
Identification of UDP-Glucuronosyltransferase 2B15 (UGT2B15) as a Target for IGF1 and Insulin Action


 ,
, 
 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Treatments
2.2. Real-Time Quantitative Polymerase Chain Reactions (RT-QPCR)
2.3. Western Blot Analysis
2.4. Small-Interfering RNA (siRNA) UGT2B15 Knockdown
2.5. Cell Proliferation Assays
2.6. Co-Immunoprecipitation (Co-IP) Assays
2.7. Animal Studies
2.8. Statistical Analyses
3. Results
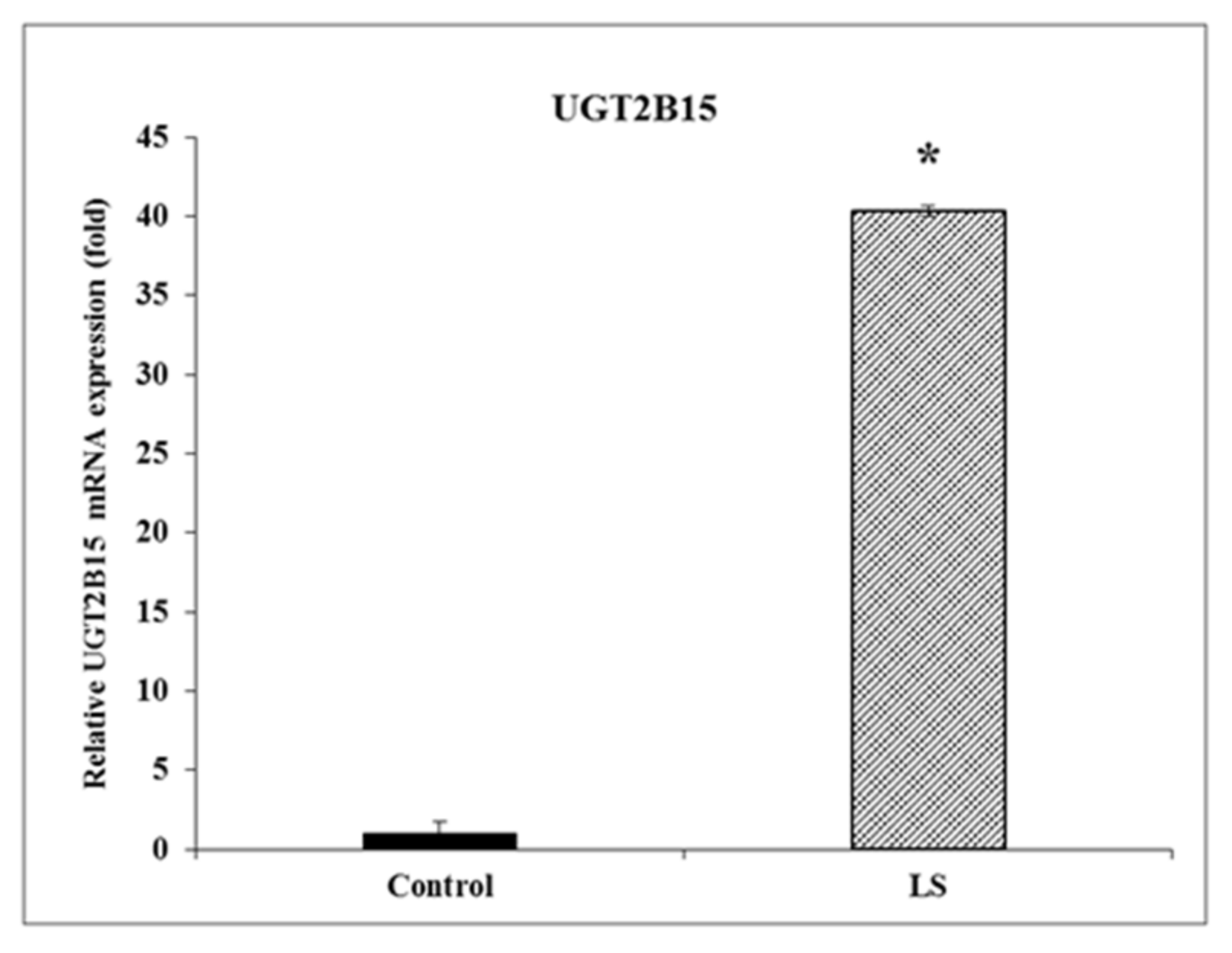
3.1. Identification of UGT2B15 as a Differentially Expressed Gene in LS
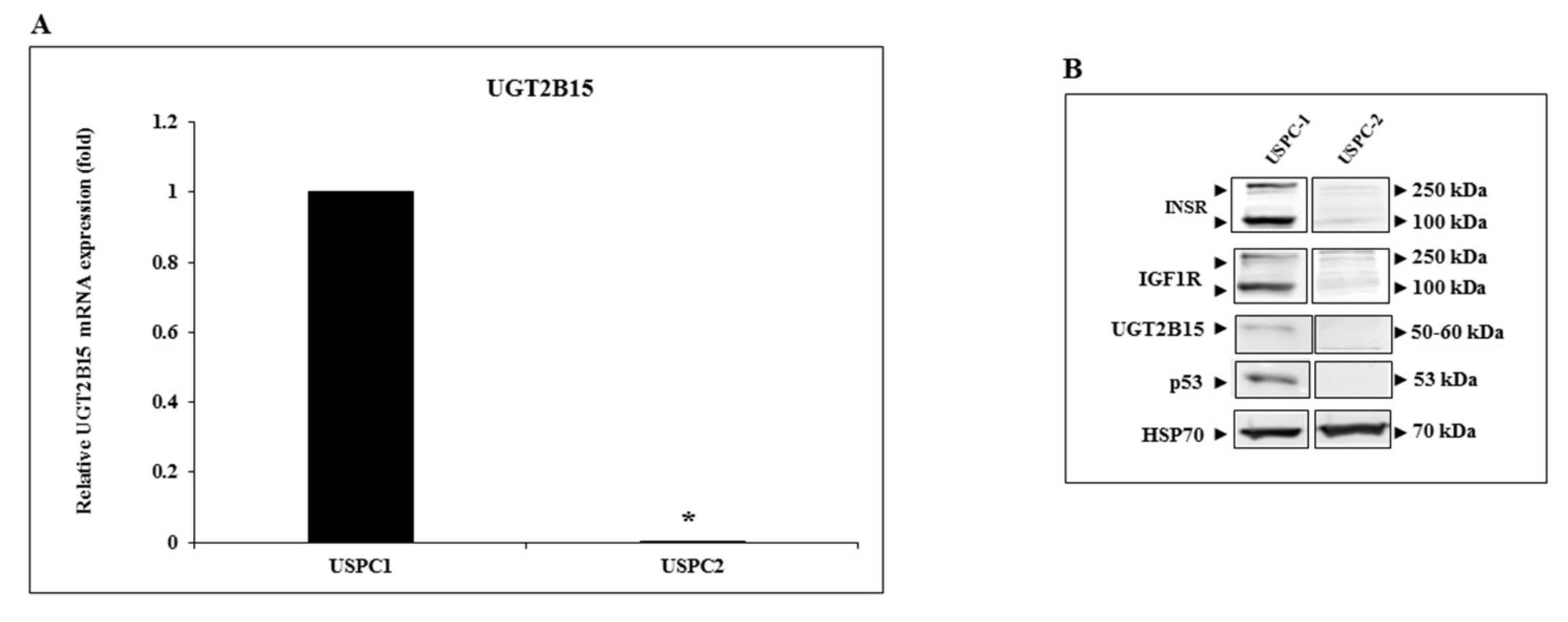
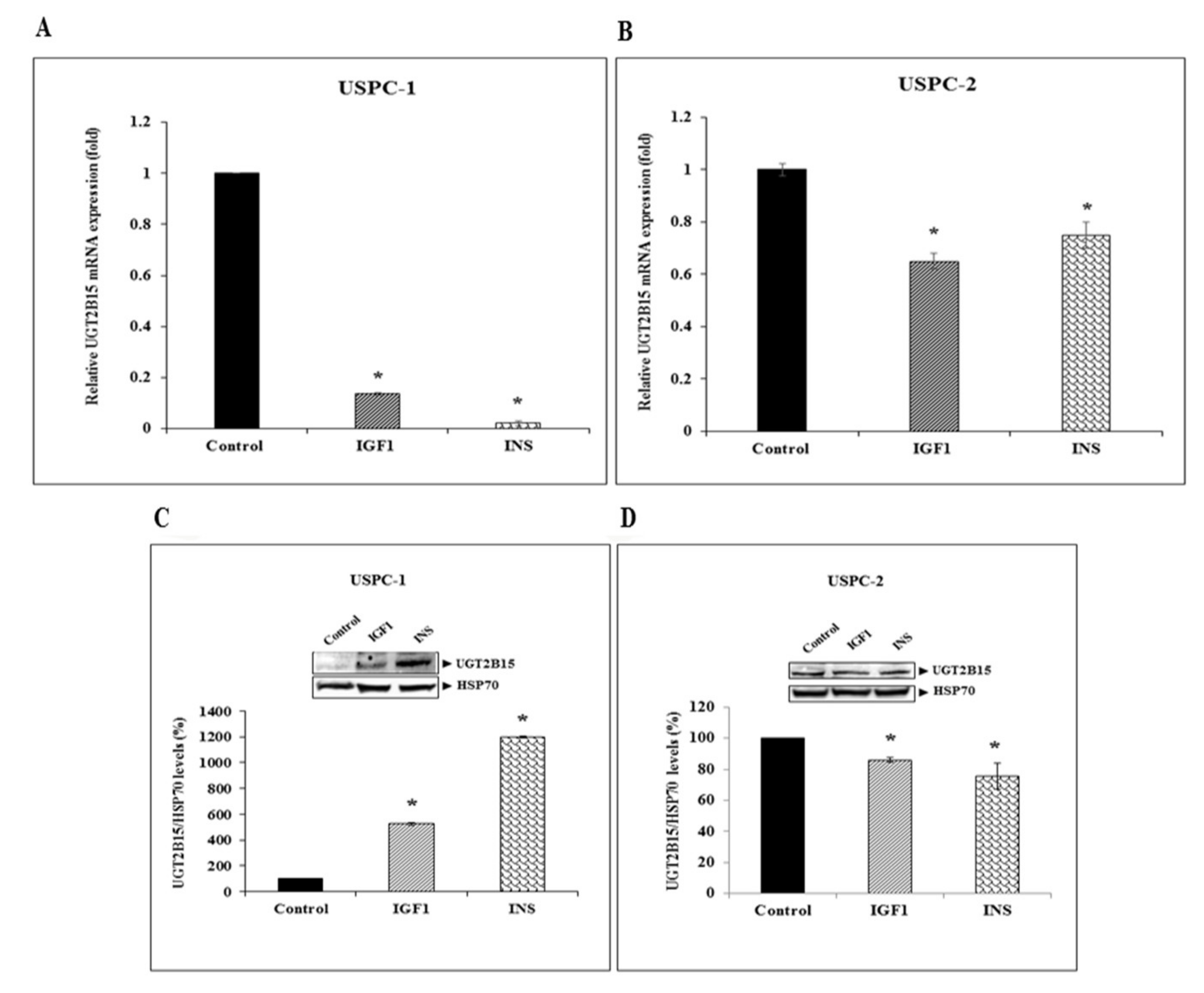
3.2. Identification of UGT2B15 as a Target for Inhibitory Regulation by IGF1 and Insulin in Endometrial Cancer Cells
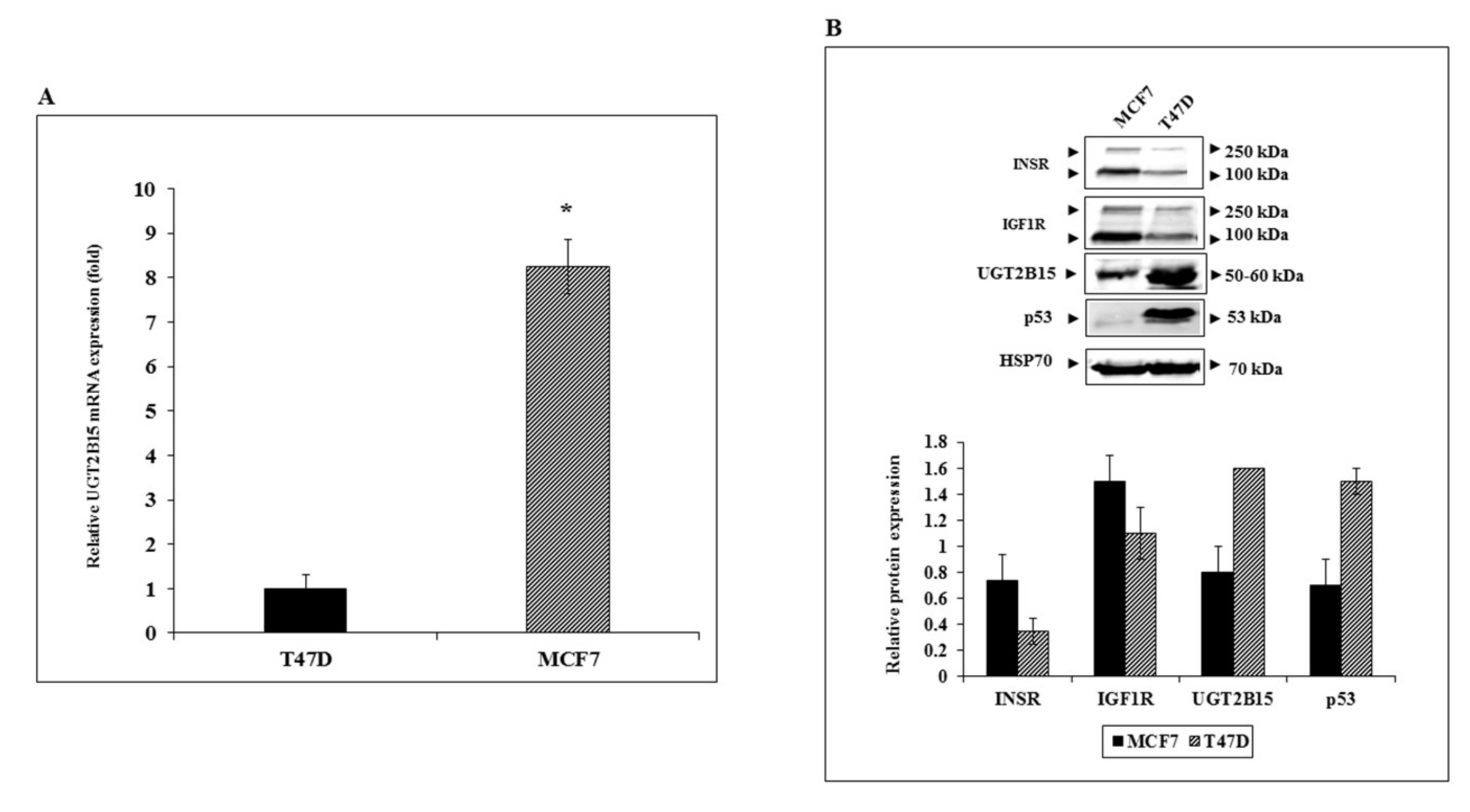
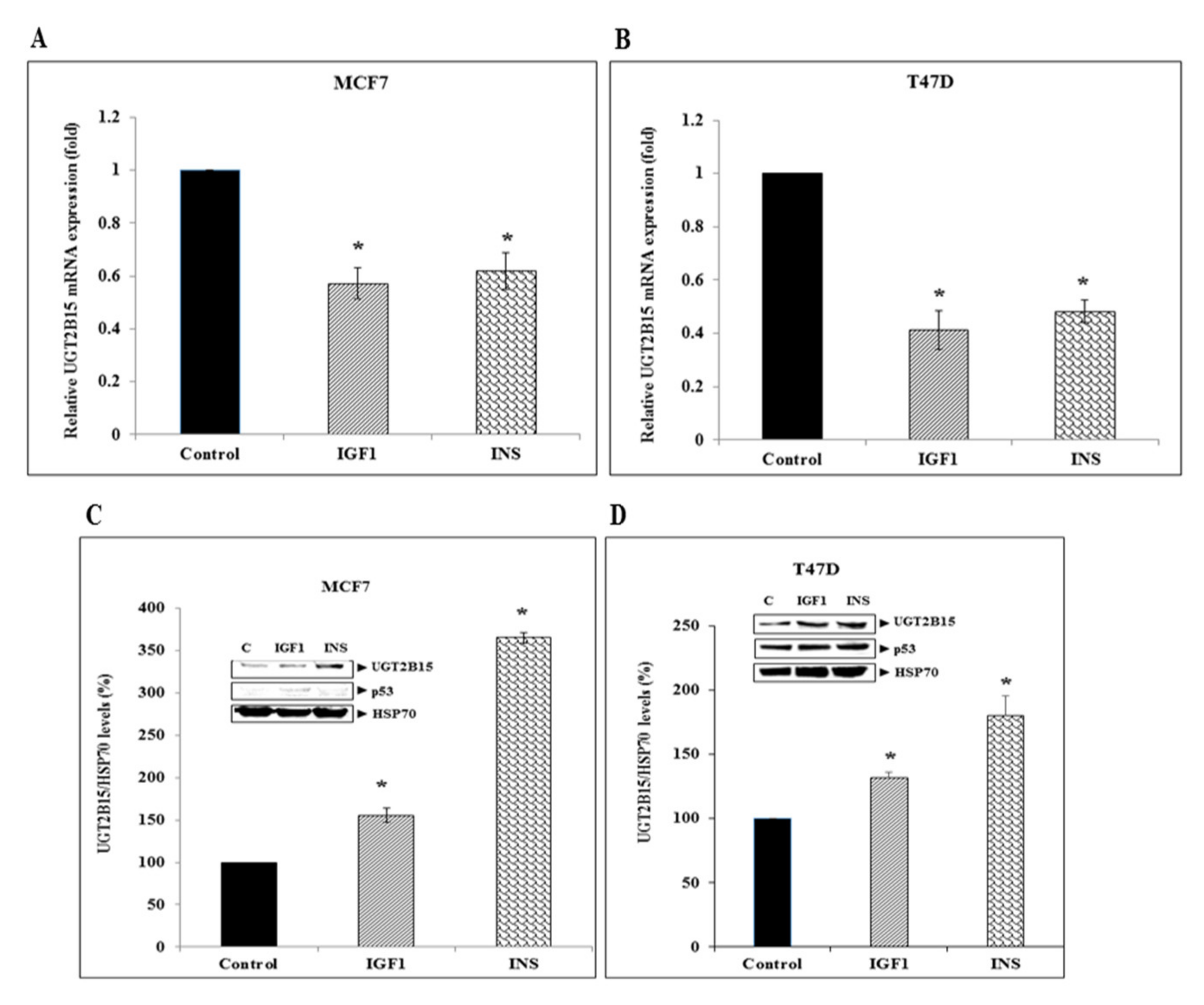
3.3. Regulation of UGT2B15 Gene Expression by IGF1 and Insulin in Breast Cancer Cells
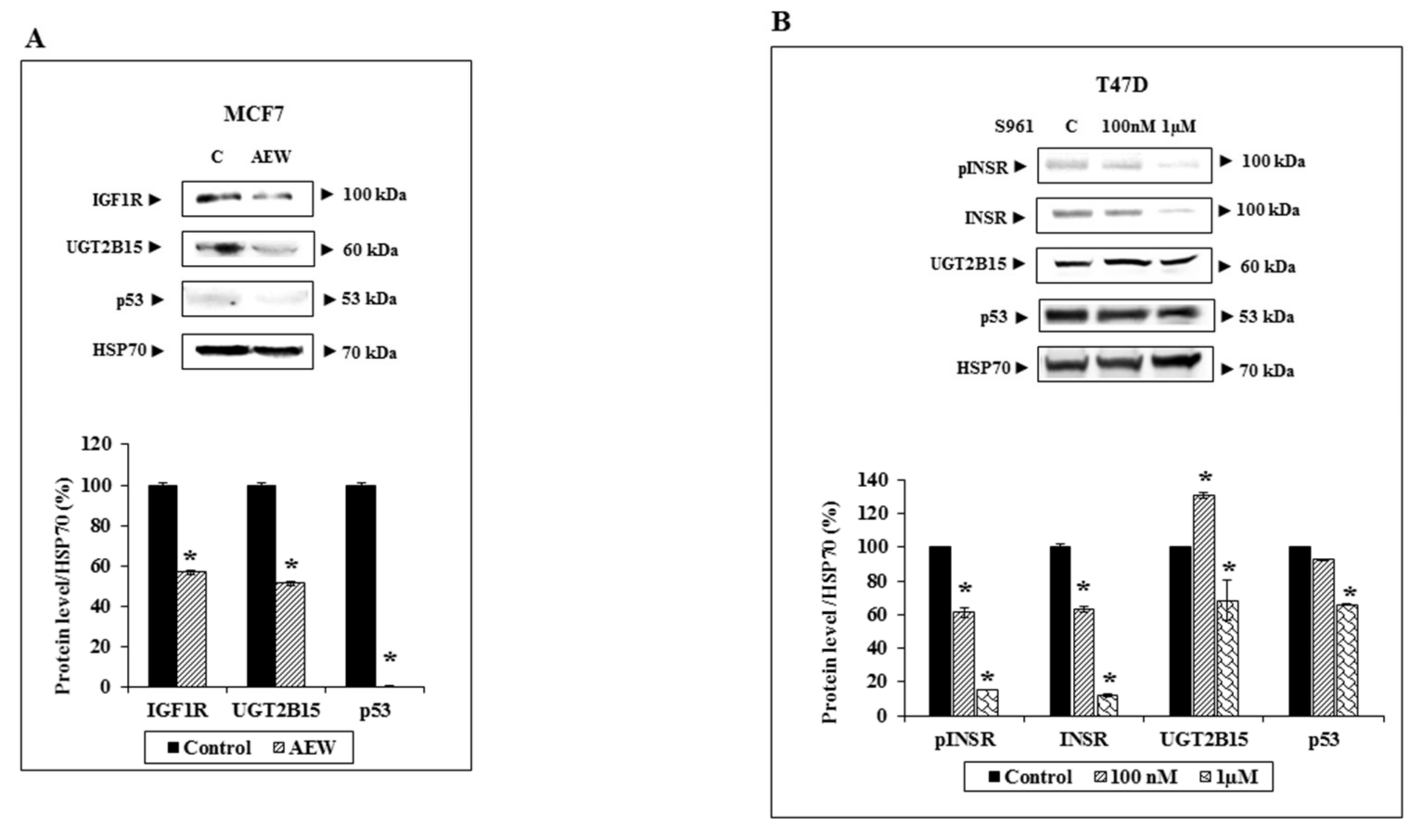
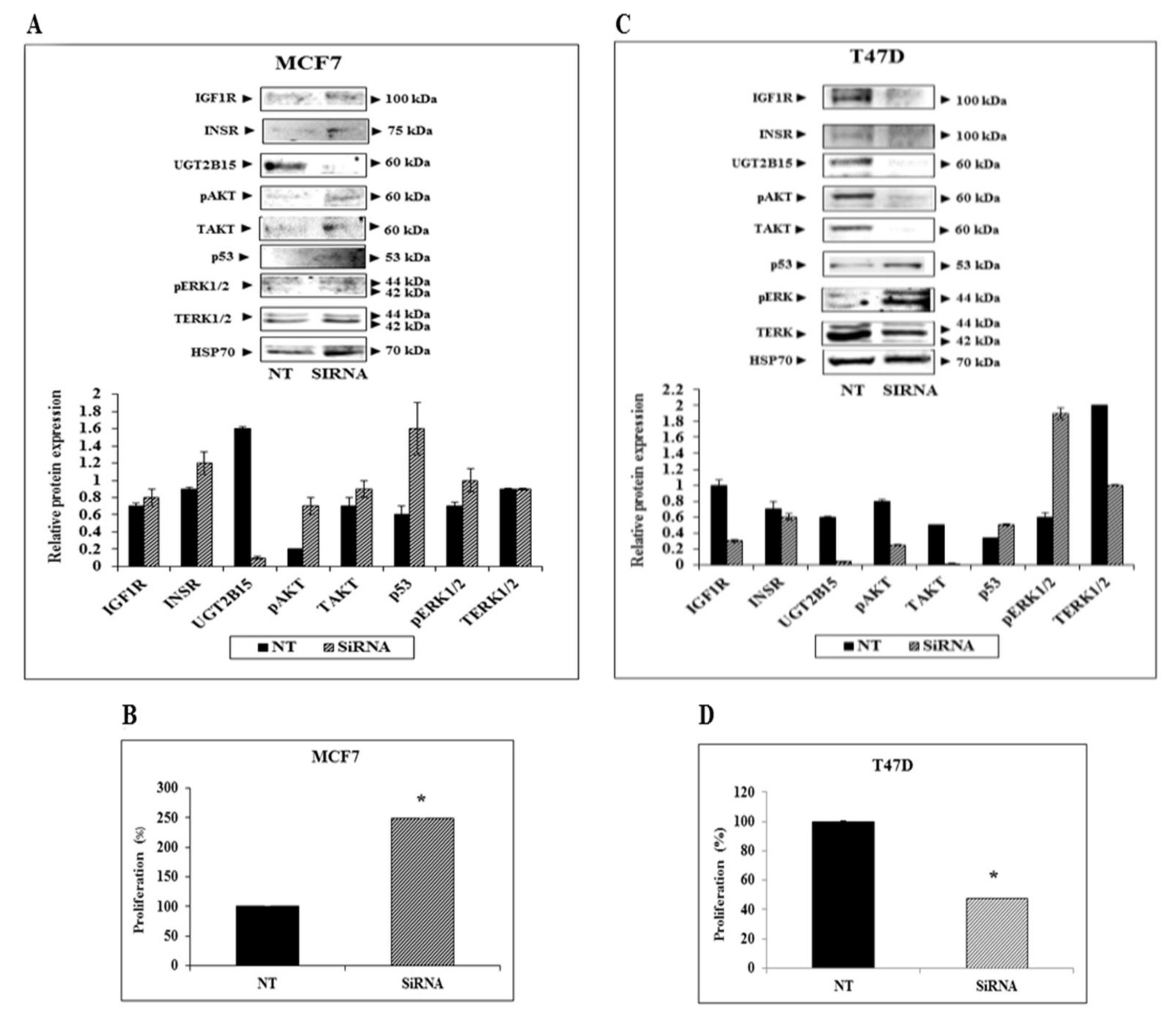
3.4. Paradoxical Effects of UGT2B15 Abrogation on IGF1R Signaling and Biological Action in MCF7 and T47D Cells
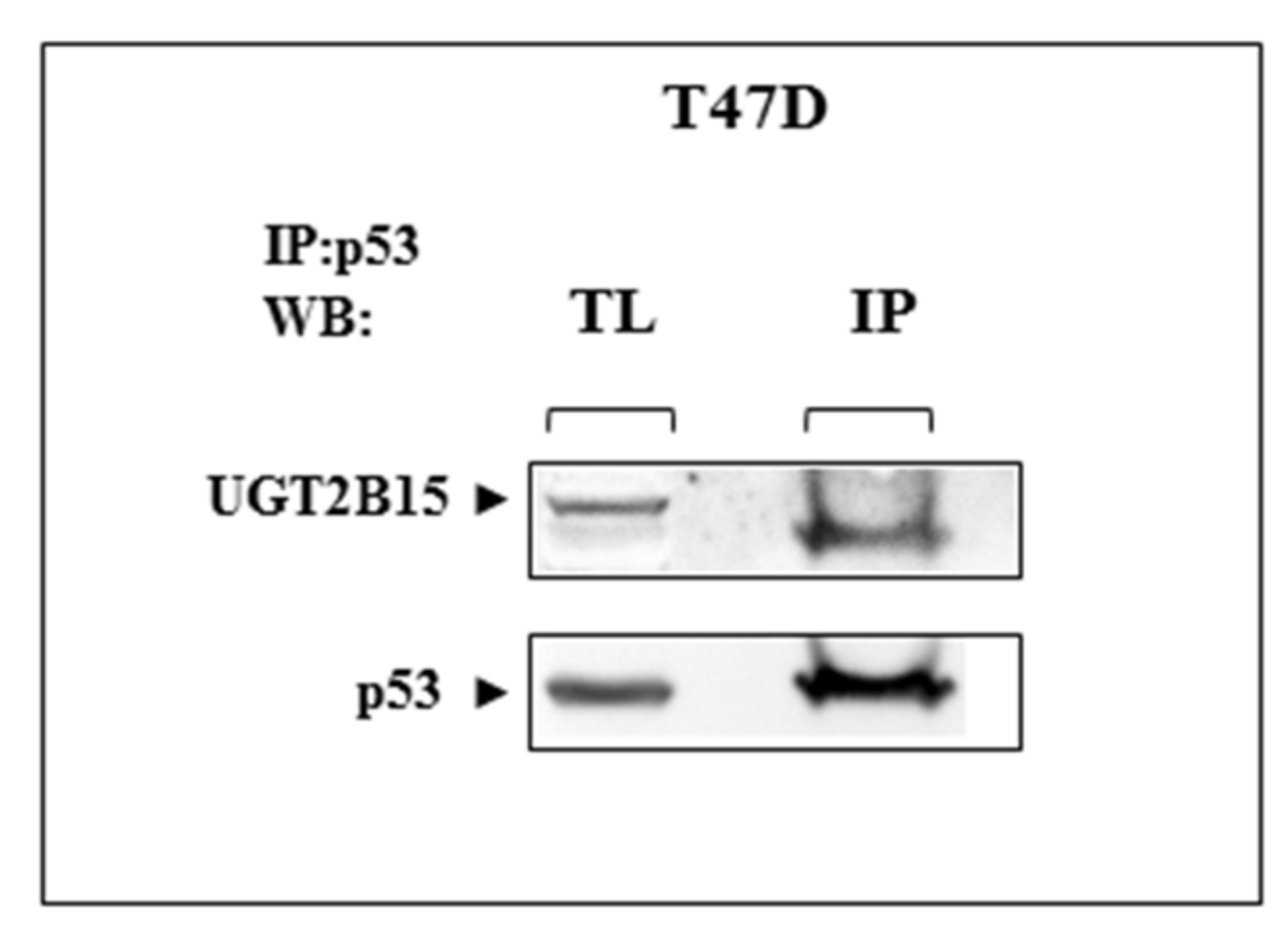
3.5. Co-Immunoprecipitation of UGT2B15 and p53
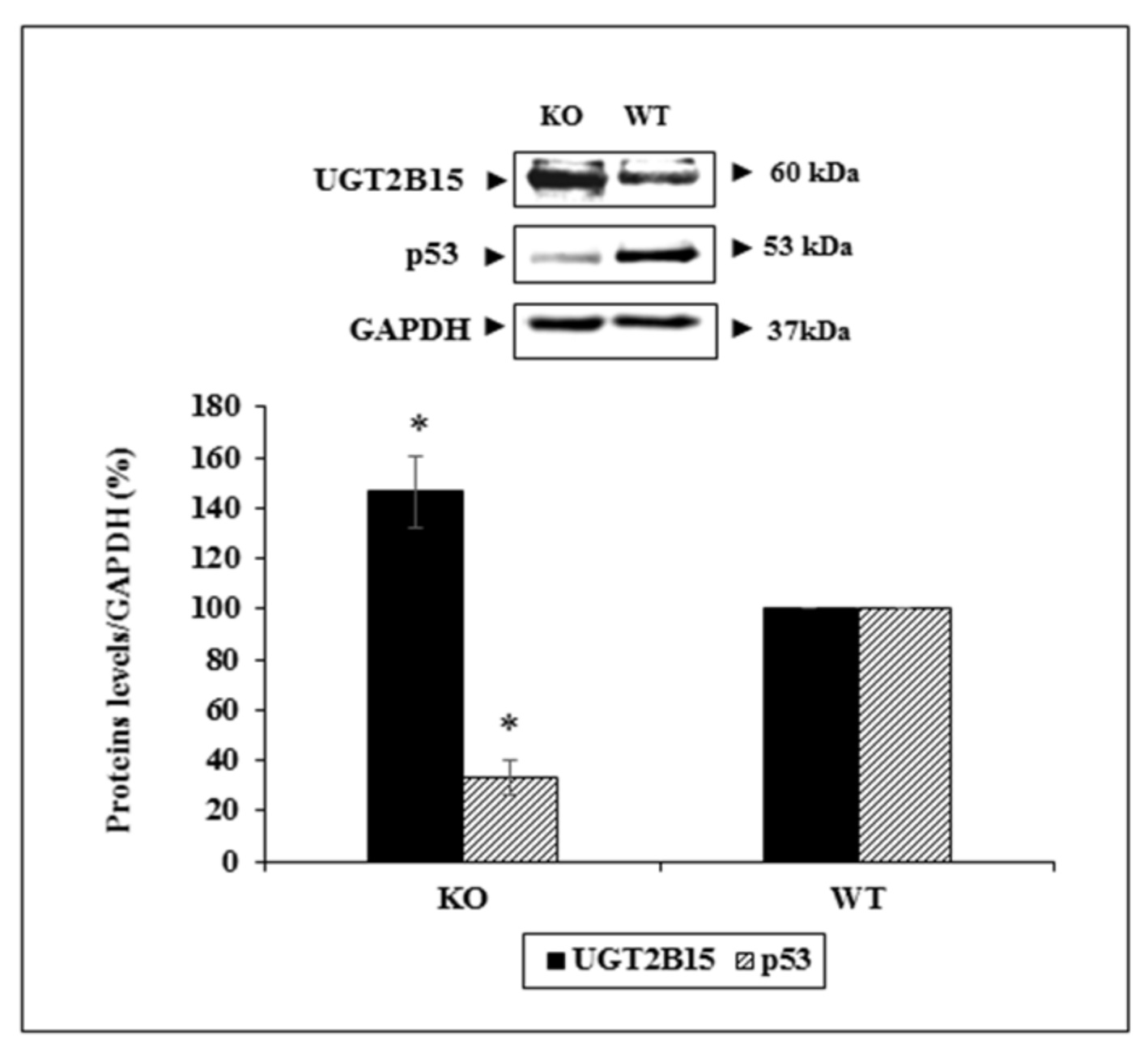
3.6. Animal Studies
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salmon, W.D.; Daughaday, W.H. A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J. Lab. Clin. Med. 1957, 49, 825–836. [Google Scholar]
- LeRoith, D.; Yakar, S. Mechanisms of disease: Metabolic effects of growth hormone and insulin-like growth factor-1. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, R.G. Insulin-like growth factors and the basis of growth. N. Engl. J. Med. 2003, 349, 2184–2186. [Google Scholar] [CrossRef] [PubMed]
- Yakar, S.; Werner, H.; Rosen, C.J. Insulin-like growth factors: Actions on the skeleton. J. Mol. Endocrinol. 2018, 61, T115–T137. [Google Scholar] [CrossRef] [PubMed]
- Milman, S.; Huffman, D.M.; Barzilai, N. The somatotropic axis in human aging: Framework for the current state of knowledge and future research. Cell Metab. 2016, 23, 980–989. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.; Campisi, J. Ras proteins are essential and selective for the action of insulin-like growth factor 1 late in the G1 phase of the cell cycle in BALB/c murine fibroblasts. Proc. Natl. Acad. Sci. USA 1992, 89, 3889–3893. [Google Scholar] [CrossRef] [Green Version]
- Baserga, R.; Rubin, R. Cell cycle and growth control. Crit. Rev. Eukaryot. Gene Expr. 1993, 3, 47–61. [Google Scholar]
- Werner, H.; Sarfstein, R.; LeRoith, D.; Bruchim, I. Insulin-like growth factor 1 signaling axis meets p53 genome protection pathways. Front. Oncol. 2016, 6, 159. [Google Scholar] [CrossRef]
- Chan, J.M.; Stampfer, M.J.; Giovannucci, E.; Gann, P.H.; Ma, J.; Wilkinson, P.; Hennekens, C.H.; Pollak, M. Plasma insulin-like growth factor-I and prostate cancer risk: A prospective study. Science 1998, 279, 563–566. [Google Scholar] [CrossRef]
- Hankinson, S.E.; Willett, W.C.; Colditz, G.A.; Hunter, D.J.; Michaud, D.S.; Deroo, B.; Rosner, B.; Speizer, F.E.; Pollak, M. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet 1998, 351, 1393–1396. [Google Scholar] [CrossRef]
- Werner, H.; Laron, Z. Role of the GH-IGF1 system in progression of cancer. Mol. Cell. Endocrinol. 2020, 518, 111003. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Zwahlen, M.; Minder, C.; O’Dwyer, S.T.; Shalet, S.M.; Egger, M. Insulin-like growth factor-I, IGF binding protein-3, and cancer risk: Systematic review and meta-regression analysis. Lancet 2004, 363, 1346–1353. [Google Scholar] [CrossRef]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. Controversy in clinical endocrinology: Problems with reclassification of insulin-like growth factor I production and action disorders. J. Clin. Endocrinol. Metab. 2008, 91, 4235–4236. [Google Scholar] [CrossRef] [PubMed]
- Klammt, J.; Pfaffle, R.; Werner, H.; Kiess, W. IGF signaling defects as causes of growth failure and IUGR. Trends Endocrinol. Metab. 2008, 19, 197–205. [Google Scholar] [CrossRef]
- Cohen, P.; Rogol, A.D.; Deal, C.L.; Saenger, P.; Reiter, E.O.; Ross, J.L.; Chernausek, S.D.; Savage, M.O.; Wit, J.M.; ISS Consensus Workshop Participants. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: A summary of the Growth Hormone Research Society, The Lawson Wilkins Pediatric Endocrine Society and the European Society for Pediatric Endocrinology Workshop. J. Clin. Endocrinol. Metab. 2008, 93, 4210–4217. [Google Scholar] [CrossRef] [Green Version]
- Woods, K.A.; Camacho-Hubner, C.; Savage, M.O.; Clark, A.J.L. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N. Engl. J. Med. 1996, 335, 1363–1367. [Google Scholar] [CrossRef]
- Domené, S.; Domené, H.M. Genetic mutations in the GH/IGF axis. Pediatr. Endocrinol. Rev. 2018, 16, 39–62. [Google Scholar]
- Wit, J.M.; Oostdijk, W.; Losekoot, M. Spectrum of insulin-like growth factor deficiency. Endocr. Dev. 2012, 23, 30–41. [Google Scholar]
- Laron, Z.; Kopchik, J.J. Laron Syndrome-From Man to Mouse; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Laron, Z. Extensive personal experience. Laron syndrome (primary growth hormone resistance or insensitivity): The personal experience 1958–2003. J. Clin. Endocrinol. Metab. 2004, 89, 1031–1044. [Google Scholar] [CrossRef] [Green Version]
- Laron, Z.; Kauli, R.; Lapkina, L.; Werner, H. IGF-I deficiency, longevity and cancer protection of patients with Laron syndrome. Mutat. Res. Rev. Mutat. Res. 2017, 772, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Amselem, S.; Duquesnoy, P.; Attree, O.; Novelli, G.; Bousnina, S.; Postel-Vinay, M.C.; Goossens, M. Laron dwarfism and mutations of the growth hormone-receptor gene. N. Engl. J. Med. 1989, 321, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Godowski, P.J.; Leung, D.W.; Meacham, L.R.; Galgani, J.P.; Hellmiss, R.; Keret, R.; Rotwein, P.S.; Parks, J.S.; Laron, Z.; Wood, W.I. Characterization of the human growth hormone receptor gene and demonstration of a partial gene deletion in two patients with Laron-type dwarfism. Proc. Natl. Acad. Sci. USA 1989, 86, 8083–8087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shevah, O.; Laron, Z. Patients with congenital deficiency of IGF-I seem protected from the development of malignancies: A preliminary report. Growth Horm. IGF Res. 2007, 17, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Steuerman, R.; Shevah, O.; Laron, Z. Congenital IGF1 deficiency tends to confer protection against post-natal development of malignancies. Eur. J. Endocrinol. 2011, 164, 485–489. [Google Scholar] [CrossRef] [Green Version]
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl. Med. 2011, 3, 70ra13. [Google Scholar] [CrossRef] [Green Version]
- Werner, H.; Lapkina-Gendler, L.; Laron, Z. Fifty years on: New lessons from Laron syndrome. Isr. Med. Assoc. J. 2017, 19, 6–7. [Google Scholar]
- Lapkina-Gendler, L.; Rotem, I.; Pasmanik-Chor, M.; Gurwitz, D.; Sarfstein, R.; Laron, Z.; Werner, H. Identification of signaling pathways associated with cancer protection in Laron syndrome. Endocr. Relat. Cancer 2016, 23, 399–410. [Google Scholar] [CrossRef] [Green Version]
- Werner, H.; Lapkina-Gendler, L.; Achlaug, L.; Nagaraj, K.; Somri, L.; Yaron-Saminsky, D.; Pasmanik-Chor, M.; Sarfstein, R.; Laron, Z.; Yakar, S. Genome-Wide Profiling of Laron Syndrome Patients Identifies Novel Cancer Protection Pathways. Cells 2019, 8, 596. [Google Scholar] [CrossRef] [Green Version]
- Werner, H.; Sarfstein, R.; Nagaraj, K.; Laron, Z. Laron syndrome research paves the way for new insights in oncological investigation. Cells 2020, 9, 2446. [Google Scholar] [CrossRef]
- Chouinard, S.; Barbier, O.; Bélanger, A. UDP-glucuronosyltransferase 2B15 (UGT2B15) and UGT2B17 enzymes are major determinants of the androgen response in prostate cancer LNCaP cells. J. Biol. Chem. 2007, 282, 33466–33474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bélanger, A.; Pelletier, G.; Labrie, F.; Barbier, O.; Chouinard, S. Inactivation of androgens by UDP-glucuronosyltransferase enzymes in humans. Trends Endocrinol. Metab. 2003, 14, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Grant, D.J.; Chen, Z.; Howard, L.E.; Wiggins, E.; De Hoedt, A.; Vidal, A.C.; Carney, S.T.; Squires, J.; Magyar, C.E.; Huang, J.; et al. UDP-glucuronosyltransferases and biochemical recurrence in prostate cancer progression. BMC Cancer 2017, 17, 463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, R.; Yokoi, T.; Nakajima, M. Structure and protein-protein interactions of human UDP-glucuronosyltransferases. Front. Pharmacol. 2016, 7, 388. [Google Scholar] [CrossRef] [Green Version]
- Rowland, A.; Miners, J.O.; Mackenzie, P.I. The UDP-glucuronosyltransferases: Their role in drug metabolism and detoxification. Int. J. Biochem. Cell Biol. 2013, 45, 1121–1132. [Google Scholar] [CrossRef]
- Turgeon, D.; Carrier, J.S.; Lévesque, E.; Beatty, B.G.; Bélanger, A.; Hum, D.W. Isolation and characterization of the human UGT2B15 gene, localized within a cluster of UGT2B genes and pseudogenes on chromosome 4. J. Mol. Biol. 2000, 295, 489–504. [Google Scholar] [CrossRef]
- Turgeon, D.; Carrier, J.S.; Lévesque, E.; Hum, D.W.; Bélanger, A. Relative enzymatic activity, protein stability, and tissue distribution of human steroid-metabolizing UGT2B subfamily members. Endocrinology 2001, 142, 778–787. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Parikh, R.; Sorek, E.; Parikh, S.; Michael, K.; Bikovski, L.; Tshori, S.; Shefer, G.; Mingelgreen, S.; Zornitzki, T.; Knobler, H.; et al. Skin exposure to UVB light induces a skin-brain-gonad axis and sexual behavior. Cell Rep. 2021, 36, 109579. [Google Scholar] [CrossRef]
- Nagaraj, K.; Lapkina-Gendler, L.; Sarfstein, R.; Gurwitz, D.; Pasmanik-Chor, M.; Laron, Z.; Yakar, S.; Werner, H. Identification of thioredoxin-interacting protein (TXNIP) as a downstream target for IGF1 action. Proc. Natl. Acad. Sci. USA 2018, 115, 1045–1050. [Google Scholar] [CrossRef] [Green Version]
- Sarfstein, R.; Lapkina-Gendler, L.; Nagaraj, K.; Laron, Z.; Werner, H. Identification of nephronectin as a new target for IGF1 action. Eur. J. Cancer 2020, 141, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Shibel, R.; Sarfstein, R.; Nagaraj, K.; Lapkina-Gendler, L.; Laron, Z.; Dixit, M.; Yakar, S.; Werner, H. The olfactory receptor gene product, OR5H2, modulates endometrial cancer cells proliferation via interaction with the IGF1 signaling pathway. Cells 2021, 10, 1483. [Google Scholar] [CrossRef] [PubMed]
- Achlaug, L.; Sarfstein, R.; Nagaraj, K.; Lapkina-Gendler, L.; Bruchim, I.; Dixit, M.; Laron, Z.; Yakar, S.; Werner, H. Identification of ZYG11A as a candidate IGF1-dependent proto-oncogene in endometrial cancer. Oncotarget 2019, 10, 4437–4448. [Google Scholar] [CrossRef] [PubMed]
- Attias-Geva, Z.; Bentov, I.; Kidron, D.; Amichay, K.; Sarfstein, R.; Fishman, A.; Bruchim, I.; Werner, H. p53 regulates insulin-like growth factor-I receptor gene expression in uterine serous carcinoma and predicts responsiveness to an insulin-like growth factor-I receptor-directed targeted therapy. Eur. J. Cancer 2012, 48, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.Y.; Vidnovic, N.; Ellisen, L.W.; Leong, C.-O. Mutant p53 mediates survival of breast cancer cells. Br. J. Cancer 2009, 101, 1606–1612. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef]
- Dates, C.R.; Fahmi, T.; Pyrek, S.J.; Yao-Borengasser, A.; Borowa-Mazgaj, B.; Bratton, S.M.; Kadlubar, S.A.; Mackenzie, P.I.; Haun, R.S.; Radominska-Pandya, A. Human UDP-Glucuronosyltransferases: Effects of altered expression in breast and pancreatic cancer cell lines. Cancer Biol. Ther. 2015, 16, 714–723. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.G.; Selth, L.A.; Tarulli, G.A.; Meech, R.; Wijayakumara, D.; Chanawong, A.; Russell, R.; Caldas, C.; Robinson, J.L.; Carroll, J.S.; et al. Androgen and estrogen receptors in breast cancer coregulate human UDP-glucuronosyltransferases 2B15 and 2B17. Cancer Res. 2016, 76, 5881–5893. [Google Scholar] [CrossRef] [Green Version]
- Bao, B.Y.; Chuang, B.F.; Wang, Q.; Sartor, O.; Balk, S.P.; Brown, M.; Kantoff, P.W.; Lee, G.S. Androgen receptor mediates the expression of UDP-glucuronosyltransferase 2 B15 and B17 genes. Prostate 2008, 68, 839–848. [Google Scholar] [CrossRef] [Green Version]
- Pâquet, S.; Fazli, L.; Grosse, L.; Verreault, M.; Têtu, B.; Rennie, P.S.; Bélanger, A.; Barbier, O. Differential expression of the androgen-conjugating UGT2B15 and UGT2B17 enzymes in prostate tumor cells during cancer progression. J. Clin. Endocrinol. Metab. 2012, 97, E428–E432. [Google Scholar] [CrossRef] [Green Version]
- Vidal, A.C.; Tucker, C.; Schildkraut, J.M.; Richardson, R.M.; McPhail, M.; Freedland, S.J.; Hoyo, C.; Grant, D.J. Novel associations of UDP-glucuronosyltransferase 2B gene variants with prostate cancer risk in a multiethnic study. BMC Cancer 2013, 13, 556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margaillan, G.; Lévesque, É.; Guillemette, C. Epigenetic regulation of steroid inactivating UDP-glucuronosyltransferases by microRNAs in prostate cancer. J. Steroid Biochem. Mol. Biol. 2016, 155, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Wijayakumara, D.D.; Hu, D.G.; Meech, R.; McKinnon, R.A.; Mackenzie, P.I. Regulation of human UGT2B15 and UGT2B17 by miR-376c in prostate cancer cell lines. J. Pharmacol. Exp. Ther. 2015, 354, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafiee-Kermani, F.; Carney, S.T.; Jima, D.; Utin, U.C.; Farrar, L.B.; Oputa, M.O.; Hines, M.R.; Kinyamu, H.K.; Trotter, K.W.; Archer, T.K.; et al. Expression of UDP Glucuronosyltransferases 2B15 and 2B17 is associated with methylation status in prostate cancer cells. Epigenetics 2021, 16, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.G.; Marri, S.; Mackenzie, P.I.; Hulin, J.A.; McKinnon, R.A.; Meech, R. The expression profiles and deregulation of UDP-glycosyltransferase (UGT) genes in human cancers and their association with clinical outcomes. Cancers 2021, 13, 4491. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Sequences (5′–3′) | Product Size |
|---|---|---|
| GAPDH-F | GCGCACCGTCAAGGCTGAGAAC | 142 bp |
| GAPDH-R | AATGGTGGTGAAGACGCCAGT | |
| β-ACTIN-F | CCTGGCACCCAGCACAAT | 144 bp |
| β-ACTIN-R | GGGCCGGACTCGTCATACT | |
| UGTB15-F | GTGTTACCCCAGAATGACC | 101 bp |
| UGTB15-R | GGGATCCCATGGTAGATCG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarfstein, R.; Nagaraj, K.; Parikh, S.; Levy, C.; Laron, Z.; Benayahu, D.; Werner, H. Identification of UDP-Glucuronosyltransferase 2B15 (UGT2B15) as a Target for IGF1 and Insulin Action. Cells 2022, 11, 1627. https://doi.org/10.3390/cells11101627
Sarfstein R, Nagaraj K, Parikh S, Levy C, Laron Z, Benayahu D, Werner H. Identification of UDP-Glucuronosyltransferase 2B15 (UGT2B15) as a Target for IGF1 and Insulin Action. Cells. 2022; 11(10):1627. https://doi.org/10.3390/cells11101627
Chicago/Turabian StyleSarfstein, Rive, Karthik Nagaraj, Shivang Parikh, Carmit Levy, Zvi Laron, Dafna Benayahu, and Haim Werner. 2022. "Identification of UDP-Glucuronosyltransferase 2B15 (UGT2B15) as a Target for IGF1 and Insulin Action" Cells 11, no. 10: 1627. https://doi.org/10.3390/cells11101627