
A Novel Role of the TRPM4 Ion Channel in Exocytosis


Abstract
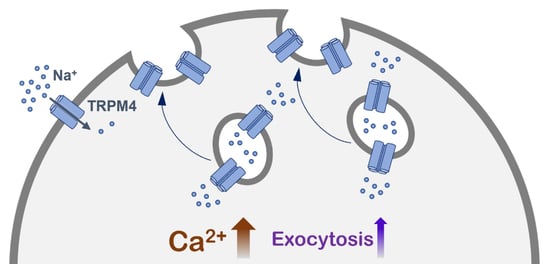
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Transient Transfection of HCT116 Cells with Tetanus Toxin Light Chain
2.3. Electrophysiology
2.4. Biotinylation Assay
3. Results
3.1. Exocytosis in HCT116 Cells Is Dependent on TRPM4
3.2. TRPM4-Dependent Exocytosis in Prostate Cancer Cells
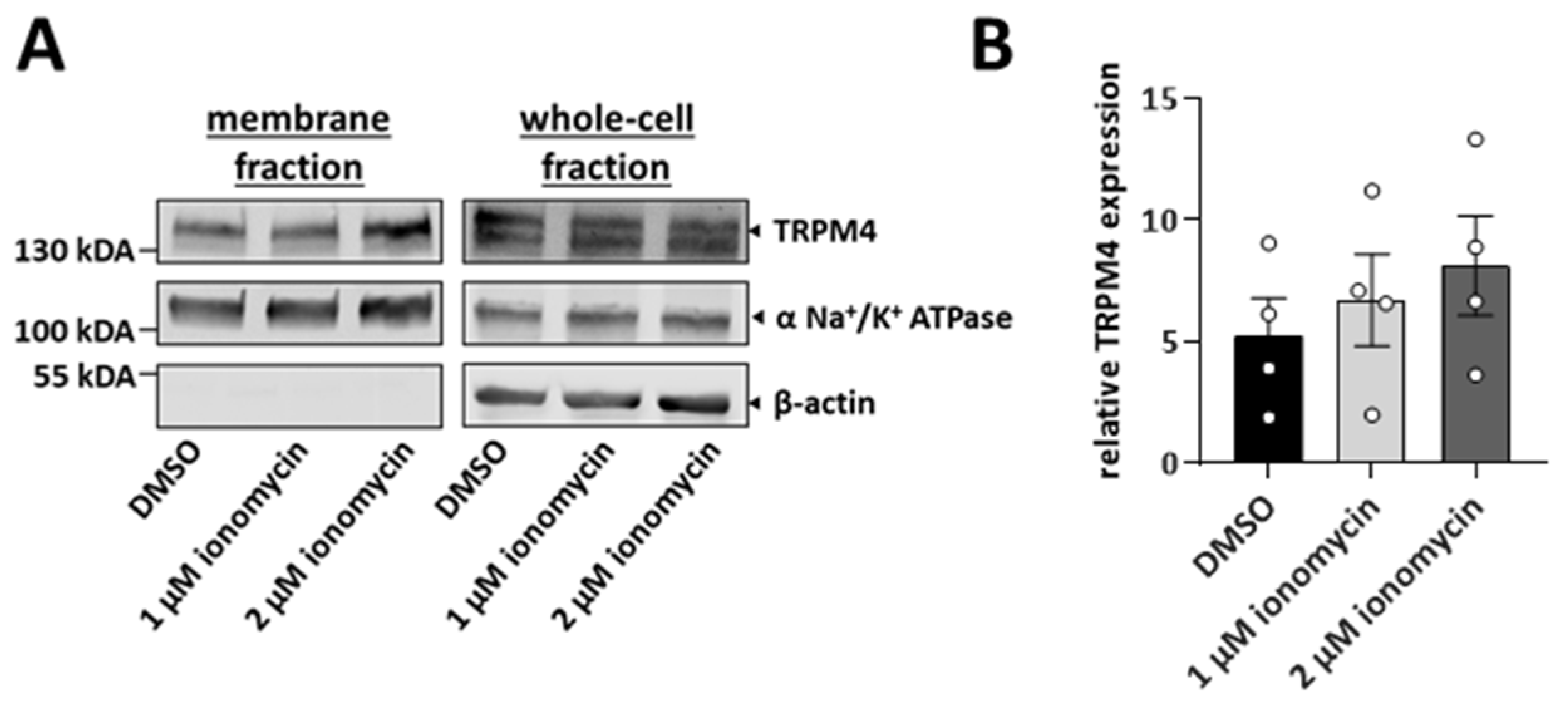
3.3. TRPM4 Is Delivered to the Plasma Membrane upon the Increase in Intracellular Calcium
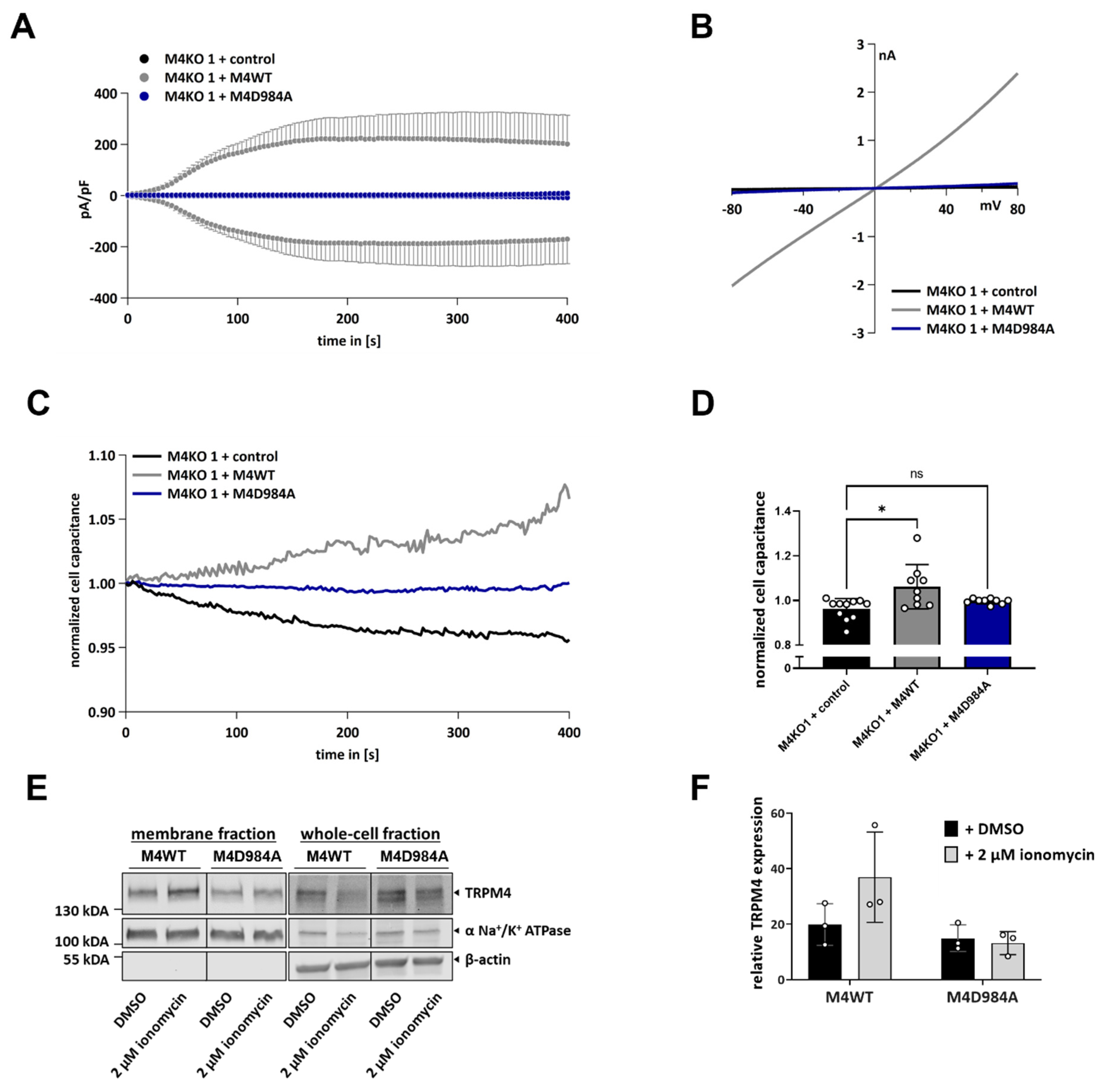
3.4. TRPM4 Ion Conductivity Plays a Role in Exocytosis
3.5. TRPM4-Dependent Exocytosis Is Mediated by SNARE Proteins
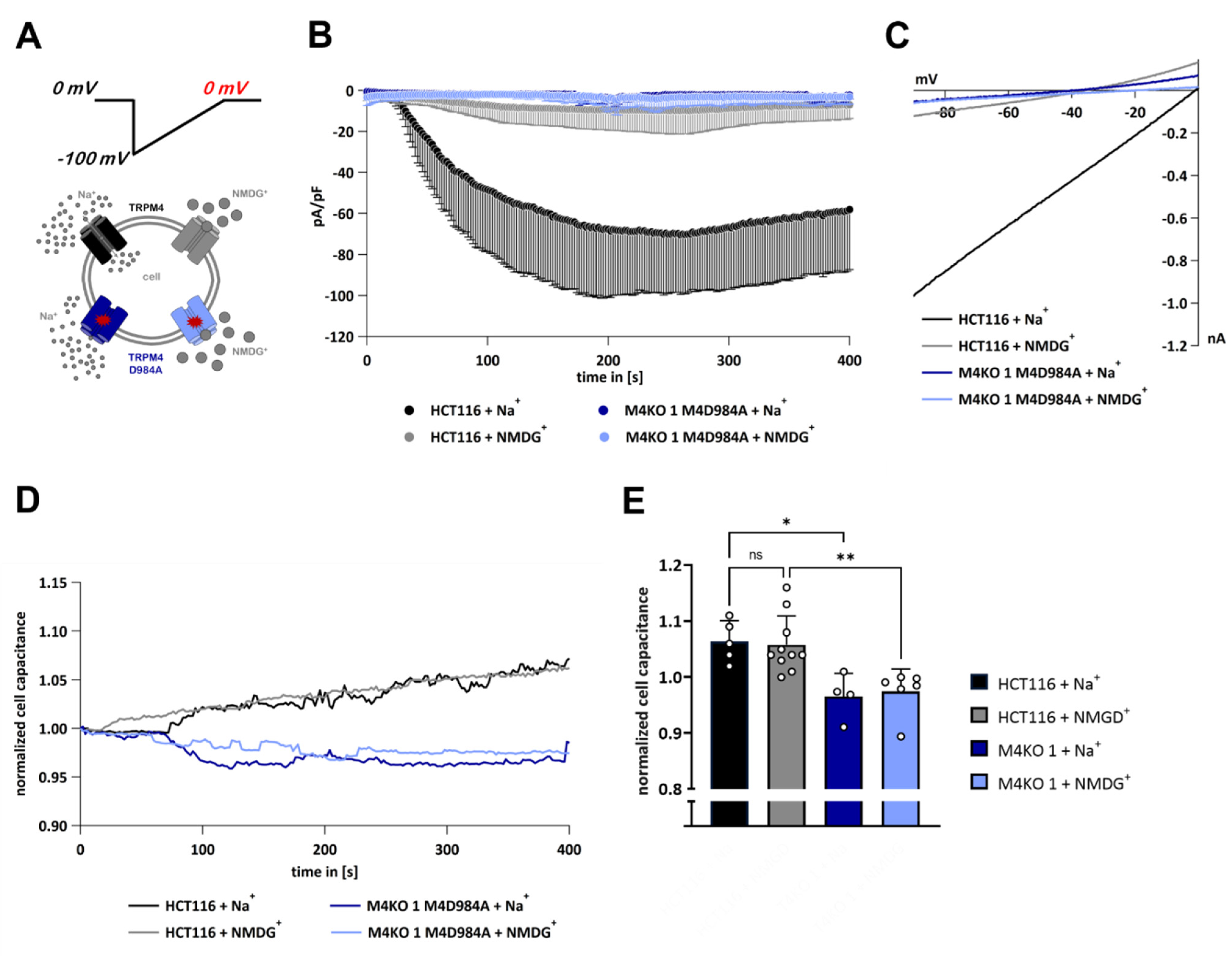
3.6. TRPM4-Dependent Exocytosis Does Not Require Plasma Membrane TRPM4 Conductivity
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Launay, P.; Fleig, A.; Perraud, A.L.; Scharenberg, A.M.; Penner, R.; Kinet, J.P. TRPM4 Is a Ca2+-Activated Nonselective Cation Channel Mediating Cell Membrane Depolarization. Cell 2002, 109, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Nilius, B.; Prenen, J.; Janssens, A.; Owsianik, G.; Wang, C.; Zhu, M.X.; Voets, T. The Selectivity Filter of the Cation Channel TRPM4. J. Biol. Chem. 2005, 280, 22899–22906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Launay, P.; Cheng, H.; Srivatsan, S.; Penner, R.; Fleig, A.; Kinet, J.P. TRPM4 Regulates Calcium Oscillations after T Cell Activation. Science 2004, 306, 1374–1377. [Google Scholar] [CrossRef] [Green Version]
- Holzmann, C.; Kappel, S.; Kilch, T.; Jochum, M.M.; Urban, S.K.; Jung, V.; Stöckle, M.; Rother, K.; Greiner, M.; Peinelt, C. Transient Receptor Potential Melastatin 4 Channel Contributes to Migration of Androgen-Insensitive Prostate Cancer Cells. Oncotarget 2015, 6, 41783–41793. [Google Scholar] [CrossRef] [PubMed]
- Kecskés, M.; Jacobs, G.; Kerselaers, S.; Syam, N.; Menigoz, A.; Vangheluwe, P.; Freichel, M.; Flockerzi, V.; Voets, T.; Vennekens, R. The Ca2+-Activated Cation Channel TRPM4 Is a Negative Regulator of Angiotensin II-Induced Cardiac Hypertrophy. Basic Res. Cardiol. 2015, 110, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilch, T.; Kappel, S.; Peinelt, C. Regulation of Ca2+ Signaling in Prostate Cancer Cells. Channels 2016, 10, 170–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbet, G.; Demion, M.; Moura, I.C.; Serafini, N.; Léger, T.; Vrtovsnik, F.; Monteiro, R.C.; Guinamard, R.; Kinet, J.-P.; Launay, P. The Calcium-Activated Nonselective Cation Channel TRPM4 Is Essential for the Migration but Not the Maturation of Dendritic Cells. Nat. Immunol. 2008, 9, 1148–1156. [Google Scholar] [CrossRef] [Green Version]
- Ngoc Tran, T.D.; Stovall, K.E.; Suantawee, T.; Hu, Y.; Yao, S.; Yang, L.-J.; Adisakwattana, S.; Cheng, H. Transient Receptor Potential Melastatin 4 Channel Is Required for Rat Dental Pulp Stem Cell Proliferation and Survival. Cell Prolif. 2017, 50, e12360. [Google Scholar] [CrossRef]
- Diszházi, G.; Magyar, Z.É.; Lisztes, E.; Tóth-Molnár, E.; Nánási, P.P.; Vennekens, R.; Tóth, B.I.; Almássy, J. TRPM4 Links Calcium Signaling to Membrane Potential in Pancreatic Acinar Cells. J. Biol. Chem. 2021, 297, 101015. [Google Scholar] [CrossRef]
- Weber, K.S.; Hildner, K.; Murphy, K.M.; Allen, P.M. Trpm4 Differentially Regulates Th1 and Th2 Function by Altering Calcium Signaling and NFAT Localization. J. Immunol. 2010, 185, 2836–2846. [Google Scholar] [CrossRef]
- Vennekens, R.; Olausson, J.; Meissner, M.; Bloch, W.; Mathar, I.; Philipp, S.E.; Schmitz, F.; Weissgerber, P.; Nilius, B.; Flockerzi, V.; et al. Increased IgE-Dependent Mast Cell Activation and Anaphylactic Responses in Mice Lacking the Calcium-Activated Nonselective Cation Channel TRPM4. Nat. Immunol. 2007, 8, 312–320. [Google Scholar] [CrossRef] [Green Version]
- Cantero-Recasens, G.; Butnaru, C.M.; Brouwers, N.; Mitrovic, S.; Valverde, M.A.; Malhotra, V. Sodium Channel TRPM4 and Sodium/Calcium Exchangers (NCX) Cooperate in the Control of Ca2+-Induced Mucin Secretion from Goblet Cells. J. Biol. Chem. 2019, 294, 816–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fliegert, R.; Glassmeier, G.; Schmid, F.; Cornils, K.; Genisyuerek, S.; Harneit, A.; Schwarz, J.R.; Guse, A.H. Modulation of Ca2+ Entry and Plasma Membrane Potential by Human TRPM4b. FEBS J. 2007, 274, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Sagredo, A.I.; Sagredo, E.A.; Cappelli, C.; Báez, P.; Andaur, R.E.; Blanco, C.; Tapia, J.C.; Echeverría, C.; Cerda, O.; Stutzin, A.; et al. TRPM4 Regulates Akt/GSK3-β Activity and Enhances β-Catenin Signaling and Cell Proliferation in Prostate Cancer Cells. Mol. Oncol. 2018, 12, 151–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palladino, A.; Papa, A.A.; Petillo, R.; Scutifero, M.; Morra, S.; Passamano, L.; Nigro, V.; Politano, L. The Role of TRPM4 Gene Mutations in Causing Familial Progressive Cardiac Conduction Disease: A Further Contribution. Genes 2022, 13, 258. [Google Scholar] [CrossRef]
- Ozhathil, L.C.; Rougier, J.-S.; Arullampalam, P.; Essers, M.C.; Ross-Kaschitza, D.; Abriel, H. Deletion of Trpm4 Alters the Function of the Na(v)1.5 Channel in Murine Cardiac Myocytes. Int. J. Mol. Sci. 2021, 22, 3401. [Google Scholar] [CrossRef]
- Dienes, C.; Kovács, Z.M.; Hézső, T.; Almássy, J.; Magyar, J.; Bányász, T.; Nánási, P.P.; Horváth, B.; Szentandrássy, N. Pharmacological Modulation and (Patho)Physiological Roles of TRPM4 Channel-Part 2: TRPM4 in Health and Disease. Pharmaceuticals 2021, 15, 40. [Google Scholar] [CrossRef]
- Schattling, B.; Steinbach, K.; Thies, E.; Kruse, M.; Menigoz, A.; Ufer, F.; Flockerzi, V.; Brück, W.; Pongs, O.; Vennekens, R.; et al. TRPM4 Cation Channel Mediates Axonal and Neuronal Degeneration in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. Nat. Med. 2012, 18, 1805–1811. [Google Scholar] [CrossRef]
- Wang, H.; Xu, Z.; Lee, B.H.; Vu, S.; Hu, L.; Lee, M.; Bu, D.; Cao, X.; Hwang, S.; Yang, Y. Gain-of-Function Mutations in TRPM4 Activation Gate Cause Progressive Symmetric Erythrokeratodermia. J. Investig. Dermatol. 2019, 139, 1089–1097. [Google Scholar] [CrossRef]
- Zhu, L.; Miao, B.; Dymerska, D.; Kuswik, M.; Bueno-Martínez, E.; Sanoguera-Miralles, L.; Velasco, E.A.; Paramasivam, N.; Schlesner, M.; Kumar, A.; et al. Germline Variants of CYBA and TRPM4 Predispose to Familial Colorectal Cancer. Cancers 2022, 14, 670. [Google Scholar] [CrossRef]
- Borgström, A.; Peinelt, C.; Stokłosa, P. TRPM4 in Cancer—A New Potential Drug Target. Biomolecules 2021, 11, 229. [Google Scholar] [CrossRef]
- Borgström, A.; Hauert, B.; Kappel, S.; Zoni, E.; Kiener, M.; Stokłosa, P.; Baur, R.; Spahn, M.; Kruithof-de Julio, M.; Peinelt, C. Small Molecular Inhibitors Block TRPM4 Currents in Prostate Cancer Cells, with Limited Impact on Cancer Hallmark Functions. J. Mol. Biol. 2021, 433, 166665. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.D.; Soldini, D.; Jung, M.; Dietrich, D.; Stephan, C.; Jung, K.; Dietel, M.; Vainer, B.; Kristiansen, G. TRPM4 Protein Expression in Prostate Cancer: A Novel Tissue Biomarker Associated with Risk of Biochemical Recurrence Following Radical Prostatectomy. Virchows Arch. 2016, 468, 345–355. [Google Scholar] [CrossRef] [Green Version]
- Armisén, R.; Marcelain, K.; Simon, F.; Tapia, J.C.; Toro, J.; Quest, A.F.G.; Stutzin, A. TRPM4 Enhances Cell Proliferation through Up-Regulation of the β-Catenin Signaling Pathway. J. Cell. Physiol. 2011, 226, 103–109. [Google Scholar] [CrossRef]
- Hong, X.; Yu, J.-J. MicroRNA-150 Suppresses Epithelial-Mesenchymal Transition, Invasion, and Metastasis in Prostate Cancer through the TRPM4-Mediated β-Catenin Signaling Pathway. Am. J. Physiol. Cell Physiol. 2019, 316, C463–C480. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Hussain, F.A. TRPM4 Is Overexpressed in Breast Cancer Associated with Estrogen Response and Epithelial-Mesenchymal Transition Gene Sets. PLoS ONE 2020, 15, e0233884. [Google Scholar] [CrossRef] [PubMed]
- Verigos, J.; Kordias, D.; Papadaki, S.; Magklara, A. Transcriptional Profiling of Tumorspheres Reveals TRPM4 as a Novel Stemness Regulator in Breast Cancer. Biomedicines 2021, 9, 1368. [Google Scholar] [CrossRef]
- Stokłosa, P.; Borgström, A.; Kappel, S.; Peinelt, C. TRP Channels in Digestive Tract Cancers. Int. J. Mol. Sci. 2020, 21, 1877. [Google Scholar] [CrossRef] [Green Version]
- Stokłosa, P.; Borgström, A.; Hauert, B.; Baur, R.; Peinelt, C. Investigation of Novel Small Molecular TRPM4 Inhibitors in Colorectal Cancer Cells. Cancers 2021, 13, 5400. [Google Scholar] [CrossRef]
- Sagredo, A.I.; Sagredo, E.A.; Pola, V.; Echeverría, C.; Andaur, R.; Michea, L.; Stutzin, A.; Simon, F.; Marcelain, K.; Armisén, R. TRPM4 Channel Is Involved in Regulating Epithelial to Mesenchymal Transition, Migration, and Invasion of Prostate Cancer Cell Lines. J. Cell. Physiol. 2019, 234, 2037–2050. [Google Scholar] [CrossRef]
- Loo, S.K.; Ch’ng, E.S.; Md Salleh, M.S.; Banham, A.H.; Pedersen, L.M.; Møller, M.B.; Green, T.M.; Wong, K.K. TRPM4 Expression Is Associated with Activated B Cell Subtype and Poor Survival in Diffuse Large B Cell Lymphoma. Histopathology 2017, 71, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Kappel, S.; Stokłosa, P.; Hauert, B.; Ross-Kaschitza, D.; Borgström, A.; Baur, R.; Galván, J.A.; Zlobec, I.; Peinelt, C. TRPM4 Is Highly Expressed in Human Colorectal Tumor Buds and Contributes to Proliferation, Cell Cycle, and Invasion of Colorectal Cancer Cells. Mol. Oncol. 2019, 13, 2393–2405. [Google Scholar] [CrossRef] [PubMed]
- Çoban, G.; Yildiz, P.; Doğan, B.; Şahin, N.; Gücin, Z. Expression of Transient Receptor Potential Melastatin 4 in Differential Diagnosis of Eosinophilic Renal Tumors. Mol. Clin. Oncol. 2021, 15, 230. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Owsianik, G.; Freichel, M.; Flockerzi, V.; Nilius, B.; Vennekens, R. TRPM4 Regulates Migration of Mast Cells in Mice. Cell Calcium 2009, 45, 226–232. [Google Scholar] [CrossRef]
- Bianchi, B.; Smith, P.A.; Abriel, H. The Ion Channel TRPM4 in Murine Experimental Autoimmune Encephalomyelitis and in a Model of Glutamate-Induced Neuronal Degeneration. Mol. Brain 2018, 11, 41. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Segal, A.; Voets, T. Distinct Modes of Perimembrane TRP Channel Turnover Revealed by TIR-FRAP. Sci. Rep. 2014, 4, 7111. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Beck, A.; Launay, P.; Gross, S.A.; Stokes, A.J.; Kinet, J.-P.; Fleig, A.; Penner, R. TRPM4 Controls Insulin Secretion in Pancreatic Beta-Cells. Cell Calcium 2007, 41, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Crnich, R.; Amberg, G.C.; Leo, M.D.; Gonzales, A.L.; Tamkun, M.M.; Jaggar, J.H.; Earley, S. Vasoconstriction Resulting from Dynamic Membrane Trafficking of TRPM4 in Vascular Smooth Muscle Cells. Am. J. Physiol. Cell Physiol. 2010, 299, C682–C694. [Google Scholar] [CrossRef] [Green Version]
- Rixecker, T.; Mathar, I.; Medert, R.; Mannebach, S.; Pfeifer, A.; Lipp, P.; Tsvilovskyy, V.; Freichel, M. TRPM4-Mediated Control of FcεRI-Evoked Ca2+ Elevation Comprises Enhanced Plasmalemmal Trafficking of TRPM4 Channels in Connective Tissue Type Mast Cells. Sci. Rep. 2016, 6, 32981. [Google Scholar] [CrossRef] [Green Version]
- Thévenod, F. Ion Channels in Secretory Granules of the Pancreas and Their Role in Exocytosis and Release of Secretory Proteins. Am. J. Physiol. Physiol. 2002, 283, C651–C672. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Groos, S.; Riederer, B.; Feng, Z.; Krabbenhöft, A.; Smolka, A.; Seidler, U. KCNQ1 Is the Luminal K+ Recycling Channel during Stimulation of Gastric Acid Secretion. J. Physiol. 2009, 587 Pt 15, 3955–3965. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Groos, S.; Riederer, B.; Feng, Z.; Krabbenhöft, A.; Manns, M.P.; Smolka, A.; Hagen, S.J.; Neusch, C.; Seidler, U. Kir4.1 Channel Expression Is Essential for Parietal Cell Control of Acid Secretion. J. Biol. Chem. 2011, 286, 14120–14128. [Google Scholar] [CrossRef] [Green Version]
- Dernick, G.; Alvarez de Toledo, G.; Lindau, M. Exocytosis of Single Chromaffin Granules in Cell-Free inside-out Membrane Patches. Nat. Cell Biol. 2003, 5, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Sterea, A.M.; Almasi, S.; El Hiani, Y. The Hidden Potential of Lysosomal Ion Channels: A New Era of Oncogenes. Cell Calcium 2018, 72, 91–103. [Google Scholar] [CrossRef]
- Li, P.; Gu, M.; Xu, H. Lysosomal Ion Channels as Decoders of Cellular Signals. Trends Biochem. Sci. 2019, 44, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Grimm, C.; Chen, C.-C.; Wahl-Schott, C.; Biel, M. Two-Pore Channels: Catalyzers of Endolysosomal Transport and Function. Front. Pharmacol. 2017, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Gerndt, S.; Chen, C.-C.; Chao, Y.-K.; Yuan, Y.; Burgstaller, S.; Scotto Rosato, A.; Krogsaeter, E.; Urban, N.; Jacob, K.; Nguyen, O.N.P.; et al. Agonist-Mediated Switching of Ion Selectivity in TPC2 Differentially Promotes Lysosomal Function. eLife 2020, 9, e54712. [Google Scholar] [CrossRef]
- Krapivinsky, G.; Mochida, S.; Krapivinsky, L.; Cibulsky, S.M.; Clapham, D.E. The TRPM7 Ion Channel Functions in Cholinergic Synaptic Vesicles and Affects Transmitter Release. Neuron 2006, 52, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Brauchi, S.; Krapivinsky, G.; Krapivinsky, L.; Clapham, D.E. TRPM7 Facilitates Cholinergic Vesicle Fusion with the Plasma Membrane. Proc. Natl. Acad. Sci. USA 2008, 105, 8304–8308. [Google Scholar] [CrossRef] [Green Version]
- Samie, M.; Wang, X.; Zhang, X.; Goschka, A.; Li, X.; Cheng, X.; Gregg, E.; Azar, M.; Zhuo, Y.; Garrity, A.G.; et al. A TRP Channel in the Lysosome Regulates Large Particle Phagocytosis via Focal Exocytosis. Dev. Cell 2013, 26, 511–524. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Pinto, S.; Danglot, L.; Vandewauw, I.; Segal, A.; Van Ranst, N.; Benoit, M.; Janssens, A.; Vennekens, R.; Vanden Berghe, P.; et al. VAMP7 Regulates Constitutive Membrane Incorporation of the Cold-Activated Channel TRPM8. Nat. Commun. 2016, 7, 10489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camprubí-Robles, M.; Planells-Cases, R.; Ferrer-Montiel, A. Differential Contribution of SNARE-Dependent Exocytosis to Inflammatory Potentiation of TRPV1 in Nociceptors. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 3722–3733. [Google Scholar] [CrossRef] [PubMed]
- Jahn, R.; Fasshauer, D. Molecular Machines Governing Exocytosis of Synaptic Vesicles. Nature 2012, 490, 201–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappel, S.; Ross-Kaschitza, D.; Hauert, B.; Rother, K.; Peinelt, C. P53 Alters Intracellular Ca2+ Signaling through Regulation of TRPM4. Cell Calcium 2022, 104, 102591. [Google Scholar] [CrossRef]
- Rother, K.; Johne, C.; Spiesbach, K.; Haugwitz, U.; Tschöp, K.; Wasner, M.; Klein-Hitpass, L.; Möröy, T.; Mössner, J.; Engeland, K. Identification of Tcf-4 as a Transcriptional Target of P53 Signalling. Oncogene 2004, 23, 3376–3384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Castle, D. Regulation of Fusion Pore Closure and Compound Exocytosis in Neuroendocrine PC12 Cells by SCAMP1. Traffic 2011, 12, 600–614. [Google Scholar] [CrossRef] [PubMed]
- Ozhathil, L.C.; Delalande, C.; Bianchi, B.; Nemeth, G.; Kappel, S.; Thomet, U.; Ross-Kaschitza, D.; Simonin, C.; Rubin, M.; Gertsch, J.; et al. Identification of Potent and Selective Small Molecule Inhibitors of the Cation Channel TRPM4. Br. J. Pharmacol. 2018, 175, 2504–2519. [Google Scholar] [CrossRef] [Green Version]
- Carrillo, L.; Cucu, B.; Bandmann, V.; Homann, U.; Hertel, B.; Hillmer, S.; Thiel, G.; Bertl, A. High-Resolution Membrane Capacitance Measurements for Studying Endocytosis and Exocytosis in Yeast. Traffic 2015, 16, 760–772. [Google Scholar] [CrossRef]
- Houy, S.; Martins, J.S.; Mohrmann, R.; Sørensen, J.B. Measurements of Exocytosis by Capacitance Recordings and Calcium Uncaging in Mouse Adrenal Chromaffin Cells. Methods Mol. Biol. 2021, 2233, 233–251. [Google Scholar] [CrossRef]
- Barclay, J.W.; Morgan, A.; Burgoyne, R.D. Calcium-Dependent Regulation of Exocytosis. Cell Calcium 2005, 38, 343–353. [Google Scholar] [CrossRef]
- Sweeney, S.T.; Broadie, K.; Keane, J.; Niemann, H.; O’Kane, C.J. Targeted Expression of Tetanus Toxin Light Chain in Drosophila Specifically Eliminates Synaptic Transmission and Causes Behavioral Defects. Neuron 1995, 14, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.K.; Huynh, C.; Proux-Gillardeaux, V.; Galli, T.; Andrews, N.W. Identification of SNAREs Involved in Synaptotagmin VII-Regulated Lysosomal Exocytosis. J. Biol. Chem. 2004, 279, 20471–20479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, E.; White-Gilbertson, S.; van de Vlekkert, D.; Janke, L.; Moshiach, S.; Campos, Y.; Finkelstein, D.; Gomero, E.; Mosca, R.; Qiu, X.; et al. Regulated Lysosomal Exocytosis Mediates Cancer Progression. Sci. Adv. 2015, 1, e1500603. [Google Scholar] [CrossRef] [Green Version]
- Kajiho, H.; Kajiho, Y.; Frittoli, E.; Confalonieri, S.; Bertalot, G.; Viale, G.; Di Fiore, P.P.; Oldani, A.; Garre, M.; Beznoussenko, G.V.; et al. RAB2A Controls MT1-MMP Endocytic and E-cadherin Polarized Golgi Trafficking to Promote Invasive Breast Cancer Programs. EMBO Rep. 2016, 17, 1061–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; Feng, S.; Wu, B.; Guo, W. Polarized Exocytosis. Cold Spring Harb. Perspect. Biol. 2017, 9, a027870. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stokłosa, P.; Kappel, S.; Peinelt, C. A Novel Role of the TRPM4 Ion Channel in Exocytosis. Cells 2022, 11, 1793. https://doi.org/10.3390/cells11111793
Stokłosa P, Kappel S, Peinelt C. A Novel Role of the TRPM4 Ion Channel in Exocytosis. Cells. 2022; 11(11):1793. https://doi.org/10.3390/cells11111793
Chicago/Turabian StyleStokłosa, Paulina, Sven Kappel, and Christine Peinelt. 2022. "A Novel Role of the TRPM4 Ion Channel in Exocytosis" Cells 11, no. 11: 1793. https://doi.org/10.3390/cells11111793