
Inhibition of Metabolism as a Therapeutic Option for Tamoxifen-Resistant Breast Cancer Cells



Abstract
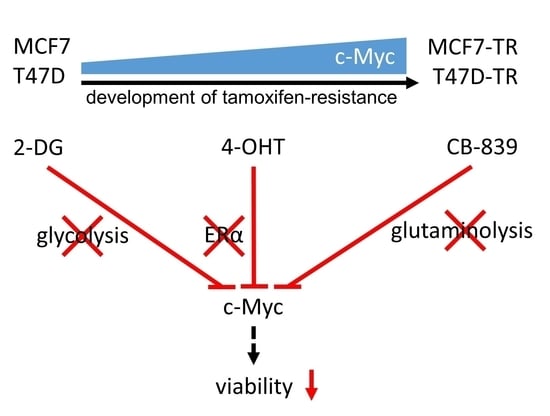
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Drugs
2.3. Viability Assay
2.4. Mitochondrial Membrane Potential
2.5. c-Myc Expression
2.6. Small Interfering RNA Transfection
2.7. Statistical Analysis
3. Results
3.1. Effects on Cell Viability
3.1.1. Single Treatments
3.1.2. Combination Treatments
3.2. Induction of Apoptosis
3.3. c-Myc
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barkhem, T.; Carlsson, B.; Nilsson, Y.; Enmark, E.; Gustafsson, J.; Nilsson, S. Differential response of estrogen receptor alpha and estrogen receptor beta to partial estrogen agonists/antagonists. Mol. Pharmacol. 1998, 54, 105–112. [Google Scholar] [CrossRef]
- Cronin-Fenton, D.P.; Damkier, P.; Lash, T.L. Metabolism and transport of tamoxifen in relation to its effectiveness: New perspectives on an ongoing controversy. Future Oncol. 2014, 10, 107–122. [Google Scholar] [CrossRef] [Green Version]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Early Breast Cancer Trialists’ Collaborative Group. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomised trials. Lancet 2011, 378, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Fisher, B.; Costantino, J.P.; Wickerham, D.L.; Cecchini, R.S.; Cronin, W.M.; Robidoux, A.; Bevers, T.B.; Kavanah, M.T.; Atkins, J.N.; Margolese, R.G.; et al. Tamoxifen for the prevention of breast cancer: Current status of the National Surgical Adjuvant Breast and Bowel Project P-1 study. J. Natl. Cancer Inst. 2005, 97, 1652–1662. [Google Scholar] [CrossRef] [Green Version]
- Early Breast Cancer Trialists’ Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 365, 1687–1717. [Google Scholar] [CrossRef]
- Warburg, O. Über den Stoffwechsel der Carcinomzelle. Naturwissenschaften 1924, 12, 1131–1137. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Lasche, M.; Emons, G.; Grundker, C. Shedding New Light on Cancer Metabolism: A Metabolic Tightrope Between Life and Death. Front. Oncol. 2020, 10, 409. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Cantley, L.C. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011, 14, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [Green Version]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Reviews. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seltzer, M.J.; Bennett, B.D.; Joshi, A.D.; Gao, P.; Thomas, A.G.; Ferraris, D.V.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Rabinowitz, J.D.; et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010, 70, 8981–8987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Heuvel, A.P.J.; Jing, J.; Wooster, R.F.; Bachman, K.E. Analysis of glutamine dependency in non-small cell lung cancer: GLS1 splice variant GAC is essential for cancer cell growth. Cancer Biol. Ther. 2012, 13, 1185–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, L.A.; Holton, T.; Yuneva, M.; Louie, R.J.; Padró, M.; Daemen, A.; Hu, M.; Chan, D.A.; Ethier, S.P.; van ‘t Veer, L.J.; et al. Glutamine Sensitivity Analysis Identifies the xCT Antiporter as a Common Triple-Negative Breast Tumor Therapeutic Target. Cancer Cell 2013, 24, 450–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demas, D.M.; Demo, S.; Fallah, Y.; Clarke, R.; Nephew, K.P.; Althouse, S.; Sandusky, G.; He, W.; Shajahan-Haq, A.N. Glutamine Metabolism Drives Growth in Advanced Hormone Receptor Positive Breast Cancer. Front Oncol 2019, 9, 686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Study of the Glutaminase Inhibitor CB-839 in Solid Tumors-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02071862 (accessed on 31 May 2021).
- Wokoun, U.; Hellriegel, M.; Emons, G.; Gründker, C. Co-treatment of breast cancer cells with pharmacologic doses of 2-deoxy-D-glucose and metformin: Starving tumors. Oncol. Rep. 2017, 37, 2418–2424. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 124. [Google Scholar] [CrossRef] [PubMed]
- Günthert, A.R.; Gründker, C.; Olota, A.; Läsche, J.; Eicke, N.; Emons, G. Analogs of GnRH-I and GnRH-II inhibit epidermal growth factor-induced signal transduction and resensitize resistant human breast cancer cells to 4OH-tamoxifen. Eur. J. Endocrinol. 2005, 153, 613–625. [Google Scholar] [CrossRef]
- Aft, R.L.; Zhang, F.W.; Gius, D. Evaluation of 2-deoxy-D-glucose as a chemotherapeutic agent: Mechanism of cell death. Br. J. Cancer 2002, 87, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Leung, E.Y.; Kim, J.E.; Askarian-Amiri, M.; Joseph, W.R.; McKeage, M.J.; Baguley, B.C. Hormone Resistance in Two MCF-7 Breast Cancer Cell Lines is Associated with Reduced mTOR Signaling, Decreased Glycolysis, and Increased Sensitivity to Cytotoxic Drugs. Front. Oncol. 2014, 4, 221. [Google Scholar] [CrossRef] [Green Version]
- Rosendahl, A.H.; Puig Blasco, L.; Borgquist, S. Towards breast cancer prevention through reduced breast density: Suppressive effects of tamoxifen on normal breast epithelial cells [abstract]. In Cancer Research, Proceedings of the 2017 San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 5–9 December 2017; AACR: Philadelphia, PA, USA, 2018; Volume 78, p. P5-14-06. [Google Scholar] [CrossRef]
- Daurio, N.A.; Tuttle, S.W.; Worth, A.J.; Song, E.Y.; Davis, J.M.; Snyder, N.W.; Blair, I.A.; Koumenis, C. AMPK activation and metabolic reprogramming by tamoxifen through estrogen receptor-independent mechanisms suggests new uses for this therapeutic modality in cancer treatment. Cancer Res. 2016, 76, 3295–3306. [Google Scholar] [CrossRef] [Green Version]
- Ambrosio, M.R.; D’Esposito, V.; Costa, V.; Liguoro, D.; Collina, F.; Cantile, M.; Prevete, N.; Passaro, C.; Mosca, G.; De Laurentiis, M.; et al. Glucose impairs tamoxifen responsiveness modulating connective tissue growth factor in breast cancer cells. Oncotarget 2017, 8, 109000–109017. [Google Scholar] [CrossRef]
- Attia, Y.M.; EL-Abhar, H.S.; Al Marzabani, M.M.; Shouman, S.A. Targeting glycolysis by 3-bromopyruvate improves tamoxifen cytotoxicity of breast cancer cell lines. BMC Cancer 2015, 15, 838. [Google Scholar] [CrossRef] [Green Version]
- Woo, Y.M.; Shin, Y.; Lee, E.J.; Lee, S.; Jeong, S.H.; Kong, H.K.; Park, E.Y.; Kim, H.K.; Han, J.; Chang, M.; et al. Inhibition of Aerobic Glycolysis Represses Akt/mTOR/HIF-1α Axis and Restores Tamoxifen Sensitivity in Antiestrogen-Resistant Breast Cancer Cells. PLoS ONE 2015, 10, e0132285. [Google Scholar] [CrossRef] [Green Version]
- Dong, N.; Xu, B.; Xu, J. EGF-Mediated Overexpression of Myc Attenuates miR-26b by Recruiting HDAC3 to Induce Epithelial-Mesenchymal Transition of Lens Epithelial Cells. BioMed Res. Int. 2018, 2018, 7148023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubik, D.; Shiu, R.P. Mechanism of estrogen activation of c-myc oncogene expression. Oncogene 1992, 7, 1587–1594. [Google Scholar] [PubMed]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Amati, B.; Alevizopoulos, K.; Vlach, J. Myc and the cell cycle. Front. Biosci. A J. Virtual Libr. 1998, 3, d250–d268. [Google Scholar] [CrossRef]
- Klefstrom, J.; Arighi, E.; Littlewood, T.; Jäättelä, M.; Saksela, E.; Evan, G.I.; Alitalo, K. Induction of TNF-sensitive cellular phenotype by c-Myc involves p53 and impaired NF-kappaB activation. EMBO J. 1997, 16, 7382–7392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zindy, F.; Eischen, C.M.; Randle, D.H.; Kamijo, T.; Cleveland, J.L.; Sherr, C.J.; Roussel, M.F. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998, 12, 2424–2433. [Google Scholar] [CrossRef] [Green Version]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC Activation Is a Hallmark of Cancer Initiation and Maintenance. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef] [Green Version]
- Escot, C.; Theillet, C.; Lidereau, R.; Spyratos, F.; Champeme, M.H.; Gest, J.; Callahan, R. Genetic alteration of the c-myc protooncogene (MYC) in human primary breast carcinomas. Proc. Natl. Acad. Sci. USA 1986, 83, 4834–4838. [Google Scholar] [CrossRef] [Green Version]
- Cappellen, D.; Schlange, T.; Bauer, M.; Maurer, F.; Hynes, N.E. Novel c-MYC target genes mediate differential effects on cell proliferation and migration. EMBO Rep. 2007, 8, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.; Chun, Y.S.; Lewis, B.C.; Dang, C.V. A unique glucose-dependent apoptotic pathway induced by c-Myc. Proc. Natl. Acad. Sci. USA 1998, 95, 1511–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 2007, 178, 93–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, P.H.; Safneck, J.R.; Le, K.; Dubik, D.; Shiu, R.P. Relationship of c-myc amplification to progression of breast cancer from in situ to invasive tumor and lymph node metastasis. J. Natl. Cancer Inst. 1993, 85, 902–907. [Google Scholar] [CrossRef]
- McNeil, C.M.; Sergio, C.M.; Anderson, L.R.; Inman, C.K.; Eggleton, S.A.; Murphy, N.C.; Millar, E.K.A.; Crea, P.; Kench, J.G.; Alles, M.C.; et al. c-Myc overexpression and endocrine resistance in breast cancer. J. Steroid Biochem. Mol. Biol. 2006, 102, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Balko, J.M.; Ghazoui, Z.; Dunbier, A.; Anderson, H.; Dowsett, M.; González-Angulo, A.M.; Mills, G.B.; Miller, W.R.; Wu, H.; et al. A gene expression signature from human breast cancer cells with acquired hormone independence identifies MYC as a mediator of antiestrogen resistance. Clin. Cancer Res. 2011, 17, 2024–2034. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Wang, L.; Mao, C.; Duraki, D.; Kim, J.E.; Huang, R.; Helferich, W.G.; Nelson, E.R.; Park, B.H.; Shapiro, D.J. Estrogen-independent Myc overexpression confers endocrine therapy resistance on breast cancer cells expressing ERαY537S and ERαD538G mutations. Cancer Lett. 2019, 442, 373–382. [Google Scholar] [CrossRef]
- Green, A.R.; Aleskandarany, M.A.; Agarwal, D.; Elsheikh, S.; Nolan, C.C.; Diez-Rodriguez, M.; Macmillan, R.D.; Ball, G.R.; Caldas, C.; Madhusudan, S.; et al. MYC functions are specific in biological subtypes of breast cancer and confers resistance to endocrine therapy in luminal tumours. Br. J. Cancer 2016, 114, 917–928. [Google Scholar] [CrossRef] [Green Version]
- Fallah, Y.; Brundage, J.; Allegakoen, P.; Shajahan-Haq, A.N. MYC-Driven Pathways in Breast Cancer Subtypes. Biomolecules 2017, 7, 53. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steifensand, F.; Gallwas, J.; Bauerschmitz, G.; Gründker, C. Inhibition of Metabolism as a Therapeutic Option for Tamoxifen-Resistant Breast Cancer Cells. Cells 2021, 10, 2398. https://doi.org/10.3390/cells10092398
Steifensand F, Gallwas J, Bauerschmitz G, Gründker C. Inhibition of Metabolism as a Therapeutic Option for Tamoxifen-Resistant Breast Cancer Cells. Cells. 2021; 10(9):2398. https://doi.org/10.3390/cells10092398
Chicago/Turabian StyleSteifensand, Friederike, Julia Gallwas, Gerd Bauerschmitz, and Carsten Gründker. 2021. "Inhibition of Metabolism as a Therapeutic Option for Tamoxifen-Resistant Breast Cancer Cells" Cells 10, no. 9: 2398. https://doi.org/10.3390/cells10092398