
Cigarette Smoke Extract Disturbs Mitochondria-Regulated Airway Epithelial Cell Responses to Pneumococci

 , , , ,
, , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cigarette Smoke Extract (CSE) Preparation
2.3. Streptococcus Pneumoniae Culture
2.4. Experimental Design
2.5. MTT Assay
2.6. RealTime-Glo MT Assay
2.7. Intracellular Reactive Oxygen Species (ROS) Levels
2.8. FACS Staining for MitoSOX
2.9. Mitochondrial Membrane Potential Analysis
2.10. Immunoblotting
2.11. Immunofluorescence Staining
2.12. Mitochondrial Morphology (TEM Analysis)
2.13. Gene Expression Microarray
2.14. Statistical Analysis
3. Results
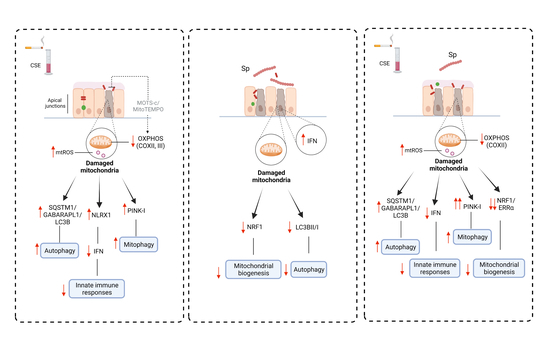
3.1. Optimization of the CSE-Induced Mitochondrial Dysfunction Model
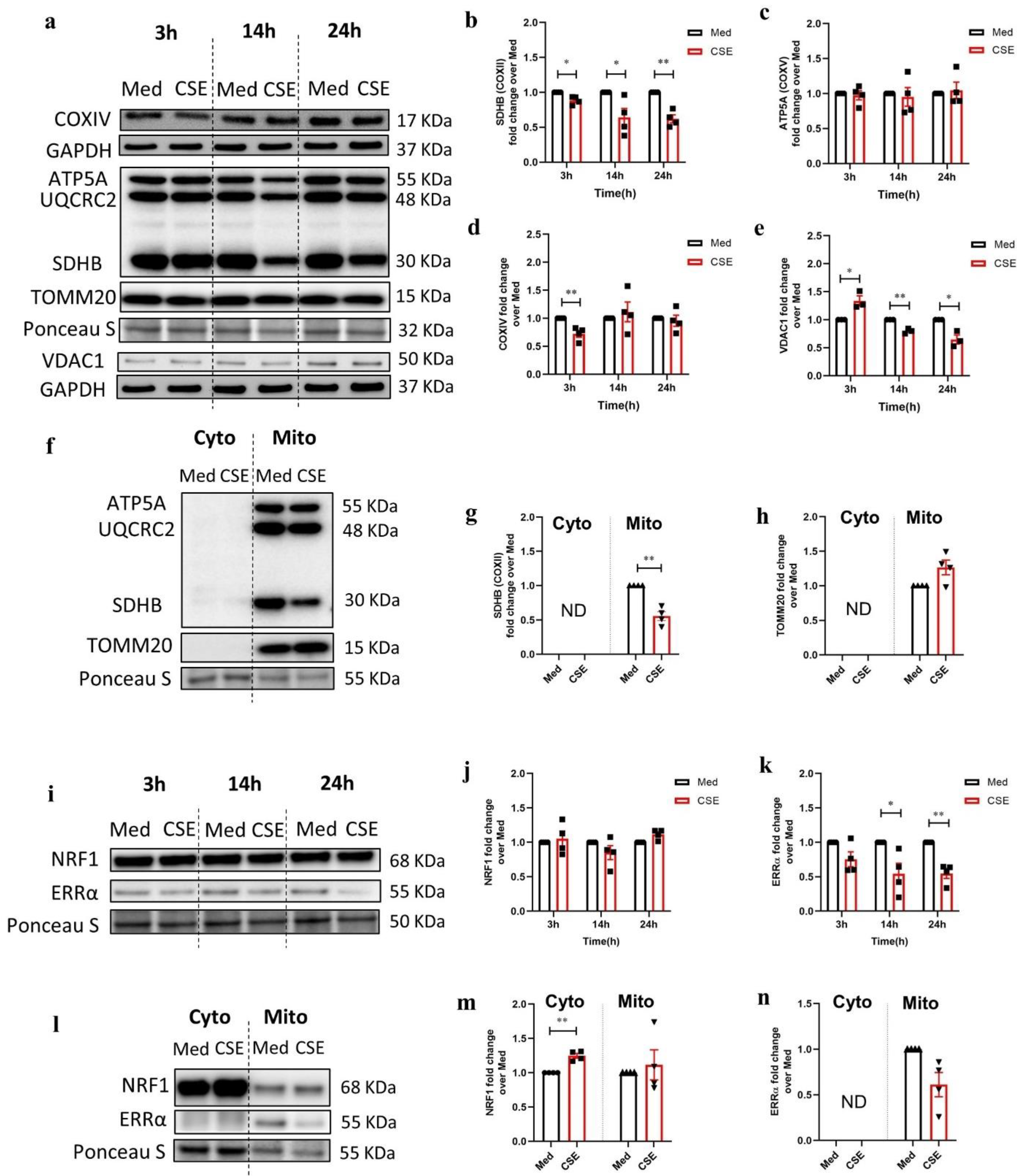
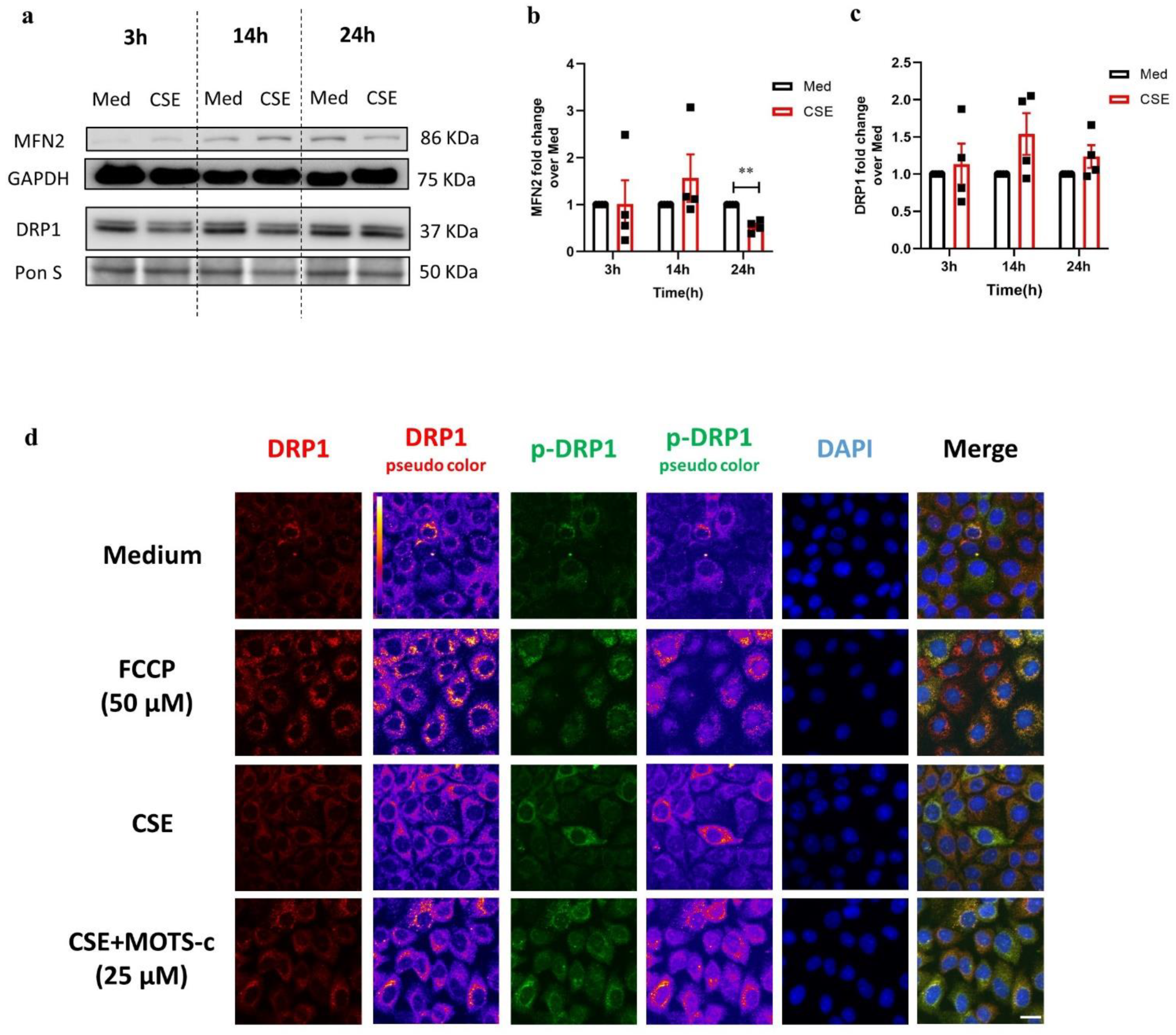
3.2. CSE Reduces Protein Expression of Key Regulators Involved in Mitochondrial Function in 16HBE Cells
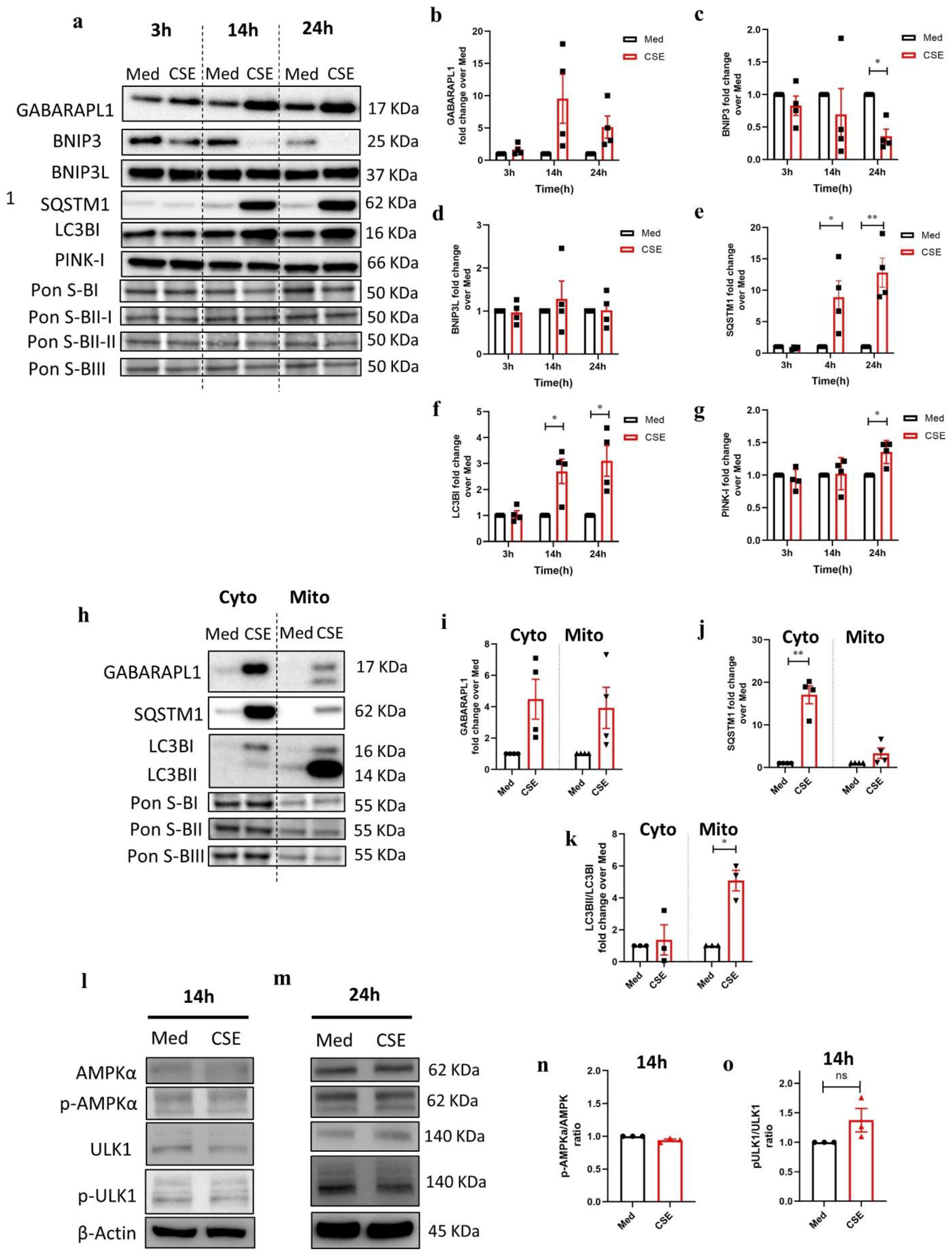
3.3. CSE Affects the Abundance of Regulators Associated with Mitochondrial Quality Control Processes
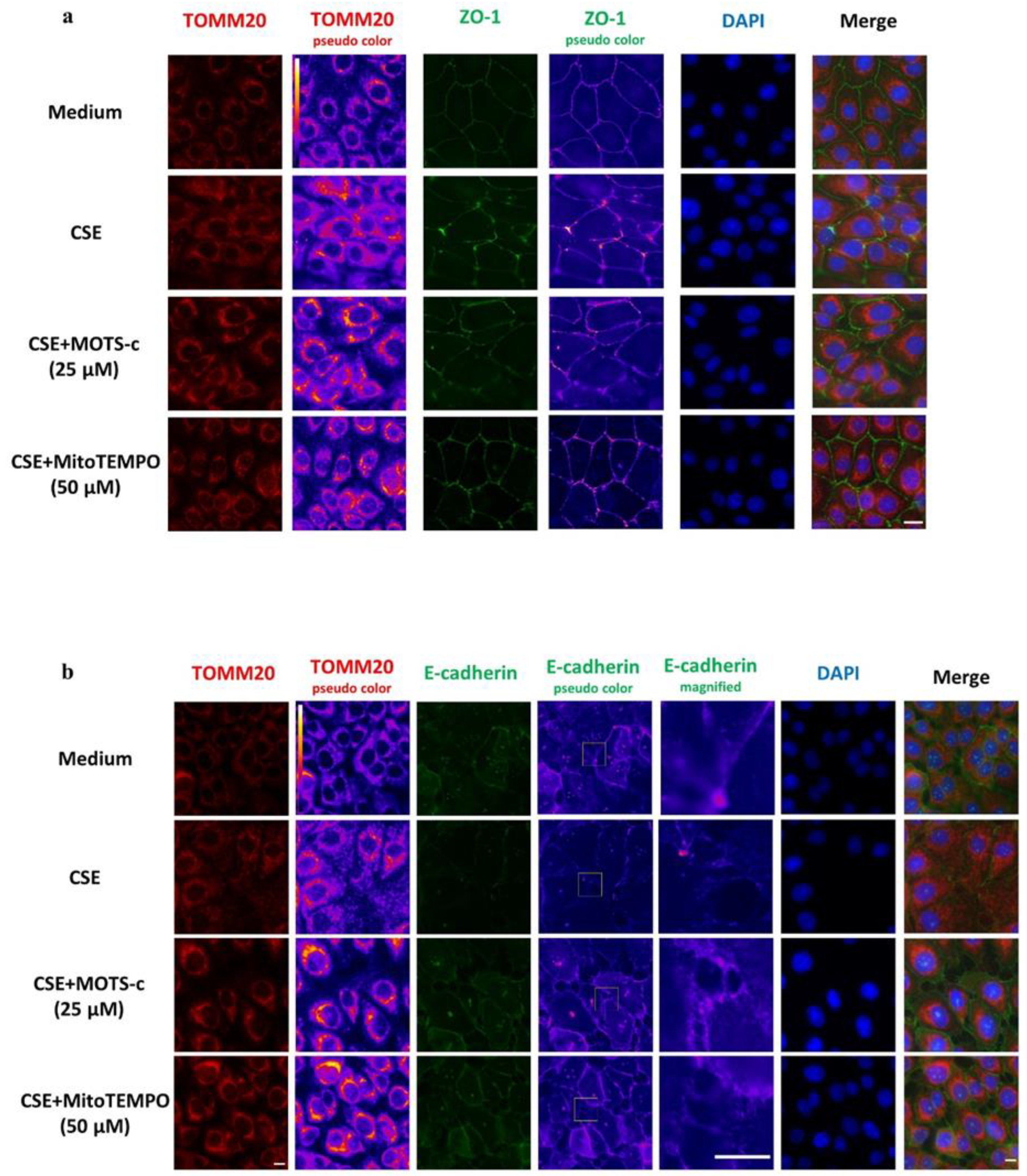
3.4. CSE-Induced Mitochondrial Dysfunction Affects Epithelial Barrier Integrity
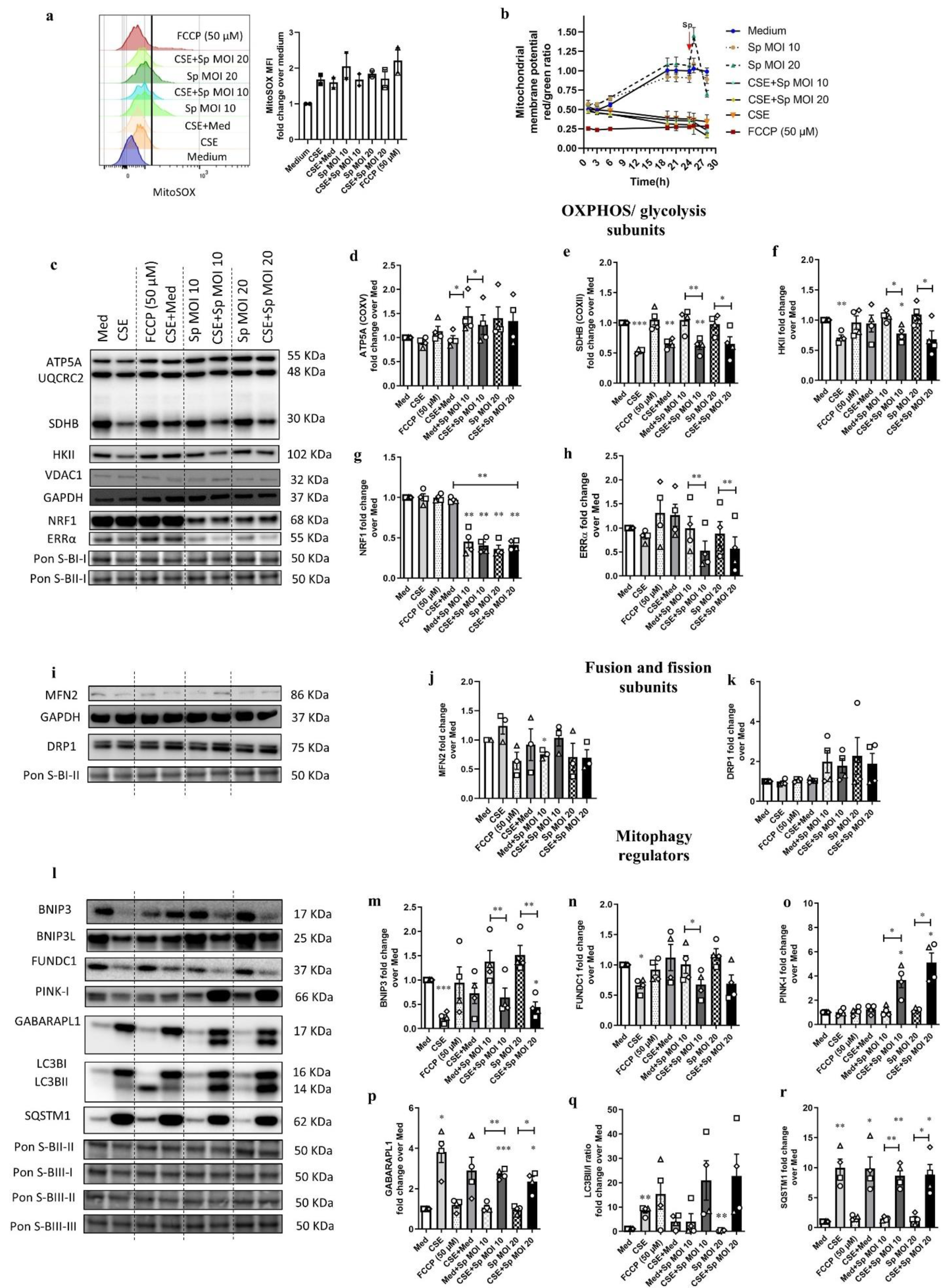
3.5. Streptococcus Pneumoniae Infection with Prior CSE Exposure Induces Mitochondrial Dysfunctions and Ultrastructure Damage in 16HBE Cells
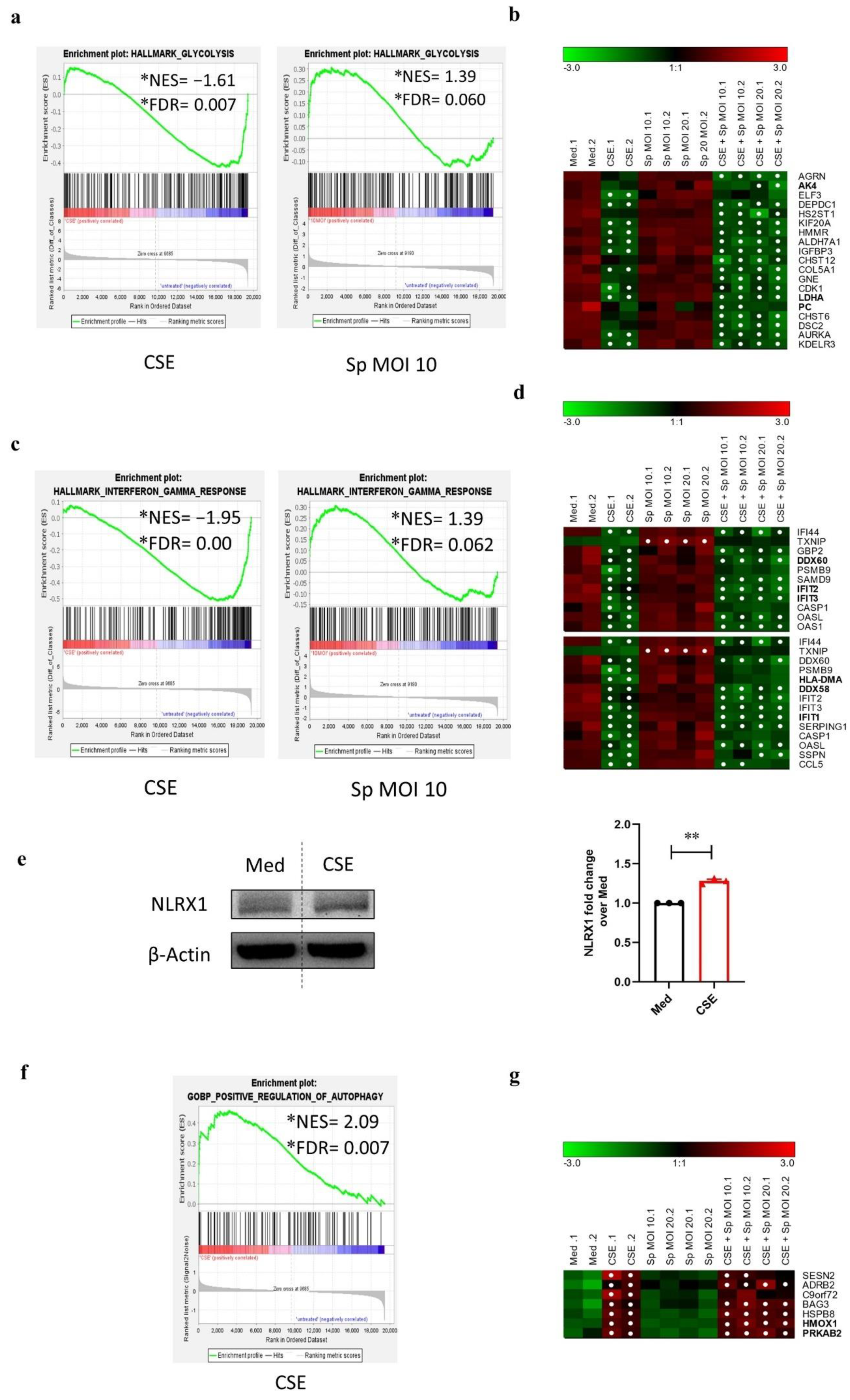
3.6. CSE Pre-Exposure Changes Expression Profiles of Genes Involved in Glycolysis, Innate Immune Responses, and Autophagy upon Sp Infection

4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aghapour, M.; Raee, P.; Moghaddam, S.J.; Hiemstra, P.S.; Heijink, I.H. Airway epithelial barrier dysfunction in chronic obstructive pulmonary disease: Role of cigarette smoke exposure. Am. J. Respir. Cell Mol. Biol. 2018, 58, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, P.S.; McCray, P.B.J.; Bals, R. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur. Respir. J. 2015, 45, 1150–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghapour, M.; Remels, A.H.V.; Pouwels, S.D.; Bruder, D.; Hiemstra, P.S.; Cloonan, S.M.; Heijink, I.H. Mitochondria: At the crossroads of regulating lung epithelial cell function in chronic obstructive pulmonary disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L149–L164. [Google Scholar] [CrossRef] [PubMed]
- Mizumura, K.; Cloonan, S.M.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Glass, K.; Owen, C.A.; Mahmood, A.; Washko, G.R.; et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Investig. 2014, 124, 3987–4003. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Araya, J.; Kurita, Y.; Kobayashi, K.; Takasaka, N.; Yoshida, M.; Hara, H.; Minagawa, S.; Wakui, H.; Fujii, S.; et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy 2015, 11, 547–559. [Google Scholar] [CrossRef]
- Wu, N.N.; Zhang, Y.; Ren, J. Mitophagy, mitochondrial dynamics, and homeostasis in cardiovascular aging. Oxid. Med. Cell. Longev. 2019, 2019, 9825061. [Google Scholar] [CrossRef] [Green Version]
- Man, W.H.; de Steenhuijsen Piters, W.A.A.; Bogaert, D. The microbiota of the respiratory tract: Gatekeeper to respiratory health. Nat. Rev. Microbiol. 2017, 15, 259–270. [Google Scholar] [CrossRef]
- Voss, M.; Wonnenberg, B.; Honecker, A.; Kamyschnikow, A.; Herr, C.; Bischoff, M.; Tschernig, T.; Bals, R.; Beisswenger, C. Cigarette smoke-promoted acquisition of bacterial pathogens in the upper respiratory tract leads to enhanced inflammation in mice. Respir. Res. 2015, 16, 41. [Google Scholar] [CrossRef] [Green Version]
- Maurice, N.M.; Bedi, B.; Yuan, Z.; Goldberg, J.B.; Koval, M.; Hart, C.M.; Sadikot, R.T. Pseudomonas aeruginosa induced host epithelial cell mitochondrial dysfunction. Sci. Rep. 2019, 9, 11929. [Google Scholar] [CrossRef]
- Tiku, V.; Tan, M.-W.; Dikic, I. Mitochondrial functions in infection and immunity. Trends Cell Biol. 2020, 30, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, R.F.; Zarrintan, S.; Brandenburg, S.M.; Kol, A.; de Bruin, H.G.; Jafari, S.; Dijk, F.; Kalicharan, D.; Kelders, M.; Gosker, H.R.; et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir. Res. 2013, 14, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturn, A.; Quackenbush, J.; Trajanoski, Z. Genesis: Cluster analysis of microarray data. Bioinformatics 2002, 18, 207–208. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, I.C.; Moore, C.B.; Schneider, M.; Lei, Y.; Davis, B.K.; Scull, M.A.; Gris, D.; Roney, K.E.; Zimmermann, A.G.; Bowzard, J.B.; et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-ΚB signaling pathways. Immunity 2011, 34, 854–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundar, I.K.; Maremanda, K.P.; Rahman, I. Mitochondrial dysfunction is associated with Miro1 reduction in lung epithelial cells by cigarette smoke. Toxicol. Lett. 2019, 317, 92–101. [Google Scholar] [CrossRef]
- Lee, J.W.; Park, S.; Takahashi, Y.; Wang, H.-G. The association of AMPK with ULK1 regulates autophagy. PLoS ONE 2010, 5, e15394. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.-Y.; Li, Y.-Y.; Huang, C.; Li, J.; Yao, H.-W. AMP-activated protein kinase reduces inflammatory responses and cellular senescence in pulmonary emphysema. Oncotarget 2017, 8, 22513–22523. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, J.; Zhou, J.-S.; Huang, H.-Q.; Li, Z.-Y.; Xu, X.-C.; Lai, T.-W.; Hu, Y.; Zhou, H.-B.; Chen, H.-P.; et al. MTOR suppresses cigarette smoke-induced epithelial cell death and airway inflammation in chronic obstructive pulmonary disease. J. Immunol. 2018, 200, 2571–2580. [Google Scholar] [CrossRef]
- Morsch, A.L.B.C.; Wisniewski, E.; Luciano, T.F.; Comin, V.H.; Silveira, G.D.B.; Marques, S.D.O.; Thirupathi, A.; Silveira Lock, P.C.; De Souza, C.T. Cigarette smoke exposure induces ROS-mediated autophagy by regulating Sestrin, AMPK, and MTOR level in mice. Redox Rep. 2019, 24, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Cui, W.; Zhang, Z.; Zhang, P.; Qu, J.; Zheng, C.; Mo, X.; Zhou, W.; Xu, L.; Yao, H.; Gao, J. Nrf2 attenuates inflammatory response in COPD/emphysema: Crosstalk with Wnt3a/β-Catenin and AMPK pathways. J. Cell. Mol. Med. 2018, 22, 3514–3525. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Park, S.J.; Cho, Y.S.; Huh, J.W.; Oh, Y.-M.; Lee, S.-D. Role of AMP-activated protein kinase (AMPK) in smoking-induced lung inflammation and emphysema. Tuberc. Respir. Dis. 2015, 78, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, W.; Li, W.; Chen, Y.; Yan, Z.; Huang, X.; Zhuang, H.; Zhong, W.; Chen, Y.; Wu, W.; Lin, C.; et al. Phosphorylation of ULK1 by AMPK regulates translocation of ULK1 to mitochondria and mitophagy. FEBS Lett. 2015, 589, 1847–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, R.; Rampersaud, R.; Aguilar, J.L.; Randis, T.M.; Kreindler, J.L.; Ratner, A.J. Cigarette smoke inhibits airway epithelial cell innate immune responses to bacteria. Infect. Immun. 2010, 78, 2146–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaschler, G.J.; Skrtic, M.; Zavitz, C.C.J.; Lindahl, M.; Onnervik, P.-O.; Murphy, T.F.; Sethi, S.; Stämpfli, M.R. Bacteria challenge in smoke-exposed mice exacerbates inflammation and skews the inflammatory profile. Am. J. Respir. Crit. Care Med. 2009, 179, 666–675. [Google Scholar] [CrossRef]
- Nurse-Lucas, M.; McGee, L.; Hawkins, P.A.; Swanston, W.H.; Akpaka, P.E. Serotypes and genotypes of Streptococcus pneumoniae isolates from Trinidad and Tobago. Int. J. Infect. Dis. 2016, 46, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Trallero, E.; Marimón, J.M.; Larruskain, J.; Alonso, M.; Ercibengoa, M. Antimicrobial susceptibilities and serotypes of Streptococcus pneumoniae isolates from elderly patients with pneumonia and acute exacerbation of chronic obstructive pulmonary disease. Antimicrob. Agents Chemother. 2011, 55, 2729–2734. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.A.T.; Fujii, H.; Vu, H.T.T.; Parry, C.M.; Dang, A.D.; Ariyoshi, K.; Yoshida, L.-M. An alarmingly high nasal carriage rate of Streptococcus pneumoniae serotype 19F non-susceptible to multiple Beta-Lactam antimicrobials among Vietnamese children. BMC Infect. Dis. 2019, 19, 241. [Google Scholar] [CrossRef] [Green Version]
- Plataki, M.; Cho, S.J.; Harris, R.M.; Huang, H.-R.; Yun, H.S.; Schiffer, K.T.; Stout-Delgado, H.W. Mitochondrial dysfunction in aged macrophages and lung during primary Streptococcus pneumoniae infection is improved with Pirfenidone. Sci. Rep. 2019, 9, 971. [Google Scholar] [CrossRef]
- Gao, Y.; Xu, W.; Dou, X.; Wang, H.; Zhang, X.; Yang, S.; Liao, H.; Hu, X.; Wang, H. Mitochondrial DNA leakage caused by Streptococcus pneumoniae hydrogen peroxide promotes type I IFN expression in lung cells. Front. Microbiol. 2019, 10, 630. [Google Scholar] [CrossRef] [Green Version]
- Nerlich, A.; Mieth, M.; Letsiou, E.; Fatykhova, D.; Zscheppang, K.; Imai-Matsushima, A.; Meyer, T.F.; Paasch, L.; Mitchell, T.J.; Tönnies, M.; et al. Pneumolysin induced mitochondrial dysfunction leads to release of mitochondrial DNA. Sci. Rep. 2018, 8, 182. [Google Scholar] [CrossRef] [PubMed]
- Thoo, L.; Noti, M.; Krebs, P. Keep calm: The intestinal barrier at the interface of peace and war. Cell Death Dis. 2019, 10, 849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amatngalim, G.D.; Schrumpf, J.A.; Henic, A.; Dronkers, E.; Verhoosel, R.M.; Ordonez, S.R.; Haagsman, H.P.; Fuentes, M.E.; Sridhar, S.; Aarbiou, J.; et al. Antibacterial defense of human airway epithelial cells from chronic obstructive pulmonary disease patients induced by acute exposure to nontypeable Haemophilus influenzae: Modulation by cigarette smoke. J. Innate Immun. 2017, 9, 359–374. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Zhang, W.; Booth, J.L.; Hutchings, D.C.; Wang, X.; White, V.L.; Youness, H.; Cross, C.D.; Zou, M.-H.; Burian, D.; et al. Human primary airway epithelial cells isolated from sctive smokers have epigenetically impaired antiviral responses. Respir. Res. 2016, 17, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modestou, M.A.; Manzel, L.J.; El-Mahdy, S.; Look, D.C. Inhibition of IFN-gamma-dependent antiviral airway epithelial defense by cigarette smoke. Respir. Res. 2010, 11, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffney, P.F.; McCarthy, C.E.; Nogales, A.; Thatcher, T.H.; Martinez-Sobrido, L.; Phipps, R.P.; Sime, P.J. Cigarette smoke dampens antiviral signaling in small airway epithelial cells by disrupting TLR3 cleavage. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L505–L513. [Google Scholar] [CrossRef] [Green Version]
- Hanada, Y.; Ishihara, N.; Wang, L.; Otera, H.; Ishihara, T.; Koshiba, T.; Mihara, K.; Ogawa, Y.; Nomura, M. MAVS is energized by Mff which senses mitochondrial metabolism via AMPK for acute antiviral immunity. Nat. Commun. 2020, 11, 5711. [Google Scholar] [CrossRef]
- Lei, Y.; Wen, H.; Yu, Y.; Taxman, D.J.; Zhang, L.; Widman, D.G.; Swanson, K.V.; Wen, K.-W.; Damania, B.; Moore, C.B.; et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity 2012, 36, 933–946. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yao, Y.; Qiu, X.; Wang, G.; Hu, Z.; Chen, S.; Wu, Z.; Yuan, N.; Gao, H.; Wang, J.; et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat. Immunol. 2019, 20, 433–446. [Google Scholar] [CrossRef]
- Lee, C.; Kim, K.H.; Cohen, P. MOTS-c: A novel mitochondrial-derived peptide regulating muscle and fat metabolism. Free Radic. Biol. Med. 2016, 100, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Michi, A.N.; Yipp, B.G.; Dufour, A.; Lopes, F.; Proud, D. PGC-1α mediates a metabolic host defense response in human airway epithelium during rhinovirus infections. Nat. Commun. 2021, 12, 3669. [Google Scholar] [CrossRef] [PubMed]
- Heijink, I.H.; Kuchibhotla, V.N.S.; Roffel, M.P.; Maes, T.; Knight, D.A.; Sayers, I.; Nawijn, M.C. Epithelial cell dysfunction, a major driver of asthma development. Allergy 2020, 75, 1902–1917. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aghapour, M.; Tulen, C.B.M.; Abdi Sarabi, M.; Weinert, S.; Müsken, M.; Relja, B.; van Schooten, F.-J.; Jeron, A.; Braun-Dullaeus, R.; Remels, A.H.; et al. Cigarette Smoke Extract Disturbs Mitochondria-Regulated Airway Epithelial Cell Responses to Pneumococci. Cells 2022, 11, 1771. https://doi.org/10.3390/cells11111771
Aghapour M, Tulen CBM, Abdi Sarabi M, Weinert S, Müsken M, Relja B, van Schooten F-J, Jeron A, Braun-Dullaeus R, Remels AH, et al. Cigarette Smoke Extract Disturbs Mitochondria-Regulated Airway Epithelial Cell Responses to Pneumococci. Cells. 2022; 11(11):1771. https://doi.org/10.3390/cells11111771
Chicago/Turabian StyleAghapour, Mahyar, Christy B. M. Tulen, Mohsen Abdi Sarabi, Sönke Weinert, Mathias Müsken, Borna Relja, Frederik-Jan van Schooten, Andreas Jeron, Rüdiger Braun-Dullaeus, Alexander H. Remels, and et al. 2022. "Cigarette Smoke Extract Disturbs Mitochondria-Regulated Airway Epithelial Cell Responses to Pneumococci" Cells 11, no. 11: 1771. https://doi.org/10.3390/cells11111771