
Is Type 2 Diabetes a Primary Mitochondrial Disorder?

1
The Hadassah Diabetes Center, Hadassah Medical Center, Jerusalem 9112102, Israel
2
The Liver Research Laboratory, Hadassah Medical Center, Jerusalem 9112102, Israel
3
Faculty of Medicine Hebrew, University of Jerusalem, Jerusalem 9112102, Israel
Cells 2022, 11(10), 1617; https://doi.org/10.3390/cells11101617
Submission received: 7 February 2022
/
Revised: 27 March 2022
/
Accepted: 20 April 2022
/
Published: 12 May 2022
(This article belongs to the Collection The Pathomechanism of Mitochondrial Diseases)
Abstract
:Diabetes mellitus is the most common endocrine disturbance in inherited mitochondrial diseases. It is essential to increase awareness of the correct diagnosis and treatment of diabetes in these patients and screen for the condition in family members, as diabetes might appear with distinctive clinical features, complications and at different ages of onset. The severity of mitochondrial-related diabetes is likely to manifest on a large scale of phenotypes depending on the location of the mutation and whether the number of affected mitochondria copies (heteroplasmy) reaches a critical threshold. Regarding diabetes treatment, the first-choice treatment for type 2 diabetes (T2D), metformin, is not recommended because of the risk of lactic acidosis. The preferred treatment for diabetes in patients with mitochondrial disorders is SGLT-2i and mitochondrial GLP-1-related substances. The tight relationship between mitochondrial dysfunction, reduced glucose-stimulated insulin secretion (GSIS), and diabetes development in human patients is acknowledged. However, despite the well-characterized role of mitochondria in GSIS, there is a relative lack of data in humans implicating mitochondrial dysfunction as a primary defect in T2D. Our recent studies have provided data supporting the significant role of the mitochondrial respiratory-chain enzyme, cytochrome c oxidase (COX), in regulating GSIS in a rodent model of T2D, the Cohen diabetic sensitive (CDs) rat. The nutritionally induced diabetic CDs rat demonstrates several features of mitochondrial diseases: markedly reduced COX activity in several tissues, increased reactive oxygen production, decreased ATP generation, and increased lactate dehydrogenase expression in islets. Moreover, our data demonstrate that reduced islet-COX activity precedes the onset of diabetes, suggesting that islet-COX deficiency is the primary defect causing diabetes in this model. This review examines the possibility of including T2D as a primary mitochondrial-related disease. Understanding the critical interdependence between diabetes and mitochondrial dysfunction, centering on the role of COX, may open novel avenues to diagnose and treat diabetes in patients with mitochondrial diseases and mitochondrial dysfunction in diabetic patients.
1. Diabetes and Pancreatic β-Cell Dysfunction
The absorption of blood glucose into the liver, fat, and skeletal muscle cells is dependent on insulin, a peptide hormone produced by β-cells in pancreatic islets (Figure 1). It is acknowledged that the actual manifestation of type 2 diabetes (T2D) occurs only when pancreatic β-cells fail to secrete sufficient insulin to stimulate glucose uptake in peripheral tissues. A combination of genetic and environmental factors results in progressive β-cell loss and dysfunction, leading to persistent hyperglycemia [1,2,3,4,5,6,7,8]. The estimated worldwide prevalence of people affected by T2D could reach 350 million by 2025 and rise [9]. Future individualized therapies for diabetes will require better characterization of the many pathways leading to β-cell dysfunction [7].
Pancreatic β-cells are sensitive glucose sensors that adjust precise quantities of insulin release to variations in blood glucose levels critical to ensure adequate entry of glucose and homeostasis. The process of glucose-stimulated insulin secretion (GSIS) is exceptionally dependent on mitochondrial function (Figure 2 and Figure 3) [3,10,11,12,13,14,15,16,17].
2. Mitochondria and Insulin Secretion in β-Cells
ATP production in β-cells is crucial for stimulus-secretion coupling, a cascade of molecular events encompassing the initial sensing and transport of glucose to β-cells, triggering the exocytosis of insulin. GSIS requires increased mitochondrial ATP generation up to the metabolic threshold required for robust insulin release [3,10,11,12,13,14,15,16,17]. Mitochondrial oxidative phosphorylation (OXPHOS) is the only process that couples glucose metabolism and generates sufficient ATP required to close KATP channels and stimulate insulin secretion (Figure 2 and Figure 4). Glucose elevation triggering OXPHOS is carried out by five mitochondrial respiratory-chain (MRC) multimeric-enzyme complexes, utilizing reduced coenzymes, reduced nicotinamide adenine dinucleotide (NADH), and reduced flavin adenine dinucleotide (FADH2) [12,15,16,17]. These four MRC complexes are located in the mitochondrial inner membrane, and coenzyme Q and cytochrome c are located in the inner membrane space. Complexes I, III, and IV (cytochrome c oxidase, COX) utilize the energy from the electron transfer to pump protons from the matrix across to the intermembrane space, creating an electrochemical gradient used to generate ATP via complex V (ATP synthase). COX catalyzes the last rate-limiting electron transport chain (ETC) step, receiving electrons from cytochrome c and transferring them to oxygen (Figure 2 and Figure 4) [11,18,19,20]. Impairment of β-cell mitochondrial ATP production leads to insufficient insulin release [2,3,5,10,11,12,13,14,15,16,21,22,23,24,25,26,27,28,29,30,31]. β-cells develop special features to ensure a tight coupling of glucose concentration to insulin secretion. β-cells perform cell-specific gene expression and repression of specific housekeeping genes. They express high metabolic sensing enzymes, including the glucose transporter GLUT2 and glucokinase, and selectively suppress enzymes such as lactate dehydrogenase A (LDHA) and the pyruvate/lactate transporter monocarboxylic acid transporter (MCT-1), which are considered genes “disallowed” to shuttle glucose, via pyruvate, to the mitochondria to fuel ATP production [6,32,33]. To replenish the cytosolic NAD+ and FADH electron donors, β-cells acquire unique shuttles, the malate–aspartate shuttle and the glycerophosphate shuttle [13,34].
Mitochondrial dysfunction in T2D: Despite the well-characterized role of mitochondria in β-cell stimulus-secretion coupling, there is a relative scarcity of data in humans implicating mitochondrial dysfunction in the pathogenesis of T2D. Observation in T2D patients supports the link between reduced ATP generation by the mitochondria and β-cell dysfunction. Abnormal mitochondrial morphology and reduced GSIS are found in β-cells from postmortem T2D patients [15,35,36,37,38,39]. Round and swollen mitochondria impact mitochondrial function, driving impaired GSIS, as shown in a study in which islets from T2D subjects failed to reverse hyperglycemia when transplanted into diabetic mice, while islets from healthy humans in equivalent numbers managed to do so [37]. T2D islets had a significantly lower selective response to glucose, whereas the response to non-nutrient secretagogues arginine and glipalamides remained, a phenomenon characteristic of β-cells with mitochondrial dysfunction [2,8,11,12,17]. Patients with mitochondrial diabetes harbor point mutations or deletions in mtDNA [40,41,42,43]. Swollen, larger mitochondria also appear in mice fed a high-fat diet for 12 weeks [44]. In our studies, we found reduced GSIS and hyperglycemia in tight association with swollen, larger mitochondria and endoplasmic reticulum (ER) in islets of the Cohen diabetic sensitive (CDs) rat, a unique, inbred, nutritionally induced T2D rat model (Figure 5A,B) [8,45,46,47]. CDs rats maintain normoglycemia on a regular diet (RD) but develop hyperglycemia after exposure to a diabetogenic diet related to markedly reduced GSIS [8,46,47,48]. Our recent observations demonstrated a significant correlation (p < 0.01) between CDs islet-COX activity and GSIS, implicating a critical role of islet-COX activity as a modulator of GSIS [10].
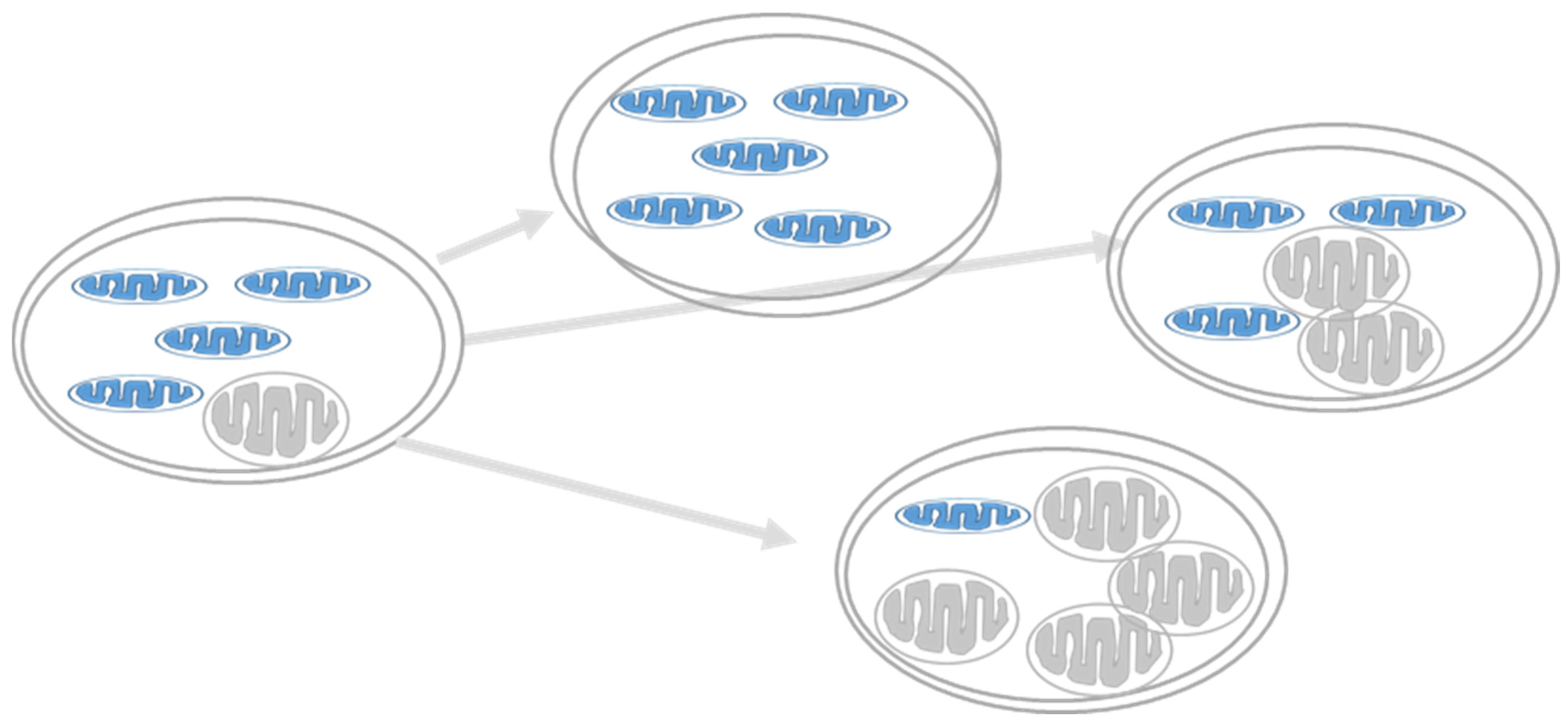
Human mitochondrial DNA (mtDNA) is a circular, double-stranded, supercoiled molecule that encodes 37 genes, essential for OXPHOS and mitochondrial protein synthesis [49]. There are approximately 1000 mitochondrial genomes in most cells, and their replication is independent of the cell cycle. mtDNA has a 10–20 times higher replication rate than nuclear DNA (ntDNA), which can explain the much higher rate of sporadic mutations in mtDNA. It is common for mutations to affect only some of the hundreds of mitochondria copies in the cell, leaving many unaffected. Heteroplasmy is the presence of more than one type of mitochondrial DNA within one cell (Figure 6). A biochemical threshold is associated with an increased mutant mtDNA percentage causing decreased OXPHOS function and phenotype onset, as described in classical mitochondrial diseases explaining the different phenotypes for comparable mutations and the other ages of onset, as well as the severity of the disorder [50,51]. In β-cells, the tight coupling between glucose metabolism and insulin secretion is exclusively dependent on the mitochondria to generate sufficient ATP production required for insulin secretion. This is a pivotal situation in β-cells because increasing mitochondrial function ensues reactive oxygen species (ROS) production, which has a deleterious effect on its function. β-cells are highly sensitive to oxidative stress due to their low antioxidant defense mechanism, the lack of protective histones, and poor DNA-repair mechanisms [52,53,54]. Thus, the accumulation of mtDNA mutations in β-cells could increase the prevalence of mitochondrial-related reduced GSIS and T2D [14,50].
3. Organization of The Electron Transport Chain
Mitochondria function relies on their structure and the organization of OXPHOS complexes. MRC complexes are arranged in the inner mitochondrial membrane as a “respirasome” [56,57]. The respirasome are supercomplexes made of complexes I, III, and IV. This arrangement allows substrate channeling or direct transfer of electrons from one enzyme to another, providing kinetic advantage, efficient electron flow, and stability, thereby reducing electron loss and ROS generation [3,56]. In accord, fused or fragmented mitochondria observed in islets of T2D donors exhibited selective impairment in GSIS [35,58,59,60,61]. Defective assembly of MRC supercomplexes was associated with diabetes in animal models [62] and the rectus abdominis muscle of obese individuals with T2D [63]. Reduced expression of a set of OXPHOS genes affecting the generation of MRC complex subunits was observed in islets of T2D patients and animal models of diabetes [64,65,66]. These observations further support a critical role of mitochondrial dysfunction and subsequent ROS overproduction in the pathogenic process leading to β-cell dysfunction. Yet, the existence and functional significance of respirasomes in islet β-cells are still under investigation.
4. Cytochrome Coxidase (COX)
COX plays a vital role in regulating mitochondrial respiration and OXPHOS to adjust ATP production to specific-cellular energy requirements, depending on nutritional or environmental stimuli. However, COX activity in pancreatic β-cells probably differs from other cell types, as it requires firm and tight regulation to ensure that the rate of insulin secretion closely mirrors the change in blood glucose levels. β-cells are totally dependent on OXPHOS for ATP production and cannot accelerate glycolysis to compensate for decreased OXPHOS due to repressed lactate dehydrogenase expression. COX adapts to changing environmental conditions [19,20]. Proper assembly of COX catalytic subunits is essential for its function. Any change or removal of nuclear subunits results in impaired COX activity and impairs the formation of supercomplexes, while mutations cause different mitochondrial diseases [67,68] (Figure 7). However, the role of COX-impairment in human T2D is not established. COX catalyzes the ETC rate-limiting step of OXPHOS, receiving electrons from cytochrome c and transferring them to oxygen. The three mitochondrial encoded COX subunits, subunits 1, 2, and 3 (COX1, COX2, COX3), build the catalytic core of the enzyme. There is a debate regarding the number of nuclear-encoded subunits that regulate, stabilize, assemble, and anchor the enzyme complex to the inner membrane of the mitochondria [19,67,68,69]. Originally, 13 subunits were reported, but recently, NDUFA4 was identified as a subunit of complex I and was suggested to be the 14th subunit of COX [19,70,71,72]. COX is usually found as a dimer, but monomeric crystal structures have also been reported to form supercomplexes, suggesting that equilibrium could exist between the dimeric and monomeric forms of COX [19,70,72]. NDUFA4 was meant to bind to the monomeric form of COX, thereby excluding the formation of COX dimers [19]. Under this proposal, NDUFA4 could bind complex I or COX under specific situations. Kadenbach et al. suggest two forms of COX: a “relaxed state” and an “active state”. The relaxed state maintains adequate membrane potential and prevents ROS generation. In this state, allosteric ATP inhibition of the phosphorylated and dimeric COX maintains a low and acceptable mitochondrial membrane. In the “active state”, dephosphorylated monomeric COX binds to NDUFA4, stabilizing it in its monomeric form. In stress situations, allosteric ATP inhibition is abolished by Ca2+-activated dephosphorylation, leading to monomerization and NDUFA4 moving from complex I to COX. The “active state” induces higher rates of COX activity and ATP synthesis but increases ROS formation, decreasing mitochondrial efficiency [19,73]. Excessively produced ROS in mitochondria participate in the generation of multiple diseases, including diabetes mellitus and mitochondrial disorders [11,16,18,52,68,74]. In a recent study, decreased COX activity and increased ROS production were observed in streptozotocin-diabetes-induced rats [74], and another study showed that in the diabetic state, COX activity was decreased in islets [64]. In a recent study, we demonstrated that COX activity was initially ~30% reduced in islets of normoglycemic CDs. When fed a diabetogenic diet, islet-COX activity reduced to ≥46% of baseline, resulting in a parallel decrease in GSIS and increase in blood glucose levels. The progressive reduction in COX activity in CDs islets positively correlated with decreasing GSIS (R2 = 0.9691, p < 0.001) and inversely with the elevation in blood glucose levels (R2 = 0.8396, p < 0.001), supporting the notion that islet-COX activity could be a major metabolic regulator of β-cell function [10]. Moreover, our study suggests that reduced islet-COX activity could be a primary inborn defect that underlies the decrease in β-cell dysfunction when exposed to environmental stressors [10]
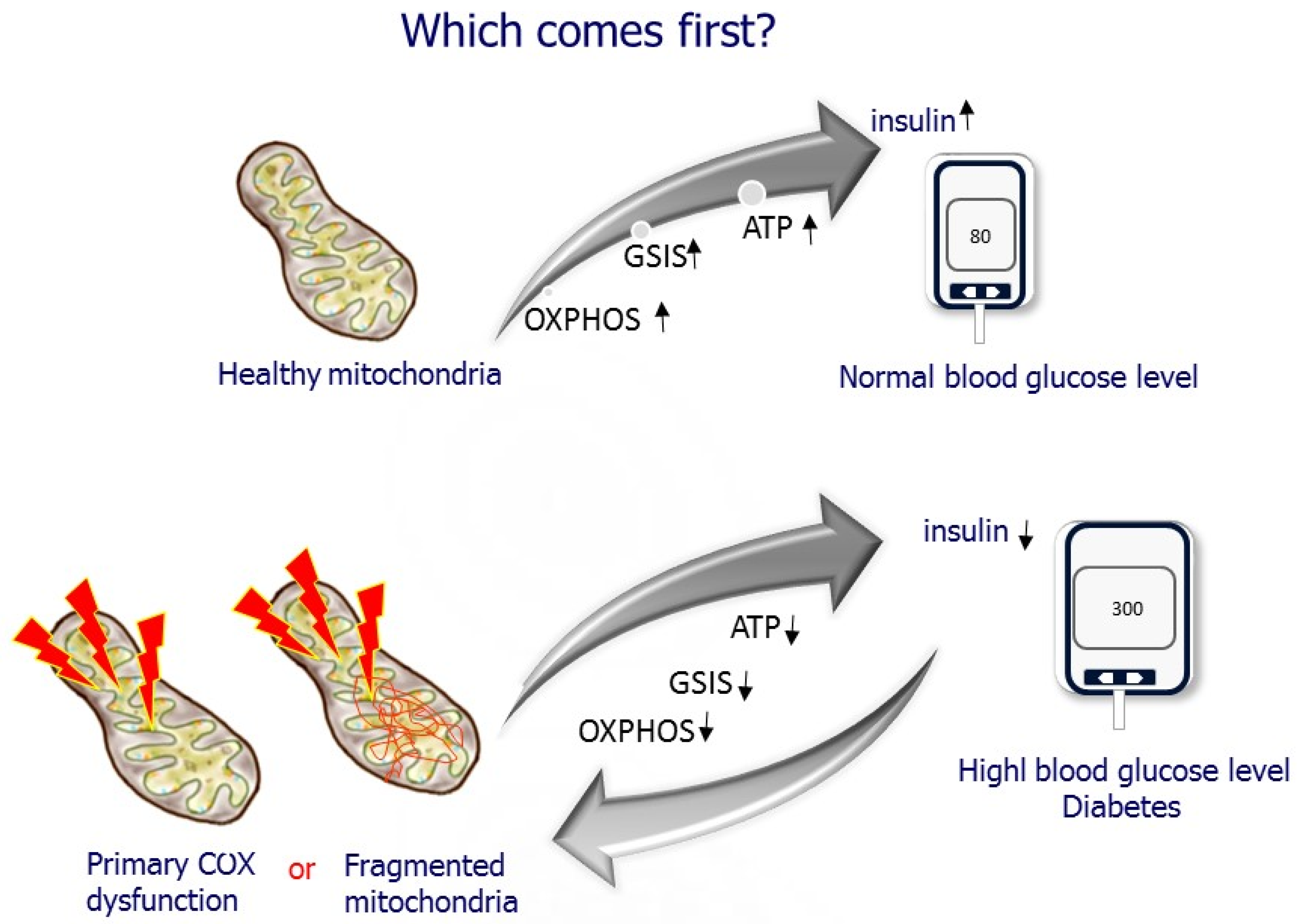
5. Which Comes First? Diabetes or Mitochondrial Dysfunction?
The presence of β-cell dysfunction as an early defining event in the onset and progress of T2D is currently the predominantly accepted view [13,15,22]. However, the cellular and molecular mechanism underlying β-cell dysfunction and what is the major contributor are unresolved. The question is, which comes first? Does hyperglycemia cause deleterious effects on β-cell mitochondria? In support of this assumption, studies have demonstrated changes in β-cell metabolism, and markedly reduced mitochondrial function has been observed in isolated islets from T2D donors, animal models of diabetes, human islets, and β-cell lines exposed to high glucose levels [22,35,75,76,77,78]. On the other hand, defective mitochondrial function decreases GSIS, as demonstrated in studies using drugs affecting the MRC and mutations and depletion of the mitochondrial genome [3,12,13,15]. However, most of those studies relate ETC dysfunction to excessive ROS generation causing cellular injury. β-cell overstimulation has recently been proposed as the starting point initiating the harmful process of β-cell dysfunction in the face of insulin resistance [13,22]. Our studies suggest the possibility of reduced islet-COX activity as a primary cause of β-cell dysfunction in T2D in a rat model that does not exhibit insulin resistance centering islet-COX dysfunction [8,10,46,47], as described in the following sections (Figure 8).
6. COX Activity as a Primary Cause of β-Cell Dysfunction in T2D
We examine whether mitochondrial dysfunction could precede hyperglycemia promoting diabetes development in an inbred, lean model of nutritionally induced diabetes, the Cohen diabetic sensitive (CDs) rat [8,47]. The CDs rat develops hyperglycemia only when fed a diabetogenic diet but maintains normoglycemia on a regular diet (RD). CDs rats do not exhibit insulin resistance and hyperglycemia due to selective impairment in GSIS. Insulin secretion induced by non-nutrient secretagogues arginine and tolbutamide is sustained, suggesting the selective GSIS impairment related to mitochondrial deficiency. Moreover, diabetes is nutritionally induced in this model, providing the opportunity to study the relationship between GSIS and COX activity at the prediabetic state, at different stages during diabetes development, and at a full diabetic condition [8,46,47]. We found that COX activity was initially reduced by 50% in the CDs rats before diabetes development. Islets isolated from hyperglycemic CDs rats exhibited markedly diminished COX activity, leaving only 15% residual activity, thereby inducing a comparable decrease in ATP production and GSIS (Figure 9) [45]
We also demonstrate a direct link between inhibition of COX activity and insulin secretion by incubating isolated islets with the COX-specific inhibitor, potassium cyanide, and demonstrating an apparent dose-dependent reduction in insulin secretion in isolated islets exposed to different doses of potassium cyanide (Figure 10).
Interestingly, we also found that reduced islet-COX activity and GSIS were highly associated with the pancreatic inflammatory process featured by peri-islet infiltration of fat and activated macrophages releasing the proinflammatory cytokine interleukin 1-beta (IL1-β). IL1-β dose-dependently diminishes GSIS by substantially increasing nitrite levels [8,45]. The increased NO production may interact and inhibit the function of several mitochondrial respiratory-chain components by reversible S-nitrosation. However, our data support the notion that NO competes with the oxygen binding, inhibiting COX. Moreover, the interference with ETC favors the production of reactive oxygen species, which by further interaction with nitric oxide (NO) form peroxynitrite, inducing cellular damage [53,79,80,81]. Hyperglycemia was fully counteracted by treating the CDs rats with specific-IL-1β antibodies [82].
In a recent study, we linked reduced mitochondrial COX activity in islets, insulin secretion, and glucose homeostasis. We demonstrated that hyperglycemia was initiated when islet-COX activity decreased below 46% of baseline, identifying a novel islet-COX activity threshold required to sustain normoglycemia (Figure 11). We demonstrated a progressive reduction in COX activity in CDs islets that correlated positively with the decreasing GSIS (R2 = 0.9691, p < 0.001) and inversely with the elevation in blood glucose levels (R2 = 0.8396, p < 0.001), suggesting that islet-COX activity could be a significant metabolic sensor in pancreatic β-cells. Our studies concluded that: (1) islet-COX activity is directly associated with GSIS; (2) pancreatic inflammation reduced GSIS via NO production triggered by IL-1β secreted from peri-islet-infiltrating macrophages; (3) COX activity was markedly reduced in hyperglycemic CDs due to a pre-existing inborn (primary) impairment in mitochondrial respiratory chain dysfunction setting the initial islet-COX activity at ≤46% of baseline, thus enabling an almost complete inhibition of islet-COX activity, diminished GSIS and increased blood glucose levels.
7. Is Diabetes a Mitochondrial Disease? What Is the Therapeutic Importance?
Diabetes mellitus is not a classical mitochondrial disease (Table 1). T2D lacks typical features of encephalopathy, lactic acidosis, and stroke-like episodes. Yet, it is the most common endocrine disease in inherited mitochondrial diseases [83,84]. So far, only variations in essential OXPHOS genes relate to mitochondrial dysfunction and reducing insulin secretion were reported in T2D [65,83,84,85]. Pancreatic β-cells maintain glucose homeostasis by “coupling”, fine-tuning glucose metabolism to insulin secretion, a process that requires OXPHOS. In accord, β-cells have unique features to ensure ATP generation by OXPHOS and adequate GSIS, including suppression of housekeeping genes (“disallowed” genes) that are usually expressed in other cells [6,32,33,86]. Yet, despite the well-characterized role of mitochondria in β-cell stimulus-secretion coupling, there is a relative lack of data in humans implicating mitochondrial dysfunction as a primary defect in T2D. Our studies provide evidence implicating reduced islet-COX activity as a primary event in the decreasing GSIS in pancreatic islets of CDs rats [10,45,46].
In conclusion, T2D is highly dependent on the mitochondria and as such could be considered a mitochondrial-associated disease. However, the question of whether it is a primary mitochondrial disease remains open. Diabetes mellitus is part of the pathologies of the mitochondrial disease exhibiting distinctive clinical features and complications that are not found in classical diabetes [41,84]. It is therefore essential to identify these patients and provide the correct treatment. Metformin, the first-choice treatment in T2D is not recommended because of the risk of lactic acidosis [87], while SGLT-2i and GLP-1-related substances improving mitochondrial functions have significant benefits for diabetic patients with identified mitochondrial mutations [88].
Funding
Ministry of Science and Culture of the State of Lower Saxony, Hannover, Germany and the AM Cohen Foundation for the Advancement of Research of the Cohen Diabetic Rat.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ashcroft, F.M.; Rorsman, P. Diabetes mellitus and the beta cell: The last ten years. Cell 2012, 148, 1160–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, D.G. The Pancreatic beta-Cell: A Bioenergetic Perspective. Physiol. Rev. 2016, 96, 1385–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parnis, J.; Rutter, G.A.; Béatrice, M.; Luc, P. Mitochondria in Obesity and Type 2 Diabetes; Academic Press: Amsterdam, The Netherlands, 2019; pp. 433–445. [Google Scholar]
- Prasad, R.B.; Groop, L. Genetics of Type 2 Diabetes—Pitfalls and Possibilities. Genes 2015, 6, 87–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prentki, M.; Nolan, C.J. Islet beta cell failure in type 2 diabetes. J. Clin. Investig. 2006, 116, 1802–1812. [Google Scholar] [CrossRef] [Green Version]
- Rutter, G.A.; Georgiadou, E.; Martinez-Sanchez, A.; Pullen, T.J. Metabolic and functional specializations of the pancreatic beta cell: Gene disallowance, mitochondrial metabolism and intercellular connectivity. Diabetologia 2020, 63, 1990–1998. [Google Scholar] [CrossRef]
- Skyler, J.S.; Bakris, G.L.; Bonifacio, E.; Darsow, T.; Eckel, R.H.; Groop, L.; Groop, P.-H.; Handelsman, Y.; Insel, R.A.; Mathieu, C.; et al. Differentiation of Diabetes by Pathophysiology, Natural History, and Prognosis. Diabetes 2016, 66, 241–255. [Google Scholar] [CrossRef] [Green Version]
- Weksler-Zangen, S.; Raz, I.; Lenzen, S.; Jorns, A.; Ehrenfeld, S.; Amir, G.; Oprescu, A.; Yagil, Y.; Yagil, C.; Zangen, D.H.; et al. Impaired Glucose-Stimulated Insulin Secretion Is Coupled with Exocrine Pancreatic Lesions in the Cohen Diabetic Rat. Diabetes 2008, 57, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Association, A.D. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2021. Diabetes Care 2020, 43, S14–S31. [Google Scholar] [CrossRef] [Green Version]
- Aharon-Hananel, G.; Romero-Afrima, L.; Saada, A.; Mantzur, C.; Raz, I.; Weksler-Zangen, S. Cytochrome c Oxidase Activity as a Metabolic Regulator in Pancreatic Beta-Cells. Cells 2022, 11, 929. [Google Scholar] [CrossRef]
- Sha, W.; Hu, F.; Bu, S. Mitochondrial dysfunction and pancreatic islet beta-cell failure (Review). Exp. Ther. Med. 2020, 20, 266. [Google Scholar] [CrossRef]
- Prasun, P. Role of mitochondria in pathogenesis of type 2 diabetes mellitus. J. Diabetes Metab. Disord. 2020, 19, 2017–2022. [Google Scholar] [CrossRef] [PubMed]
- Esser, N.; Utzschneider, K.M.; Kahn, S.E. Early beta cell dysfunction vs insulin hypersecretion as the primary event in the pathogenesis of dysglycaemia. Diabetologia 2020, 63, 2007–2021. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef]
- Fex, M.; Nicholas, L.M.; Vishnu, N.; Medina, A.; Sharoyko, V.V.; Nicholls, D.G.; Spegel, P.; Mulder, H. The pathogenetic role of beta-cell mitochondria in type 2 diabetes. J. Endocrinol. 2018, 236, R145–R159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulder, H. Transcribing beta-cell mitochondria in health and disease. Mol. Metab. 2017, 6, 1040–1051. [Google Scholar] [CrossRef]
- Wiederkehr, A.; Wollheim, C.B. Mitochondrial signals drive insulin secretion in the pancreatic beta-cell. Mol. Cell. Endocrinol. 2012, 353, 123–137. [Google Scholar] [CrossRef]
- Vogt, S.; Ramzan, R.; Grossman, L.I.; Singh, K.K.; Ferguson-Miller, S.; Yoshikawa, S.; Lee, I.; Huttemann, M. Mitochondrial respiration is controlled by Allostery, Subunit Composition and Phosphorylation Sites of Cytochrome c Oxidase: A trailblazer’s tale-Bernhard Kadenbach. Mitochondrion 2021, 60, 228–233. [Google Scholar] [CrossRef]
- Kadenbach, B. Complex IV-The regulatory center of mitochondrial oxidative phosphorylation. Mitochondrion 2021, 58, 296–302. [Google Scholar] [CrossRef]
- Timón-Gómez, A.; Bartley-Dier, E.L.; Fontanesi, F.; Barrientos, A. HIGD-Driven Regulation of Cytochrome c Oxidase Biogenesis and Function. Cells 2020, 9, 2620. [Google Scholar] [CrossRef]
- Dabravolski, S.; Orekhova, V.; Baig, M.; Bezsonov, E.; Starodubova, A.; Popkova, T.; Orekhov, A. The Role of Mitochondrial Mutations and Chronic Inflammation in Diabetes. Int. J. Mol. Sci. 2021, 22, 6733. [Google Scholar] [CrossRef]
- Haythorne, E.; Rohm, M.; van de Bunt, M.; Brereton, M.F.; Tarasov, A.I.; Blacker, T.S.; Sachse, G.; Silva Dos Santos, M.; Terron Exposito, R.; Davis, S.; et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic beta-cells. Nat. Commun. 2019, 10, 2474. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Mohammed Al-Amily, I.; Mohammed, S.; Luan, C.; Asplund, O.; Ahmed, M.; Ye, Y.; Ben-Hail, D.; Soni, A.; Vishnu, N.; et al. Preserving Insulin Secretion in Diabetes by Inhibiting VDAC1 Overexpression and Surface Translocation in beta Cells. Cell Metab. 2019, 29, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J.; Breuer, T.G.K.; Bonadonna, R.C.; Tannapfel, A.; Uhl, W.; Schmidt, W.E.; Schrader, H.; Menge, B.A. Pancreatic diabetes manifests when beta cell area declines by approximately 65% in humans. Diabetologia 2012, 55, 1346–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.A.; Zhao, Z.; Turk, J. Mitochondrial dysfunction and beta-cell failure in type 2 diabetes mellitus. Exp. Diabetes Res. 2012, 2012, 703538. [Google Scholar] [CrossRef] [Green Version]
- Pongratz, R.L.; Kibbey, R.G.; Kirkpatrick, C.; Zhao, X.; Pontoglio, M.; Yaniv, M.; Wollheim, C.B.; Shulman, G.; Cline, G.W. Mitochondrial Dysfunction Contributes to Impaired Insulin Secretion in INS-1 Cells with Dominant-negative Mutations of HNF-1α and in HNF-1α-deficient Islets. J. Biol. Chem. 2009, 284, 16808–16821. [Google Scholar] [CrossRef] [Green Version]
- Mulder, H.; Ling, C. Mitochondrial dysfunction in pancreatic beta-cells in Type 2 diabetes. Mol. Cell Endocrinol. 2009, 297, 34–40. [Google Scholar] [CrossRef] [Green Version]
- Henquin, J.C. Regulation of insulin secretion: A matter of phase control and amplitude modulation. Diabetologia 2009, 52, 739–751. [Google Scholar] [CrossRef] [Green Version]
- Wiederkehr, A.; Wollheim, C.B. Minireview: Implication of mitochondria in insulin secretion and action. Endocrinology 2006, 147, 2643–2649. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, P.; Del Prato, S.; Lupi, R.; Del Guerra, S. The pancreatic beta-cell in human Type 2 diabetes. Nutr. Metab. Cardiovasc. Dis. 2006, 16, S3–S6. [Google Scholar] [CrossRef]
- Kahn, S.E. The importance of the beta-cell in the pathogenesis of type 2 diabetes mellitus. Am. J. Med. 2000, 108, 2S–8S. [Google Scholar] [CrossRef]
- Quintens, R.; Hendrickx, N.; Lemaire, K.; Schuit, F. Why expression of some genes is disallowed in beta-cells. Biochem. Soc. Trans. 2008, 36, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Thorrez, L.; Laudadio, I.; Van Deun, K.; Quintens, R.; Hendrickx, N.; Granvik, M.; Lemaire, K.; Schraenen, A.; Van Lommel, L.; Lehnert, S.; et al. Tissue-specific disallowance of housekeeping genes: The other face of cell differentiation. Genome Res. 2010, 21, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prentki, M.; Matschinsky, F.M.; Madiraju, S.M. Metabolic Signaling in Fuel-Induced Insulin Secretion. Cell Metab. 2013, 18, 162–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anello, M.; Lupi, R.; Spampinato, D.; Piro, S.; Masini, M.; Boggi, U.; Del Prato, S.; Rabuazzo, A.M.; Purrello, F.; Marchetti, P. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 2005, 48, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Del Guerra, S.; Lupi, R.; Marselli, L.; Masini, M.; Bugliani, M.; Sbrana, S.; Torri, S.; Pollera, M.; Boggi, U.; Mosca, F.; et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes 2005, 54, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; Vatamaniuk, M.; Huang, X.; Doliba, N.; Lian, M.-M.; Frank, A.; Velidedeoglu, E.; Desai, N.M.; Koeberlein, B.; Wolf, B.; et al. Structural and Functional Abnormalities in the Islets Isolated from Type 2 Diabetic Subjects. Diabetes 2004, 53, 624–632. [Google Scholar] [CrossRef] [Green Version]
- Maechler, P.; de Andrade, P.B. Mitochondrial damages and the regulation of insulin secretion. Biochem. Soc. Trans. 2006, 34, 824–827. [Google Scholar] [CrossRef] [Green Version]
- Maechler, P.; Wollheim, C.B. Mitochondrial function in normal and diabetic beta-cells. Nature 2001, 414, 807–812. [Google Scholar] [CrossRef]
- De Andrade, P.B.; Rubi, B.; Frigerio, F.; van den Ouweland, J.M.; Maassen, J.A.; Maechler, P. Diabetes-associated mitochondrial DNA mutation A3243G impairs cellular metabolic pathways necessary for beta cell function. Diabetologia 2006, 49, 1816–1826. [Google Scholar] [CrossRef]
- Gerbitz, K.-D.; Ouweland, J.M.V.D.; Maassen, J.; Jaksch, M. Mitochondrial diabetes mellitus: A review. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 1995, 1271, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Nakanishi, K.; Nakase, H.; Kajio, H.; Okubo, M.; Murase, T.; Kosaka, K. In situ characterization of islets in diabetes with a mitochondrial DNA mutation at nucleotide position 3243. Diabetes 1997, 46, 1567–1571. [Google Scholar] [CrossRef] [PubMed]
- Velho, G.; Byrne, M.M.; Clement, K.; Sturis, J.; Pueyo, M.E.; Blanche, H.; Vionnet, N.; Fiet, J.; Passa, P.; Robert, J.J.; et al. Clinical phenotypes, insulin secretion, and insulin sensitivity in kindreds with maternally inherited diabetes and deafness due to mitochondrial tRNALeu(UUR) gene mutation. Diabetes 1996, 45, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Fex, M.; Nitert, M.D.; Wierup, N.; Sundler, F.; Ling, C.; Mulder, H. Enhanced mitochondrial metabolism may account for the adaptation to insulin resistance in islets from C57BL/6J mice fed a high-fat diet. Diabetologia 2006, 50, 74–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weksler-Zangen, S.; Aharon-Hananel, G.; Mantzur, C.; Aouizerat, T.; Gurgul-Convey, E.; Raz, I.; Saada, A. IL-1beta hampers glucose-stimulated insulin secretion in Cohen diabetic rat islets through mitochondrial cytochrome c oxidase inhibition by nitric oxide. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E648–E657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weksler-Zangen, S.; Jorns, A.; Tarsi-Chen, L.; Vernea, F.; Aharon-Hananel, G.; Saada, A.; Lenzen, S.; Raz, I. Dietary copper supplementation restores beta-cell function of Cohen diabetic rats: A link between mitochondrial function and glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1023–E1034. [Google Scholar] [CrossRef] [Green Version]
- Weksler-Zangen, S.; Yagil, C.; Zangen, D.H.; Ornoy, A.; Jacob, H.J.; Yagil, Y. The newly inbred cohen diabetic rat: A nonobese normolipidemic genetic model of diet-induced type 2 diabetes expressing sex differences. Diabetes 2001, 50, 2521–2529. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.M. The Cohen Diabetic Rat; Cohen, A.M., Rosenmann, E., Eds.; Karger: Basel, Switzerland, 1990; pp. 1–9. [Google Scholar]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; De Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA heteroplasmy in disease and targeted nuclease-based therapeutic approaches. EMBO Rep. 2020, 21, e49612. [Google Scholar] [CrossRef]
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.-P.; Letellier, T. Mitochondrial threshold effects. Biochem. J. 2003, 370, 751–762. [Google Scholar] [CrossRef] [Green Version]
- Leenders, F.; Groen, N.; de Graaf, N.; Engelse, M.A.; Rabelink, T.J.; de Koning, E.J.P.; Carlotti, F. Oxidative Stress Leads to beta-Cell Dysfunction Through Loss of beta-Cell Identity. Front. Immunol. 2021, 12, 690379. [Google Scholar] [CrossRef]
- Gurgul-Convey, E.; Mehmeti, I.; Lortz, S.; Lenzen, S. Cytokine toxicity in insulin-producing cells is mediated by nitro-oxidative stress-induced hydroxyl radical formation in mitochondria. Klin. Wochenschr. 2011, 89, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Lenzen, S. Oxidative stress: The vulnerable beta-cell. Biochem. Soc. Trans. 2008, 36, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Grady, J.P.; Pickett, S.J.; Ng, Y.S.; Alston, C.L.; Blakely, E.L.; Hardy, S.A.; Feeney, C.L.; Bright, A.A.; Schaefer, A.M.; Gorman, G.S.; et al. mtDNA heteroplasmy level and copy number indicate disease burden in m.3243A>G mitochondrial disease. EMBO Mol. Med. 2018, 10, e8262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letts, J.; Fiedorczuk, K.; Sazanov, L. The architecture of respiratory supercomplexes. Nature 2016, 537, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Schagger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennings, T.G.; Chopra, D.G.; DeLeon, E.R.; VanDeusen, H.R.; Sesaki, H.; Merrins, M.J.; Ku, G.M. In Vivo Deletion of beta-Cell Drp1 Impairs Insulin Secretion Without Affecting Islet Oxygen Consumption. Endocrinology 2018, 159, 3245–3256. [Google Scholar] [CrossRef]
- Jezek, P.; Dlaskova, A. Dynamic of mitochondrial network, cristae, and mitochondrial nucleoids in pancreatic beta-cells. Mitochondrion 2019, 49, 245–258. [Google Scholar] [CrossRef]
- Las, G.; Oliveira, M.F.; Shirihai, O.S. Emerging roles of beta-cell mitochondria in type-2-diabetes. Mol. Aspects Med. 2020, 71, 100843. [Google Scholar] [CrossRef]
- Park, K.-S.; Wiederkehr, A.; Kirkpatrick, C.; Mattenberger, Y.; Martinou, J.-C.; Marchetti, P.; Demaurex, N.; Wollheim, C.B. Selective Actions of Mitochondrial Fission/Fusion Genes on Metabolism-Secretion Coupling in Insulin-releasing Cells. J. Biol. Chem. 2008, 283, 33347–33356. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.P.; Kohler, M.; Graff, C.; Oldfors, A.; Magnuson, M.A.; Berggren, P.O.; Larsson, N.G. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat. Genet. 2000, 26, 336–340. [Google Scholar] [CrossRef]
- Antoun, G.; McMurray, F.; Thrush, A.B.; Patten, D.A.; Peixoto, A.C.; Slack, R.; McPherson, R.; Dent, R.; Harper, M.-E. Impaired mitochondrial oxidative phosphorylation and supercomplex assembly in rectus abdominis muscle of diabetic obese individuals. Diabetologia 2015, 58, 2861–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Koshkin, V.; Allister, E.M.; Gyulkhandanyan, A.V.; Wheeler, M.B. Molecular and metabolic evidence for mitochondrial defects associated with beta-cell dysfunction in a mouse model of type 2 diabetes. Diabetes 2010, 59, 448–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, A.H.; Rönn, T.; Ladenvall, C.; Parikh, H.; Isomaa, B.; Groop, L.; Ling, C. Two common genetic variants near nuclear-encoded OXPHOS genes are associated with insulin secretion in vivo. Eur. J. Endocrinol. 2011, 164, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Wollheim, C.B. Beta-cell mitochondria in the regulation of insulin secretion: A new culprit in Type II diabetes. Diabetologia 2000, 43, 265–277. [Google Scholar] [CrossRef]
- Kadenbach, B.; Huttemann, M. The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion 2015, 24, 64–76. [Google Scholar] [CrossRef]
- Sinkler, C.A.; Kalpage, H.; Shay, J.; Lee, I.; Malek, M.H.; Grossman, L.I.; Huttemann, M. Tissue- and Condition-Specific Isoforms of Mammalian Cytochrome c Oxidase Subunits: From Function to Human Disease. Oxid. Med. Cell. Longev. 2017, 2017, 1534056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadenbach, B.; Stroh, A.; Ungibauer, M.; Kuhn-Nentwig, L.; Büge, U.; Jarausch, J. [4] Isozymes of cytochrome-c oxidase: Characterization and isolation from different tissues. In Methods in enzymology; Academic Press: New York, NY, USA, 1986; Volume 126, pp. 32–45. [Google Scholar] [CrossRef]
- Balsa, E.; Marco, R.; Perales-Clemente, E.; Szklarczyk, R.; Calvo, E.; Landázuri, M.O.; Enríquez, J.A. NDUFA4 Is a Subunit of Complex IV of the Mammalian Electron Transport Chain. Cell Metab. 2012, 16, 378–386. [Google Scholar] [CrossRef] [Green Version]
- Gladyck, S.; Aras, S.; Hüttemann, M.; Grossman, L.I. Regulation of COX Assembly and Function by Twin CX9C Proteins—Implications for Human Disease. Cells 2021, 10, 197. [Google Scholar] [CrossRef]
- Pitceathly, R.D.; Taanman, J.-W. NDUFA4 (Renamed COXFA4) Is a Cytochrome-c Oxidase Subunit. Trends Endocrinol. Metab. 2018, 29, 452–454. [Google Scholar] [CrossRef]
- Kadenbach, B. Regulation of cytochrome c oxidase contributes to health and optimal life. World J. Biol. Chem. 2020, 11, 52–61. [Google Scholar] [CrossRef]
- Friday, D.P.; Alleyneb, T.A.; Ignaciob, D.N.; Arrindellb, D.; Raoa, S.R.; Legall, G. The Impact of Diabetes Mellitus on Oxygen Utilization by Complex IV: Preliminary Insights. J. Endocrinol. Metab. 2017, 7, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Brereton, M.F.; Rohm, M.; Shimomura, K.; Holland, C.; Tornovsky-Babeay, S.; Dadon, D.; Iberl, M.; Chibalina, M.V.; Lee, S.; Glaser, B.; et al. Hyperglycaemia induces metabolic dysfunction and glycogen accumulation in pancreatic beta-cells. Nat. Commun. 2016, 7, 13496. [Google Scholar] [CrossRef] [PubMed]
- Gohring, I.; Sharoyko, V.V.; Malmgren, S.; Andersson, L.E.; Spegel, P.; Nicholls, D.G.; Mulder, H. Chronic high glucose and pyruvate levels differentially affect mitochondrial bioenergetics and fuel-stimulated insulin secretion from clonal INS-1 832/13 cells. J. Biol. Chem. 2014, 289, 3786–3798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, J.; Li, Z.; Zhong, W.; Hao, Q.; Lei, L.; Wang, L.; Zhao, D.; Xu, P.; Zhou, Y.; Wang, Y.; et al. Temporal Transcriptomic and Proteomic Landscapes of Deteriorating Pancreatic Islets in Type 2 Diabetic Rats. Diabetes 2017, 66, 2188–2200. [Google Scholar] [CrossRef] [Green Version]
- Segerstolpe, Å.; Palasantza, A.; Eliasson, P.; Andersson, E.-M.; Andréasson, A.-C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.C.; Borutaite, V. Nitric oxide inhibition of mitochondrial respiration and its role in cell death. Free Radic. Biol. Med. 2002, 33, 1440–1450. [Google Scholar] [CrossRef]
- Cnop, M.; Welsh, N.; Jonas, J.C.; Jorns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes. 2005, 54, S97–S107. [Google Scholar] [CrossRef] [Green Version]
- Cooper, C.E.; Davies, N.A.; Psychoulis, M.; Canevari, L.; Bates, T.E.; Dobbie, M.S.; Casley, C.S.; Sharpe, M.A. Nitric oxide and peroxynitrite cause irreversible increases in the K(m) for oxygen of mitochondrial cytochrome oxidase: In vitro and in vivo studies. Biochim. Biophys. Acta. 2003, 1607, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Aharon-Hananel, G.; Jorns, A.; Lenzen, S.; Raz, I.; Weksler-Zangen, S. Antidiabetic Effect of Interleukin-1beta Antibody Therapy Through beta-Cell Protection in the Cohen Diabetes-Sensitive Rat. Diabetes 2015, 64, 1780–1785. [Google Scholar] [CrossRef] [Green Version]
- Chow, J.; Rahman, J.; Achermann, J.; Dattani, M.T.; Rahman, S. Mitochondrial disease and endocrine dysfunction. Nat. Rev. Endocrinol. 2016, 13, 92–104. [Google Scholar] [CrossRef]
- Yee, M.L.; Wong, R.; Datta, M.; Fazio, T.N.; Ebrahim, M.M.; McNamara, E.C.; De Jong, G.; Gilfillan, C. Mitochondrial disease: An uncommon but important cause of diabetes mellitus. Endocrinol. Diabetes Metab. Case Rep. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.H.; Yang, B.T.; Hall, E.; Taneera, J.; Salehi, A.; Nitert, M.D.; Ling, C. Decreased expression of genes involved in oxidative phosphorylation in human pancreatic islets from patients with type 2 diabetes. Eur. J. Endocrinol. 2011, 165, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Pullen, T.; Khan, A.; Barton, G.; Butcher, S.A.; Sun, G.; Rutter, G. Identification of genes selectively disallowed in the pancreatic islet. Islets 2010, 2, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Keidai, Y.; Iwasaki, Y.; Honjo, S.; Aizawa-Abe, M.; Iwasaki, K.; Hamasaki, A. “Switched” metabolic acidosis in mitochondrial diabetes mellitus. J. Diabetes Investig. 2019, 10, 1116–1117. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.L.; Roddick, A.J.; Aghar-Jaffar, R.; Shun-Shin, M.J.; Francis, D.; Oliver, N.; Meeran, K. Association Between Use of Sodium-Glucose Cotransporter 2 Inhibitors, Glucagon-like Peptide 1 Agonists, and Dipeptidyl Peptidase 4 Inhibitors with All-Cause Mortality in Patients With Type 2 Diabetes: A Systematic Review and Meta-analysis. JAMA 2018, 319, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The pancreas is a mixed gland that contains both an endocrine and an exocrine digestive function. The pancreatic islets of Langerhans are irregularly shaped patches of endocrine tissue scattered within the pancreas. The islet includes five types of cells secreting different hormones: α cells produce glucagon released to the blood when glucose concentration is low (20% of total islet cells); β-cells, the only source of insulin in the body, release insulin when blood glucose concentration increases (≈55–70% of total islet cells); Δ cells produce somatostatin that inhibits insulin and glucagon secretion (<10% of total islet cells); ε cells produce ghrelin, the “hunger hormone” (<1% of total islet cells); PP cells or γ cells produce pancreatic polypeptide that regulates pancreatic secretion activities (<5% of total islet cells).
Figure 1.
The pancreas is a mixed gland that contains both an endocrine and an exocrine digestive function. The pancreatic islets of Langerhans are irregularly shaped patches of endocrine tissue scattered within the pancreas. The islet includes five types of cells secreting different hormones: α cells produce glucagon released to the blood when glucose concentration is low (20% of total islet cells); β-cells, the only source of insulin in the body, release insulin when blood glucose concentration increases (≈55–70% of total islet cells); Δ cells produce somatostatin that inhibits insulin and glucagon secretion (<10% of total islet cells); ε cells produce ghrelin, the “hunger hormone” (<1% of total islet cells); PP cells or γ cells produce pancreatic polypeptide that regulates pancreatic secretion activities (<5% of total islet cells).

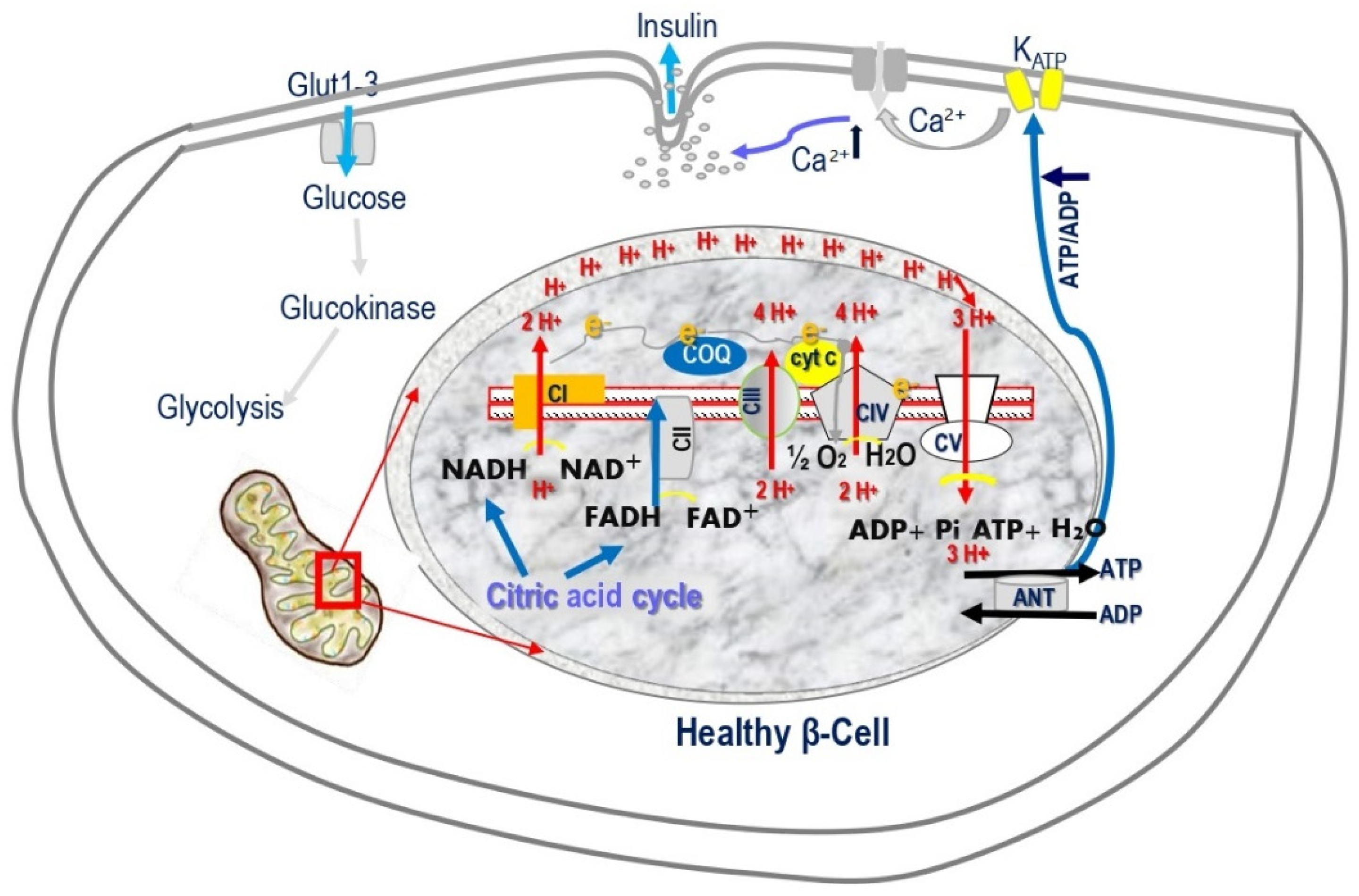
Figure 2.
Glucose-stimulated insulin secretion (GSIS), the triggering pathway, includes entry of glucose into β-cells, acceleration of glucose metabolism, increase in ATP content and ATP/ADP ratio, closure of ATP-sensitive K+ channels (KATP channels), membrane depolarization, opening of voltage-dependent Ca2+ channels (VDCCs), increase in Ca2+ influx through VDCCs, raised intracellular Ca2+ concentration ([Ca2+] i), and exocytosis of insulin granules.
Figure 2.
Glucose-stimulated insulin secretion (GSIS), the triggering pathway, includes entry of glucose into β-cells, acceleration of glucose metabolism, increase in ATP content and ATP/ADP ratio, closure of ATP-sensitive K+ channels (KATP channels), membrane depolarization, opening of voltage-dependent Ca2+ channels (VDCCs), increase in Ca2+ influx through VDCCs, raised intracellular Ca2+ concentration ([Ca2+] i), and exocytosis of insulin granules.

Figure 3.
Glucose depends on insulin to enter the peripheral tissues, muscles, and fat. Without insulin, glucose stays in the blood, causing diabetes. The increase in blood glucose concentration triggers insulin secretion. Blood glucose levels, brown graph; the increase in blood glucose level triggers a prompt and parallel increase in insulin secretion, pink graph. With time, blood glucose levels reduce due to insulin’s facilitated entry into fat and muscles. In accord, reducing blood glucose levels is paralleled by lowering insulin levels.
Figure 3.
Glucose depends on insulin to enter the peripheral tissues, muscles, and fat. Without insulin, glucose stays in the blood, causing diabetes. The increase in blood glucose concentration triggers insulin secretion. Blood glucose levels, brown graph; the increase in blood glucose level triggers a prompt and parallel increase in insulin secretion, pink graph. With time, blood glucose levels reduce due to insulin’s facilitated entry into fat and muscles. In accord, reducing blood glucose levels is paralleled by lowering insulin levels.

Figure 4.
Mitochondrial oxidative phosphorylation (OXPHOS) is the only process that generates sufficient ATP to induce insulin secretion. OXPHOS is carried out by the mitochondrial respiration chain (MRC) located in the mitochondrial inner membrane. Electrons move between the four complexes and are transferred to oxygen by complex IV, cytochrome coxidase (COX). The free energy released pumps protons at complexes I, III, and IV to the intermembrane space, creating a proton gradient. At complex V, ATP synthase, protons diffuse along this electrochemical gradient to generate ATP.
Figure 4.
Mitochondrial oxidative phosphorylation (OXPHOS) is the only process that generates sufficient ATP to induce insulin secretion. OXPHOS is carried out by the mitochondrial respiration chain (MRC) located in the mitochondrial inner membrane. Electrons move between the four complexes and are transferred to oxygen by complex IV, cytochrome coxidase (COX). The free energy released pumps protons at complexes I, III, and IV to the intermembrane space, creating a proton gradient. At complex V, ATP synthase, protons diffuse along this electrochemical gradient to generate ATP.

Figure 5.
Photomicrographs of pancreas sections of hyperglycemic Cohen diabetic rats [8,10,47]. (A) The β-cells in the center of the islets exhibited well-preserved mitochondria (arrow) and normal ER (arrowhead). The β-cells in the periphery of the islets (B) showed swollen mitochondria (arrowhead) and dilated ER. EM magnification 18,000×.
Figure 5.
Photomicrographs of pancreas sections of hyperglycemic Cohen diabetic rats [8,10,47]. (A) The β-cells in the center of the islets exhibited well-preserved mitochondria (arrow) and normal ER (arrowhead). The β-cells in the periphery of the islets (B) showed swollen mitochondria (arrowhead) and dilated ER. EM magnification 18,000×.

Figure 6.
Heteroplasmy and diabetes. Heteropalsmy is the relative proportion of mtDNA copies carrying the mutation in a specific tissue. A specific phenotype develops when a certain threshold level of mutation is reached (usually ~60%) [55].
Figure 6.
Heteroplasmy and diabetes. Heteropalsmy is the relative proportion of mtDNA copies carrying the mutation in a specific tissue. A specific phenotype develops when a certain threshold level of mutation is reached (usually ~60%) [55].

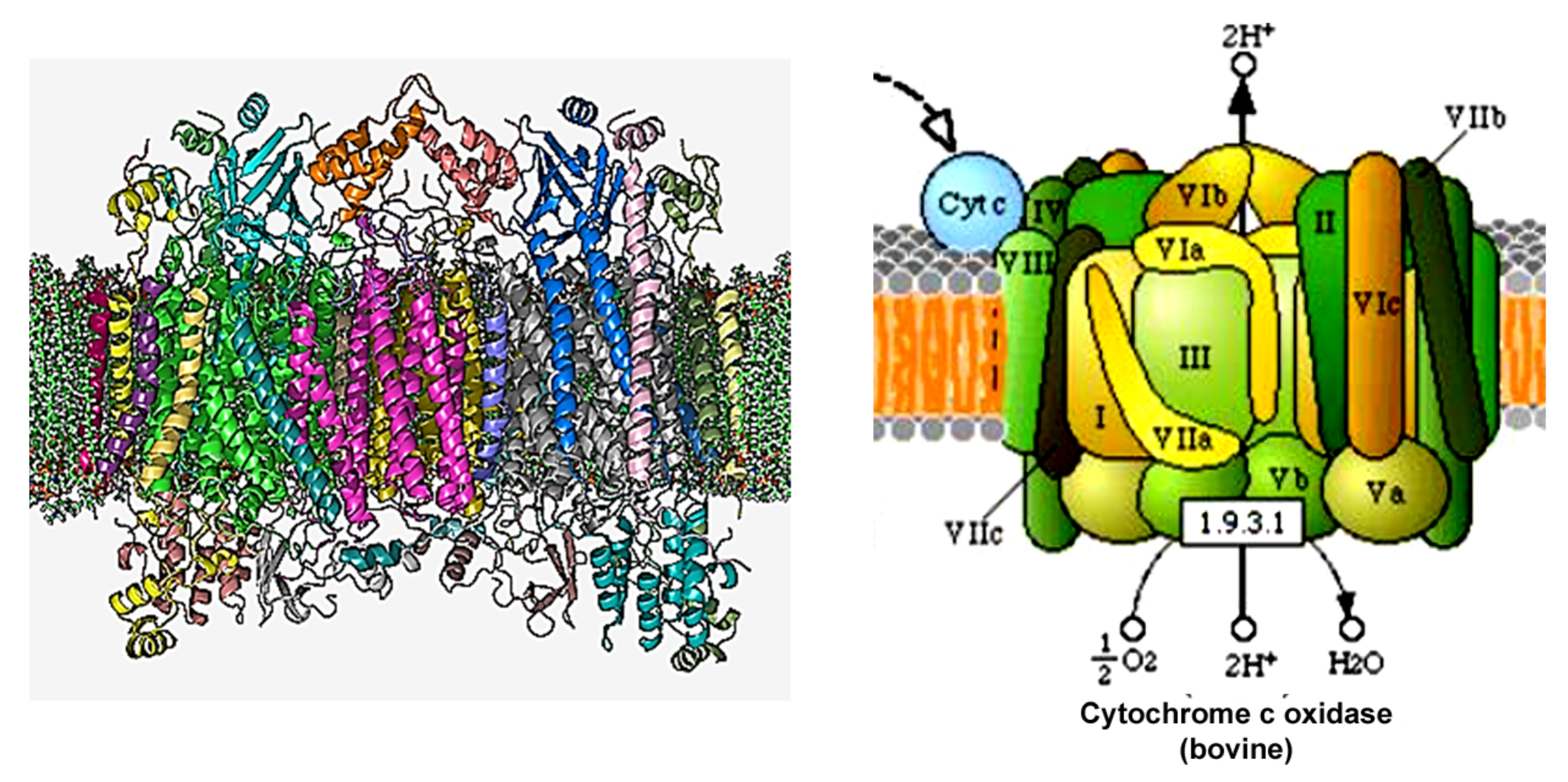
Figure 7.
(Left) The crystal structure of bovine homodimer cytochrome c oxidase in a phospholipid bilayer. Adapted from The Protein Data Bank (PDB) PDB: 1OCC. PDB structures: RCSB PDB PDBe PDBsum. Diagrammatic representation of COX's subunits in different colors. (Right) A diagram of COX taken from the KEGG pathways (https://www.genome.jp/kegg-bin/show_pathway?map00190 accessed on 5 February 2022).
Figure 7.
(Left) The crystal structure of bovine homodimer cytochrome c oxidase in a phospholipid bilayer. Adapted from The Protein Data Bank (PDB) PDB: 1OCC. PDB structures: RCSB PDB PDBe PDBsum. Diagrammatic representation of COX's subunits in different colors. (Right) A diagram of COX taken from the KEGG pathways (https://www.genome.jp/kegg-bin/show_pathway?map00190 accessed on 5 February 2022).

Figure 8.
Which comes first? Does mitochondrial dysfunction cause diabetes, or does diabetes hamper mitochondrial function?
Figure 8.
Which comes first? Does mitochondrial dysfunction cause diabetes, or does diabetes hamper mitochondrial function?

Figure 9.
(A) Glucose-stimulated insulin secretion (GSIS), (B) cytochrome c oxidase (COX) activity, and ATP content (C) in islets isolated from Cohen diabetic sensitive (CDs) rats or control rats fed either a regular diet (black bars) or a diabetogenic diet (gray bars). (A) GSIS measured at 1.7 (baseline) and 16.7 mmol/L (stimulatory) glucose concentrations. (B) COX activity normalized to citrate synthase (CS) activity (COX/CS). (C) ATP content (pmol/islet) determined by luciferin luciferase. Data are means ± SE for at least three separate experiments. * p < 0.01. Figure is used and modified from [45] with permission from the publisher.
Figure 9.
(A) Glucose-stimulated insulin secretion (GSIS), (B) cytochrome c oxidase (COX) activity, and ATP content (C) in islets isolated from Cohen diabetic sensitive (CDs) rats or control rats fed either a regular diet (black bars) or a diabetogenic diet (gray bars). (A) GSIS measured at 1.7 (baseline) and 16.7 mmol/L (stimulatory) glucose concentrations. (B) COX activity normalized to citrate synthase (CS) activity (COX/CS). (C) ATP content (pmol/islet) determined by luciferin luciferase. Data are means ± SE for at least three separate experiments. * p < 0.01. Figure is used and modified from [45] with permission from the publisher.

Figure 10.
Glucose-stimulated insulin secretion of islets isolated from control rats fed a regular diet and incubated for 1 h with either 0, 0.4, 0.8, or 8 mol/L potassium cyanide (KCN) in the presence of 1.7 (baseline) or 16.7 mmol/L (stimulatory) glucose. The figure expresses insulin secretions measured as insulin secreted under stimulatory glucose minus insulin secreted under baseline glucose concentrations per islet of insulin content. Data are means ± SE for at least three separate experiments, * p < 0.01. Figure is used from [45] with permission from the publisher.
Figure 10.
Glucose-stimulated insulin secretion of islets isolated from control rats fed a regular diet and incubated for 1 h with either 0, 0.4, 0.8, or 8 mol/L potassium cyanide (KCN) in the presence of 1.7 (baseline) or 16.7 mmol/L (stimulatory) glucose. The figure expresses insulin secretions measured as insulin secreted under stimulatory glucose minus insulin secreted under baseline glucose concentrations per islet of insulin content. Data are means ± SE for at least three separate experiments, * p < 0.01. Figure is used from [45] with permission from the publisher.

Figure 11.
The islet-COX activity threshold. In CDs rats, islet-COX activity of ≥46% is mandatory to maintain normoglycemia [10].
Figure 11.
The islet-COX activity threshold. In CDs rats, islet-COX activity of ≥46% is mandatory to maintain normoglycemia [10].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
T2D as mitochondrial disease.
| Pros | Cons |
|---|---|
| mtDNA mutations or deletion of mitochondrial genes reduce GSIS capacity and causes diabetes | Most mtDNA mutations found were not causatively related to diabetes |
| Deficient mitochondrial function are found in many diabetes patients | Mitochondrial dysfunction might be caused by hyperglycemia |
| Mitochondrial dysfunction or deletion reduced or inhibit GSIS | No sufficient clinical data to implicate mitochondrial dysfunction as a primary cause of T2D |
| During the development of overt T2D, mitochondria appear round and swollen | Mitochondrial structure abnormalities could be ensued by diabetes |
| Comparable to mitochondrial diseases, GSIS is markedly reduced but insulin secretion in response non nutrient secretagogue sustain | No clinical manifestation of classical mitochondrial disease such as encephalopathy lactic acidosis & stroke like episodes |
| ATP generation up to the metabolic threshold for robust insulin secretion release requires ATP generation by OXPHOS | |
| β-cells have unique features to ensure ATP generation by OXPHOS and adequate GSIS such as suppression of housekeeping genes that are normally expressed in other cells | |
| Islets of T2D patients exhibit a variation in the expression of OXPHOS and metabolic genes that are likely to reduce GSIS and cause diabetes | |
| Variants in the mitochondrial transcription factor TFB1M have been implicated by GWAS of T2D | |
| Loss of the mitochondrial protein frataxin impairs the activity of the electron transport chain (ETC), leading to a higher incidence of diabetes in Friedreich ataxia patients |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Weksler-Zangen, S. Is Type 2 Diabetes a Primary Mitochondrial Disorder? Cells 2022, 11, 1617. https://doi.org/10.3390/cells11101617
AMA Style
Weksler-Zangen S. Is Type 2 Diabetes a Primary Mitochondrial Disorder? Cells. 2022; 11(10):1617. https://doi.org/10.3390/cells11101617
Chicago/Turabian StyleWeksler-Zangen, Sarah. 2022. "Is Type 2 Diabetes a Primary Mitochondrial Disorder?" Cells 11, no. 10: 1617. https://doi.org/10.3390/cells11101617
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.