
Assessment and Validation of Globodera pallida as a Novel In Vivo Model for Studying Alzheimer’s Disease

 , , , , , and
, , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Highlights
- G. pallida, a plant parasitic nematode, can be used as a non-transgenic model of AD.
- G. pallida appears to be a reliable non-transgenic nematode compared to C. elegans transgenic strains, for studying AD experimentally.
- G. pallida can assimilate amyloid beta (Aβ) peptides, which co-localize with its neurological structures mimicking AD physiopathology.
- Treatment with various Aβ isoforms 1–40, 1–42, 17–42, 17–40, 1–28, or 1–16) impaired G. pallida’s chemotaxis, survival, production of ROS, and GSSG/GSH levels.
- G. pallida represents a unique model that can sensitively distinguish differences between different treatment concentrations, durations, and other modalities.
- Clinically approved neuroprotective agents were effective in protecting G. pallida from Aβ (1–42) exposure.
- G. pallida is an interesting new in vivo model with strong potential for discovery of novel bioactive compounds with anti-AD activity.
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Methods
2.2.1. Chemosensory Studies with G. pallida
2.2.2. Immunocytochemistry (ICC)
2.2.3. Measuring Effects of Aβ on the Health of G. pallida Survival Rate
Measurement of G. pallida Viability
Measurement of ROS
Determination of Total Glutathione
2.2.4. C. elegans Chemotaxis Assays
2.3. Data Analysis
3. Results
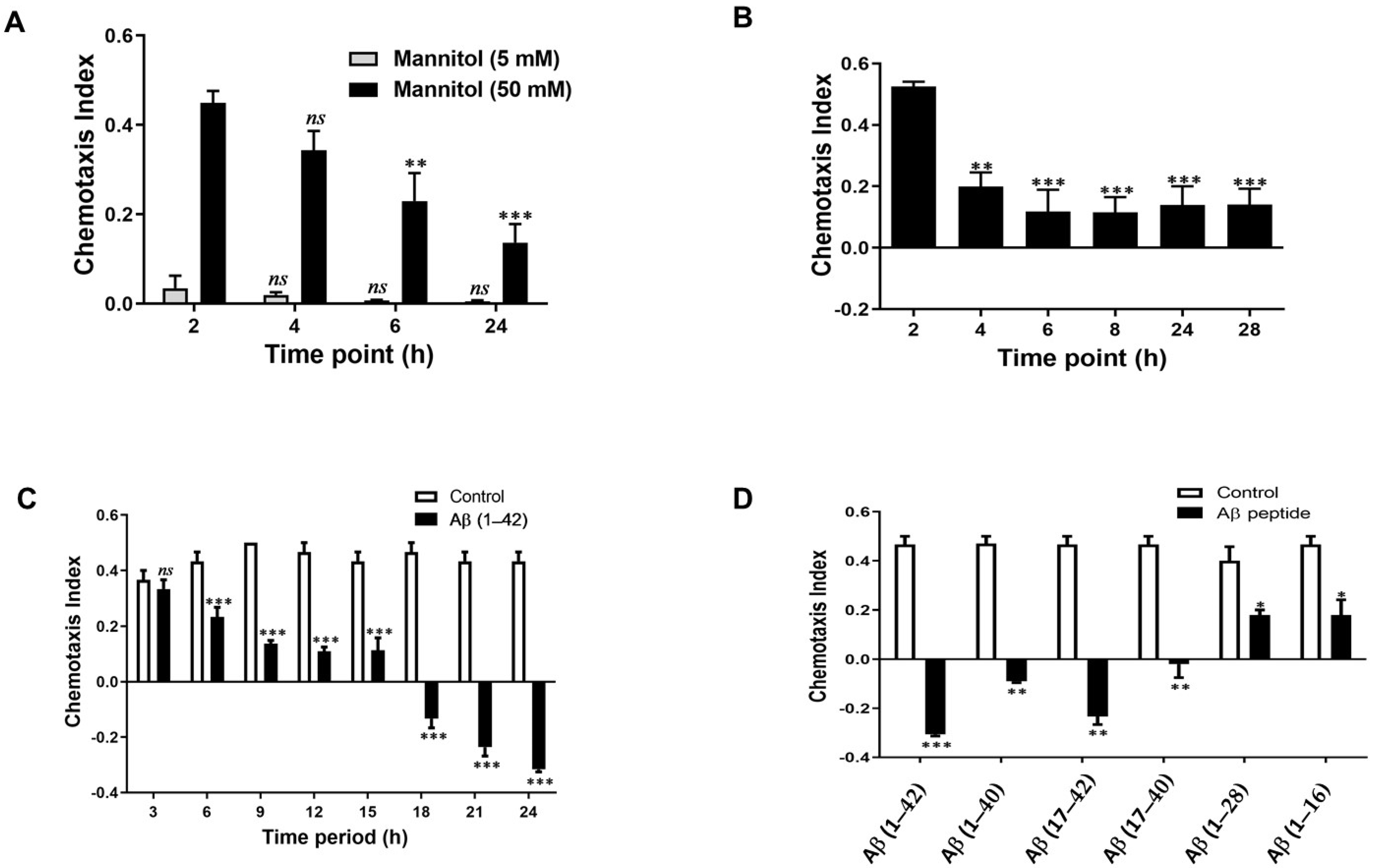
3.1. Optimisation of Chemosensory Assays
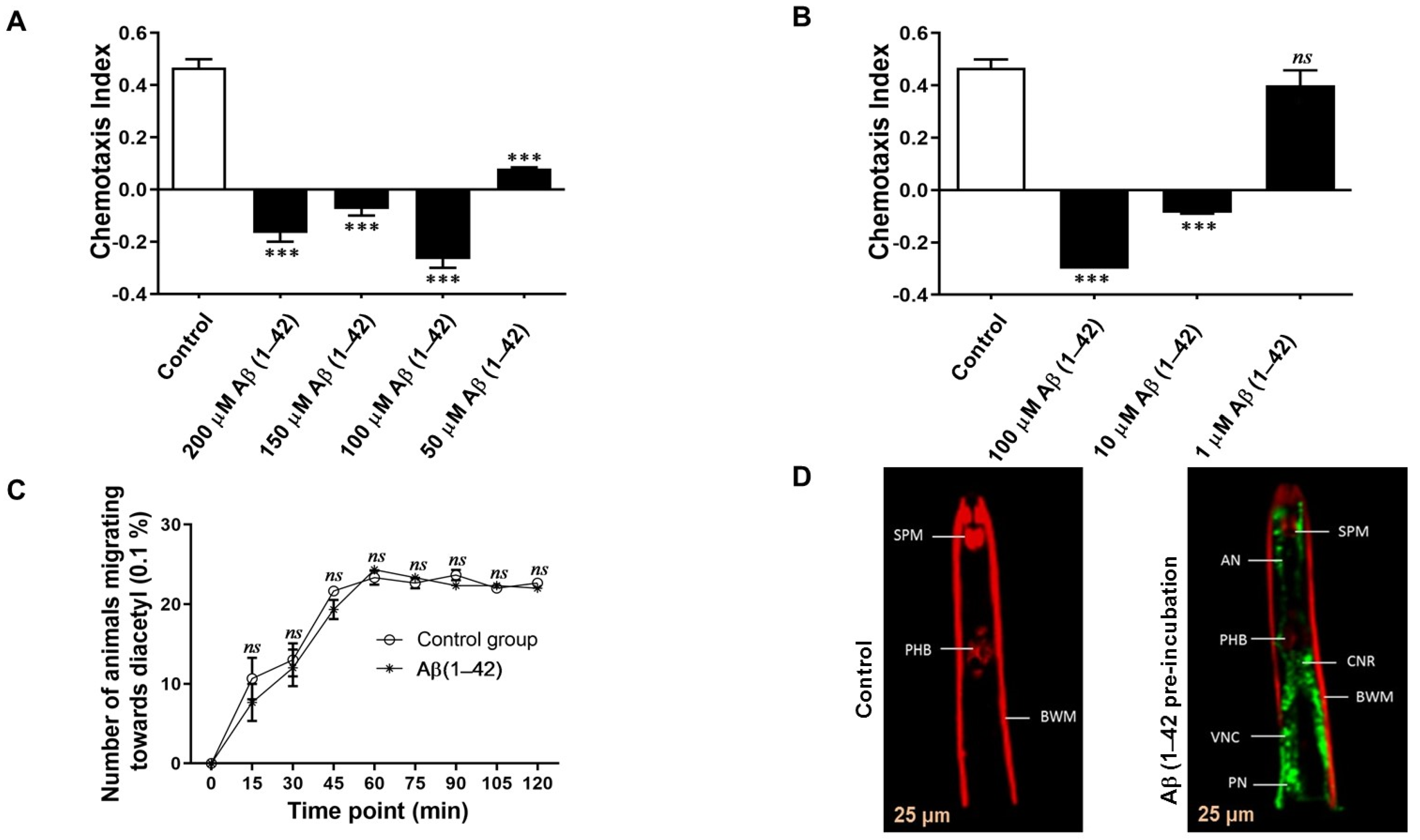
3.2. Effects of Aβ on the Chemotaxis of G. pallida
3.3. Localisation of Aβ (1–42) within G. pallida
3.4. Effects of Aβ (1–42) on the Health Parameters of G. pallida
3.4.1. Viability
3.4.2. Survival
3.4.3. ROS Production
3.4.4. GSSG/GSH Levels
3.5. Effects of Neuroprotective Agents on Aβ (1–42)-Induced Impairment of Chemosensing
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Alzheimer’s Disease International. World Alzheimer Report 2019: Attitudes to Dementia; ADI: London, UK, 2019. [Google Scholar]
- Prince, M.; Comas-Herrera, A.; Knapp, M.; Guerchet, M.; Karagiannidou, M. World Alzheimer Report 2016: Improving Healthcare for People Living with Dementia: Coverage, Quality, and Costs Now and in the Future; ADI: London, UK, 2016. [Google Scholar]
- Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimers Dement. 2020, 16, 391–460. [Google Scholar]
- Lim, C.H.; Kaur, P.; Teo, E.; Lam, V.Y.M.; Zhu, F.; Kibat, C.; Gruber, J.; Mathuru, A.S.; Tolwinski, N.S. Application of optogenetic Amyloid-β distinguishes between metabolic and physical damages in neurodegeneration. eLife 2020, 9, e52589. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Phillips, J.C. Why Aβ42 is much more toxic than Aβ40. ACS Chem. Neuroscience 2019, 10, 2843–2847. [Google Scholar]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. Aβ (1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef] [Green Version]
- Schlachetzki, J.; Saliba, S.W.; Oliveira, A.C.P.D. Studying neurodegenerative diseases in culture models. Rev. Bras. Psiquiatr. 2013, 35, S92–S100. [Google Scholar] [CrossRef] [Green Version]
- Prüßing, K.; Voigt, A.; Schulz, J.B. Drosophila melanogaster as a model organism for Alzheimer’s disease. Mol. Neurodegener. 2013, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Alexander, A.G.; Marfil, V.; Li, C. Use of Caenorhabditis elegans as a model to study Alzheimer’s disease and other neurodegenerative diseases. Front. Genet. 2014, 5, 279. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Le, W. Modeling neurodegenerative diseases in Caenorhabditis elegans. Exp. Neurol. 2013, 250, 94–103. [Google Scholar] [CrossRef]
- Gutierrez-Zepeda, A.; Luo, Y. Testing the amyloid toxicity hypothesis of Alzheimer’s disease in transgenic Caenorhabditis elegans model. Front. Biosci. 2004, 9, 3333–3338. [Google Scholar] [CrossRef] [Green Version]
- Link, C.D. Invertebrate models of Alzheimer’s disease. Genes Brain Behav. 2005, 4, 147–156. [Google Scholar] [CrossRef]
- Praitis, V.; Casey, E.; Collar, D.; Austin, J. Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics 2001, 157, 1217–1226. [Google Scholar] [CrossRef]
- Praitis, V.; Maduro, M.F. Transgenesis in C. elegans. Methods Cell Biol. 2011, 106, 161–185. [Google Scholar] [PubMed]
- Dosanjh, L.E.; Brown, M.K.; Rao, G.; Link, C.D.; Luo, Y. Behavioral phenotyping of a transgenic Caenorhabditis elegans expressing neuronal amyloid-β. J. Alzheimers Dis. 2010, 19, 681–690. [Google Scholar] [CrossRef]
- Wang, D.; Jones, L.M.; Urwin, P.E.; Atkinson, H.J. A synthetic peptide shows retro-and anterograde neuronal transport before disrupting the chemosensation of plant-pathogenic nematodes. PLoS ONE 2011, 6, e17475. [Google Scholar] [CrossRef] [Green Version]
- Winter, M.D.; McPherson, M.J.; Atkinson, H.J. Neuronal uptake of pesticides disrupts chemosensory cells of nematodes. Parasitology 2002, 125, 561–565. [Google Scholar]
- Green, J.; Wang, D.; Lilley, C.J.; Urwin, P.E.; Atkinson, H.J. Transgenic potatoes for potato cyst nematode control can replace pesticide use without impact on soil quality. PLoS ONE 2012, 7, e30973. [Google Scholar] [CrossRef] [Green Version]
- Dalzell, J.J.; McMaster, S.; Fleming, C.C.; Maule, A.G. Short interfering RNA-mediated gene silencing in Globodera pallida and Meloidogyne incognita infective stage juveniles. Int. J. Parasitol. 2010, 40, 91–100. [Google Scholar] [CrossRef]
- Warnock, N.D.; Wilson, L.; Canet-Perez, J.V.; Fleming, T.; Fleming, C.C.; Maule, A.G.; Dalzell, J.J. Exogenous RNA interference exposes contrasting roles for sugar exudation in host-finding by plant pathogens. Int. J. Parasitol. 2016, 46, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Hart, A.C. (Ed.) Behavior. Available online: https://www.ncbi.nlm.nih.gov/books/NBK19734/ (accessed on 25 July 2021).
- Coons, A.H.; Leduc, E.H.; Connolly, J.M. Studies on antibody production: I. A method for the histochemical demonstration of specific antibody and its application to a study of the hyperimmune rabbit. J. Exp. Med. 1955, 102, 49–60. [Google Scholar] [CrossRef]
- Minniti, A.N.; Rebolledo, D.L.; Grez, P.M.; Fadic, R.; Aldunate, R.; Volitakis, I.; Cherny, R.A.; Opazo, C.; Masters, C.; Bush, A.I.; et al. Intracellular amyloid formation in muscle cells of Aβ-transgenic Caenorhabditis elegans: Determinants and physiological role in copper detoxification. Mol. Neurodegener. 2009, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalzell, J.J.; Warnock, N.D.; Mcveigh, P.; Marks, N.J.; Mousley, A.; Atkinson, L.; Maule, A.G. Considering RNAi experimental design in parasitic helminths. Parasitology 2012, 139, 589. [Google Scholar] [CrossRef]
- Ng, L.F.; Gruber, J.; Cheah, I.K.; Goo, C.K.; Cheong, W.F.; Shui, G.; Sit, K.P.; Wenk, M.R.; Halliwell, B. The mitochondria-targeted antioxidant MitoQ extends lifespan and improves healthspan of a transgenic Caenorhabditis elegans model of Alzheimer disease. Free Radic. Biol. Med. 2014, 71, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Sangha, J.S.; Sun, X.; Wally, O.S.; Zhang, K.; Ji, X.; Wang, Z.; Wang, Y.; Zidichouski, J.; Prithiviraj, B.; Zhang, J. Liuwei Dihuang (LWDH), a traditional Chinese medicinal formula, protects against β-amyloid toxicity in transgenic Caenorhabditis elegans. PLoS ONE 2012, 7, e43990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, J.; Ng, L.F.; Fong, S.; Wong, Y.T.; Koh, S.A.; Chen, C.B.; Halliwell, B. Mitochondrial changes in ageing Caenorhabditis elegans–what do we learn from superoxide dismutase knockouts? PLoS ONE 2011, 6, e19444. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, T.; Izumi, J.; Hioki, M.; Nagaya, H.; Kobayashi, Y. Sensory interaction between attractant diacetyl and repellent 2-nonanone in the nematode Caenorhabditis elegans. J. Exp. Zool. A Ecol. Genet. Physiol. 2013, 319, 285–295. [Google Scholar] [CrossRef]
- Noratiqah, S.B.; Naina-Mohamed, I.; Zulfarina, M.S.; Qodriyah, H.M. Natural polyphenols in the treatment of Alzheimer’s Disease. Curr. Drug Targets 2017, 19, 927–937. [Google Scholar] [CrossRef]
- Warnock, N.D.; Wilson, L.; Patten, C.; Fleming, C.C.; Maule, A.G.; Dalzell, J.J. Nematode neuropeptides as transgenic nematicides. PLoS Pathog. 2017, 13, e1006237. [Google Scholar] [CrossRef] [Green Version]
- Amin, F.U.; Shah, S.A.; Kim, M.O. Vanillic acid attenuates Aβ1-42-induced oxidative stress and cognitive impairment in mice. Sci. Rep. 2017, 7, 40753. [Google Scholar] [CrossRef]
- Wu, Y.; Wu, Z.; Butko, P.; Christen, Y.; Lambert, M.P.; Klein, W.L.; Link, C.D.; Luo, Y. Amyloid-β-induced pathological behaviors are suppressed by Ginkgo biloba extract EGb 761 and ginkgolides in transgenic Caenorhabditis elegans. J. Neurosci. 2006, 26, 13102–13113. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid β-peptide (1–42)-induced oxidative stress in Alzheimer disease: Importance in disease pathogenesis and progression. Antioxid. Redox Signal. 2013, 19, 823–835. [Google Scholar] [CrossRef] [Green Version]
- Drake, J.; Link, C.D.; Butterfield, D.A. Oxidative stress precedes fibrillar deposition of Alzheimer’s disease amyloid β-peptide (1–42) in a transgenic Caenorhabditis elegans model. Neurobiol. Aging 2003, 24, 415–420. [Google Scholar] [CrossRef]
- Butterfield, D.A. Amyloid β-peptide (1–42)-induced oxidative stress and neurotoxicity: Implications for neurodegeneration in Alzheimer’s disease brain. A review. Free Radic. Res. 2002, 36, 1307–1313. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef] [Green Version]
- Gill, I.; Kaur, S.; Kaur, N.; Dhiman, M.; Mantha, A.K. Phytochemical ginkgolide B attenuates amyloid-β1-42 induced oxidative damage and altered cellular responses in human neuroblastoma SH-SY5Y Cells. J. Alzheimers Dis. 2017, 60, S25–S40. [Google Scholar] [CrossRef] [PubMed]
- Swomley, A.M.; Förster, S.; Keeney, J.T.; Triplett, J.; Zhang, Z.; Sultana, R.; Butterfield, D.A. Abeta, oxidative stress in Alzheimer disease: Evidence based on proteomics studies. Biochim. Biophys. Acta 2014, 1842, 1248–1257. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Reed, T.; Newman, S.F.; Sultana, R. Roles of amyloid β-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic. Biol. Med. 2007, 43, 658–677. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, A.; Laurie, C.; Mosley, R.L.; Gendelman, H.E. Oxidative stress and the pathogenesis of neurodegenerative disorders. Int. Rev. Neurobiol. 2007, 82, 297–325. [Google Scholar]
- Manton, K.G.; Volovik, S.; Kulminski, A. ROS effects on neurodegeneration in Alzheimer’s disease and related disorders: On environmental stresses of ionizing radiation. Curr. Alzheimer Res. 2004, 1, 277–293. [Google Scholar] [CrossRef]
- Joshi, G.; Sultana, R.; Perluigi, M.; Butterfield, D.A. In vivo protection of synaptosomes from oxidative stress mediated by Fe 2+/H2O2 or 2,2-azobis-(2-amidinopropane) dihydrochloride by the glutathione mimetic tricyclodecane-9-yl-xanthogenate. Free Radic. Biol. Med. 2005, 38, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Yepes, J.; Zavala-Flores, L.; Anandhan, A.; Wang, F.; Skotak, M.; Chandra, N.; Li, M.; Pappa, A.; Martinez-Fong, D.; Del Razo, L.M.; et al. Antioxidant gene therapy against neuronal cell death. Pharmacol. Ther. 2014, 142, 206–230. [Google Scholar] [CrossRef] [Green Version]
- Boyd-Kimball, D.; Sultana, R.; Mohmmad-Abdul, H.; Butterfield, D.A. Rodent Aβ (1–42) exhibits oxidative stress properties similar to those of human Aβ (1–42): Implications for proposed mechanisms of toxicity. J. Alzheimers Dis. 2004, 6, 515–525. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Chen, S.; Ma, M.; Zhang, C.; Wang, K.; Li, F.; Guo, W.; Huang, J.; Zhang, C. Do ROS really slow down aging in C. elegans? arXiv 2017, arXiv:1704.06086. [Google Scholar]
- Dai, D.F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 2014, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Arriola, E.; Cárdenas-Rodríguez, N.; Coballase-Urrutia, E.; Pedraza-Chaverri, J.; Carmona-Aparicio, L.; Ortega-Cuellar, D. Caenorhabditis elegans: A useful model for studying metabolic disorders in which oxidative stress is a contributing factor. Oxid. Med. Cell. Longev. 2014, 2014, 705253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kılıçgün, H.; Göksen, G. Life span effects of Hypericum perforatum extracts on Caenorhabditis elegans under heat stress. Pharmacogn. Mag. 2012, 8, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, K.I.; Pincus, Z.; Slack, F.J. Longevity and stress in Caenorhabditis elegans. Aging 2011, 3, 733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, A.; Vantipalli, M.C.; Lithgow, G.J. Lifespan extension of Caenorhabditis elegans following repeated mild hormetic heat treatments. Biogerontology 2006, 7, 221–230. [Google Scholar] [CrossRef]
- Du, X.T.; Wang, L.; Wang, Y.J.; Andreasen, M.; Zhan, D.W.; Feng, Y.; Li, M.; Zhao, M.; Otzen, D.; Xue, D.; et al. Aβ1-16 can aggregate and induce the production of reactive oxygen species, nitric oxide, and inflammatory cytokines. J. Alzheimers Dis. 2011, 27, 401–413. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D. The critical role of methionine 35 in Alzheimer’s amyloid β-peptide (1–42)-induced oxidative stress and neurotoxicity. Biochim. Biophys. Acta 2005, 1703, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Sultana, R. Methionine-35 of Aβ (1–42): Importance for oxidative stress in Alzheimer disease. J. Amino. Acids. 2011, 2011, 198430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, J.; Wu, Q.Y.; Ma, Y.C.; Chen, Y.L.; Zou, C.G. Antioxidant response is a protective mechanism against nutrient deprivation in C. elegans. Sci. Rep. 2017, 7, 43547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, W.M.; Dostal, V.; Huemann, B.N.; Yerg, J.E.; Link, C.D. Identifying Aβ-specific pathogenic mechanisms using a nematode model of Alzheimer’s disease. Neurobiol. Aging 2015, 36, 857–866. [Google Scholar]
- Galindo, M.F.; Ikuta, I.; Zhu, X.; Casadesus, G.; Jordán, J. Mitochondrial biology in Alzheimer’s disease pathogenesis. J. Neurochem. 2010, 114, 933–945. [Google Scholar] [CrossRef]
- Ferrer, I. Altered mitochondria, energy metabolism, voltage-dependent anion channel, and lipid rafts converge to exhaust neurons in Alzheimer’s disease. J. Bioenerg. Biomembr. 2009, 41, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Shen, H.; Zhang, J.; Zhu, Y.G.; Ransom, B.R.; Chen, X.C.; Ye, Z.C. Dual pathways mediate β-amyloid stimulated glutathione release from astrocytes. Glia 2015, 63, 2208–2219. [Google Scholar] [CrossRef]
- Saharan, S.; Mandal, P.K. The emerging role of glutathione in Alzheimer’s disease. J. Alzheimers Dis. 2014, 40, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Lasierra-Cirujeda, J.; Coronel, P.; Aza, M.J.; Gimeno, M. Beta-amyloidolysis and glutathione in Alzheimer’s disease. J. Blood Med. 2013, 4, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Li, J.K.; Liu, X.D.; Shen, L.; Zeng, W.M.; Qiu, G.Z. Natural plant polyphenols for alleviating oxidative damage in man: Current status and future perspectives. Trop. J. Pharm. Res. 2016, 15, 1089–1098. [Google Scholar] [CrossRef] [Green Version]
- Siddique, S.; Matera, C.; Radakovic, Z.S.; Hasan, M.S.; Gutbrod, P.; Rozanska, E.; Sobczak, M.; Torres, M.A.; Grundler, F.M. Parasitic worms stimulate host NADPH oxidases to produce reactive oxygen species that limit plant cell death and promote infection. Sci. Signal. 2014, 7, ra33. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.T.; Reavy, B.; Smant, G.; Prior, A.E. Glutathione peroxidases of the potato cyst nematode Globodera rostochiensis. Gene 2004, 324, 47–54. [Google Scholar] [CrossRef]
- Mehdy, M.C. Active oxygen species in plant defense against pathogens. Plant Physiol. 1994, 105, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Yang, H.; Chen, Y.; Sun, H. Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 132, 294–309. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Asaduzzaman, M.; Hossain, M.S.; Sarker, J.; Rahman, S.A.; Rashid, M.; Rahman, M.M. In vitro antioxidant and cholinesterase inhibitory activities of methanolic fruit extract of Phyllanthus acidus. BMC Complement. Altern. Med. 2015, 15, 403. [Google Scholar]
- Asaduzzaman, M.; Uddin, M.; Kader, M.A.; Alam, A.H.M.K.; Rahman, A.A.; Rashid, M.; Kato, K.; Tanaka, T.; Takeda, M.; Sadik, G. In vitro acetylcholinesterase inhibitory activity and the antioxidant properties of Aegle marmelos leaf extract: Implications for the treatment of Alzheimer’s disease. Psychogeriatrics 2014, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sang, Z.; Pan, W.; Wang, K.; Ma, Q.; Yu, L.; Yang, Y.; Bai, P.; Leng, C.; Xu, Q.; Li, X.; et al. Design, synthesis, and evaluation of novel ferulic acid-O-alkylamine derivatives as potential multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017, 130, 379–392. [Google Scholar] [CrossRef]
- Čolović, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lilienfeld, S. Galantamine—A novel cholinergic drug with a unique dual mode of action for the treatment of patients with Alzheimer’s disease. CNS Drug Rev. 2002, 8, 159–176. [Google Scholar] [CrossRef]
- Russo, P.; Frustaci, A.; Del Bufalo, A.; Fini, M.; Cesario, A. From traditional European medicine to discovery of new drug candidates for the treatment of dementia and Alzheimer’s disease: Acetylcholinesterase inhibitors. Curr. Med. Chem. 2013, 20, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Parsons, C.G.; Rammes, G.; Danysz, W. Pharmacodynamics of memantine: An update. Curr. Neuropharmacol. 2008, 6, 55–78. [Google Scholar] [CrossRef] [Green Version]
- Parsons, C.G.; Danysz, W.; Dekundy, A.; Pulte, I. Memantine and cholinesterase inhibitors: Complementary mechanisms in the treatment of Alzheimer’s disease. Neurotox. Res. 2013, 24, 358–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, L.; Yamujala, R.; Wang, Y.; Wang, H.; Wu, W.H.; Lawton, M.A.; Long, C.; Di, R. Acetylcholineestarase-inhibiting alkaloids from Lycoris radiata delay paralysis of amyloid beta-expressing transgenic C. elegans CL4176. PLoS ONE 2013, 8, e63874. [Google Scholar]
- Sutphin, G.L.; Bishop, E.; Yanos, M.E.; Moller, R.M.; Kaeberlein, M. Caffeine extends life span, improves healthspan, and delays age-associated pathology in Caenorhabditis elegans. Longev. Healthspan 2012, 1, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Dostal, V.; Roberts, C.M.; Link, C.D. Genetic mechanisms of coffee extract protection in a Caenorhabditis elegans model of β-amyloid peptide toxicity. Genetics 2010, 186, 857–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martorell, P.; Bataller, E.; Llopis, S.; Gonzalez, N.; Álvarez, B.; Montón, F.; Ortiz, P.; Ramón, D.; Genovés, S. A cocoa peptide protects Caenorhabditis elegans from oxidative stress and β-amyloid peptide toxicity. PLoS ONE 2013, 8, e63283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurk, E.; Refsum, H.; Drevon, C.A.; Tell, G.S.; Nygaard, H.A.; Engedal, K.; Smith, A.D. Intake of flavonoid-rich wine, tea, and chocolate by elderly men and women is associated with better cognitive test performance. J. Nutr. 2009, 139, 120–127. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Althobaiti, N.A.; Menaa, F.; Albalawi, A.E.; Dalzell, J.J.; Warnock, N.D.; Mccammick, E.M.; Alsolais, A.; Alkhaibari, A.M.; Green, B.D. Assessment and Validation of Globodera pallida as a Novel In Vivo Model for Studying Alzheimer’s Disease. Cells 2021, 10, 2481. https://doi.org/10.3390/cells10092481
Althobaiti NA, Menaa F, Albalawi AE, Dalzell JJ, Warnock ND, Mccammick EM, Alsolais A, Alkhaibari AM, Green BD. Assessment and Validation of Globodera pallida as a Novel In Vivo Model for Studying Alzheimer’s Disease. Cells. 2021; 10(9):2481. https://doi.org/10.3390/cells10092481
Chicago/Turabian StyleAlthobaiti, Norah A., Farid Menaa, Aishah E. Albalawi, Johnathan J. Dalzell, Neil D. Warnock, Erin M. Mccammick, Abdulellah Alsolais, Abeer M. Alkhaibari, and Brian D. Green. 2021. "Assessment and Validation of Globodera pallida as a Novel In Vivo Model for Studying Alzheimer’s Disease" Cells 10, no. 9: 2481. https://doi.org/10.3390/cells10092481