/R adult DLBCL (including DLBCL arising from indolent lymphoma) | CD19 (FMC63) | 4-1BB-CD3ζ/IgG4 and CD28 | Lentivirus | May 2021/ pending registration | TRANCEND NHL 001, n = 256 [NCT02631044] [21] | CR 53% | TRANCEND NHL 001 follow-up, n = 257 [22] | median OS 27.3 months (2.4 year) |
| R/R adult PMBCL | |||||||||
| R/R FL3B | |||||||||
| Idecabtagene Vicleucel (ABECMA) | R/R MM | BCMA | 4-1BB-CD3ζ/CD8α | Lentivirus | Mar 2021/ August 2021 | KarMMa, n = 100 (efficacy group) [NCT03361748] [23,24] | ORR 72% sCR 28% | not yet available | not yet available |
| Targeted Molecule | Physiological Functions | Expression in Non-Malignant Cells [References] | Targeted Disease | References or Numbers of Clinical Trials |
|---|---|---|---|---|
| pre-clinical studies | ||||
| CD23 | regulation of IgE responses | a subset of T and B cells, monocytes, leukocytes, follicular DCs, intestinal epithelial cells, bone marrow stromal cells [129,130,131,132,133,134,135] | CLL | [136,137] |
| CD32B | regulation of B cell activation, antibody production | B cells, DCs, granulocytes, liver endothelial cells, airway smooth muscle cells [138,139,140] | CLL | [141] |
| CD70 | T cell activation and proliferation (cytokine, CD27 ligand) | subsets of activated T cells, B cells, NK cells, mature DCs, epithelial cells of the thymic medulla [142,143] | B cell lymphoma | [144,145] |
| CD72 | B cell proliferation, B cell differentiation, negative regulation of BCR signaling | B cells [146] | B-ALL | [147] |
| CD133 | unclear, a marker of undifferentiated cells | hematopoietic stem and progenitor cells [148] | B-ALL | [149,150] |
| FcμR | IgM receptor | subsets of B cells, T cells, NK cells [151] | CLL | [152] |
| Siglec-6 | inhibition of immune response | memory B cells, exhausted B cells, placenta, mast cells [153,154,155] | CLL | [156] |
| TSLPR | T and B cell development | DCs, monocytes [157] | B-ALL | [158] |
| clinical trials | ||||
| BAFF-R | promotion of B cell survival | B cells (except plasma cells), memory T cells [159] | B-ALL | NCT04690595 |
| CD20 | B cell proliferation, B cell differentiation | pre-B cells and mature B cells [160,161] | CLL, B cell lymphoma | NCT04169932 |
| NCT04030195 | ||||
| NCT03664635 | ||||
| CD22 | regulation of BCR signaling, B cell migration | B cells [162,163] | B-ALL, B cell lymphoma | NCT04546906 |
| NCT04088864 | ||||
| NCT04088890 | ||||
| NCT04007978 | ||||
| NCT03262298 | ||||
| NCT04340167 | ||||
| NCT03244306 | ||||
| NCT04571138 | ||||
| NCT02650414 | ||||
| NCT02315612 | ||||
| NCT04150497 | ||||
| NCT03620058 | ||||
| CD30 | possibly T cell survival and/or establishment of memory responses | a subset of activated T and B lymphocytes [164,165] | DLBCL, Hodgkin lymphoma | NCT04526834 |
| NCT03049449 | ||||
| CD37 | regulation of humoral and cellular immune responses | mature B cells [166] | CLL, B cell lymphoma | NCT04136275 |
| CD38 | cell adhesion and migration, generation of nucleotide metabolites | T cells, B cells, NK cells, plasma cells, monocytes [167] | B-ALL, MM | NCT03754764 |
| NCT03464916 | ||||
| CD70 | T cell activation and proliferation (cytokine, CD27 ligand) | APCs, activated T and B cells [142,143] | MM | NCT04662294 |
| CD79B | promotion of B cell survival | B cells [168] | B-ALL, B cell NHL | NCT04609241 |
| CD123 | IL-3 signal transmission, regulation of hematopoiesis | Basophils, plasmacytoid DCs [169,170] | B-ALL | NCT04318678 |
| CD138 | cell adhesion, endocytosis | epithelial cells, hepatocytes, plasma cells [171,172] | MM | NCT03672318 |
| GPRC5D | hard keratinization | epithelial cells of the hair follicles, plasma cells, B cells [173,174,175] | MM | NCT05016778 |
| LMP1 | induction and maintenance of virus latency by modulation of host’s immune responses | EBV-infected cells [176] | B cell lymphoma | NCT04657965 |
| ROR1 | cell differentiation, proliferation and survival | various cells during embryogenesis [177] | CLL, MCL, ALL | NCT02706392 |
| SLAMF7 | inhibition of proinflammatory immune responses | B cells, T cells, DCs, NK cells, monocytes [178,179] | MM | NCT04499339 |
| NCT03958656 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marhelava, K.; Krawczyk, M.; Firczuk, M.; Fidyt, K. CAR-T Cells Shoot for New Targets: Novel Approaches to Boost Adoptive Cell Therapy for B Cell-Derived Malignancies. Cells 2022, 11, 1804. https://doi.org/10.3390/cells11111804
Marhelava K, Krawczyk M, Firczuk M, Fidyt K. CAR-T Cells Shoot for New Targets: Novel Approaches to Boost Adoptive Cell Therapy for B Cell-Derived Malignancies. Cells. 2022; 11(11):1804. https://doi.org/10.3390/cells11111804
Chicago/Turabian StyleMarhelava, Katsiaryna, Marta Krawczyk, Malgorzata Firczuk, and Klaudyna Fidyt. 2022. "CAR-T Cells Shoot for New Targets: Novel Approaches to Boost Adoptive Cell Therapy for B Cell-Derived Malignancies" Cells 11, no. 11: 1804. https://doi.org/10.3390/cells11111804