
Regulation of the Hypoxia-Inducible Factor (HIF) by Pro-Inflammatory Cytokines


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
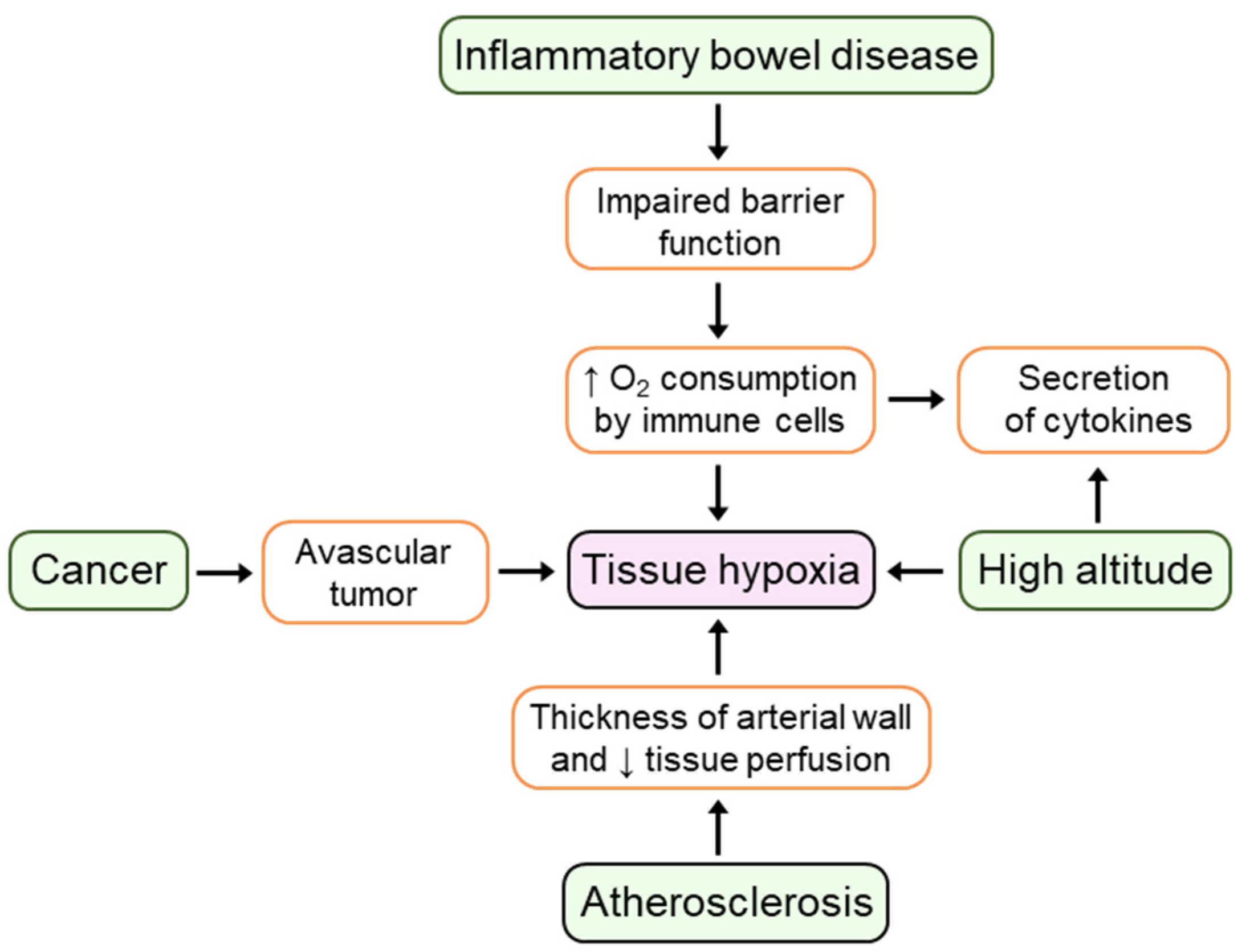
2. Hypoxia and Inflammation
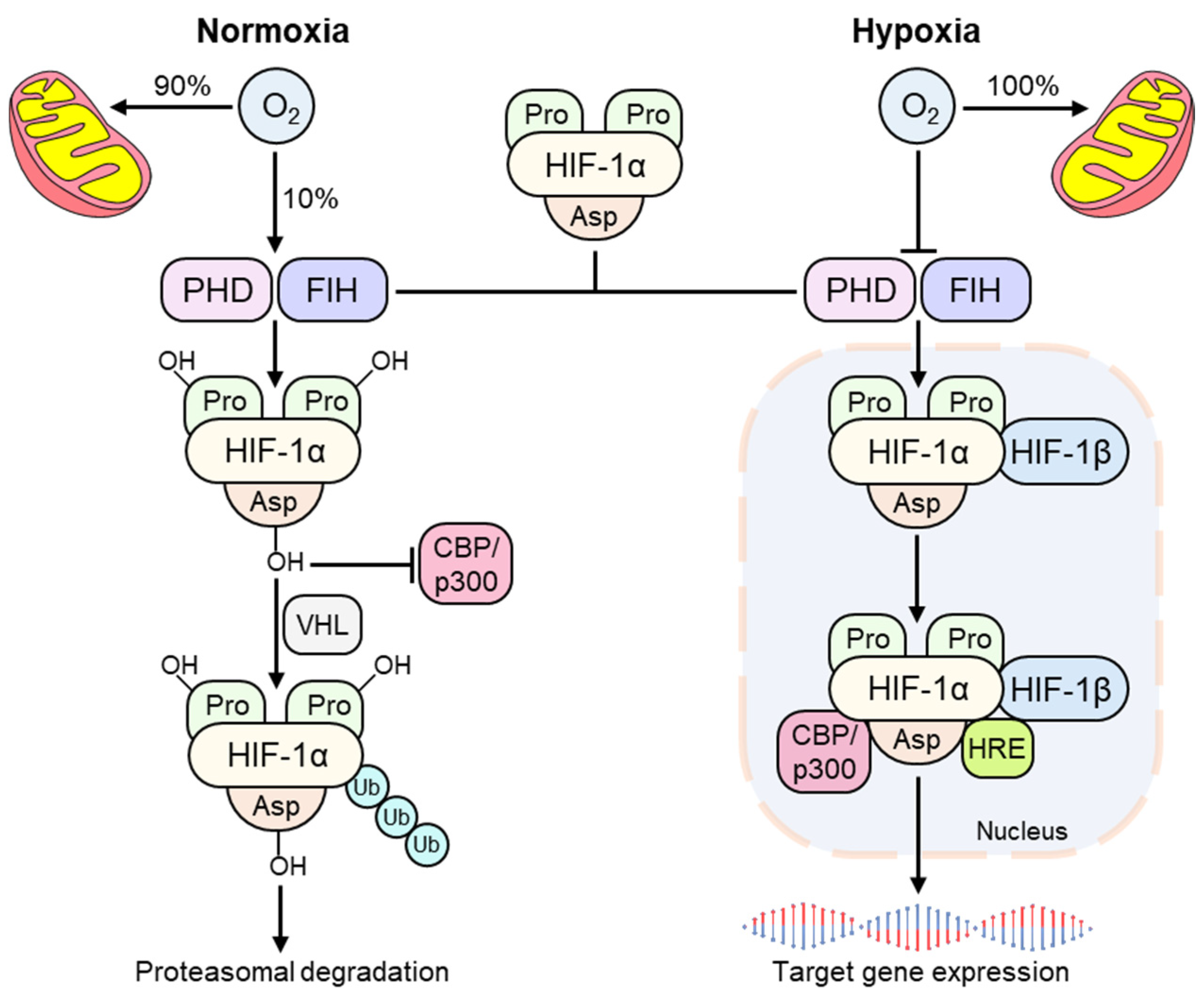
3. The Hypoxia-Inducible Factor (HIF)
4. The Involvement of HIF in Cytokine Production
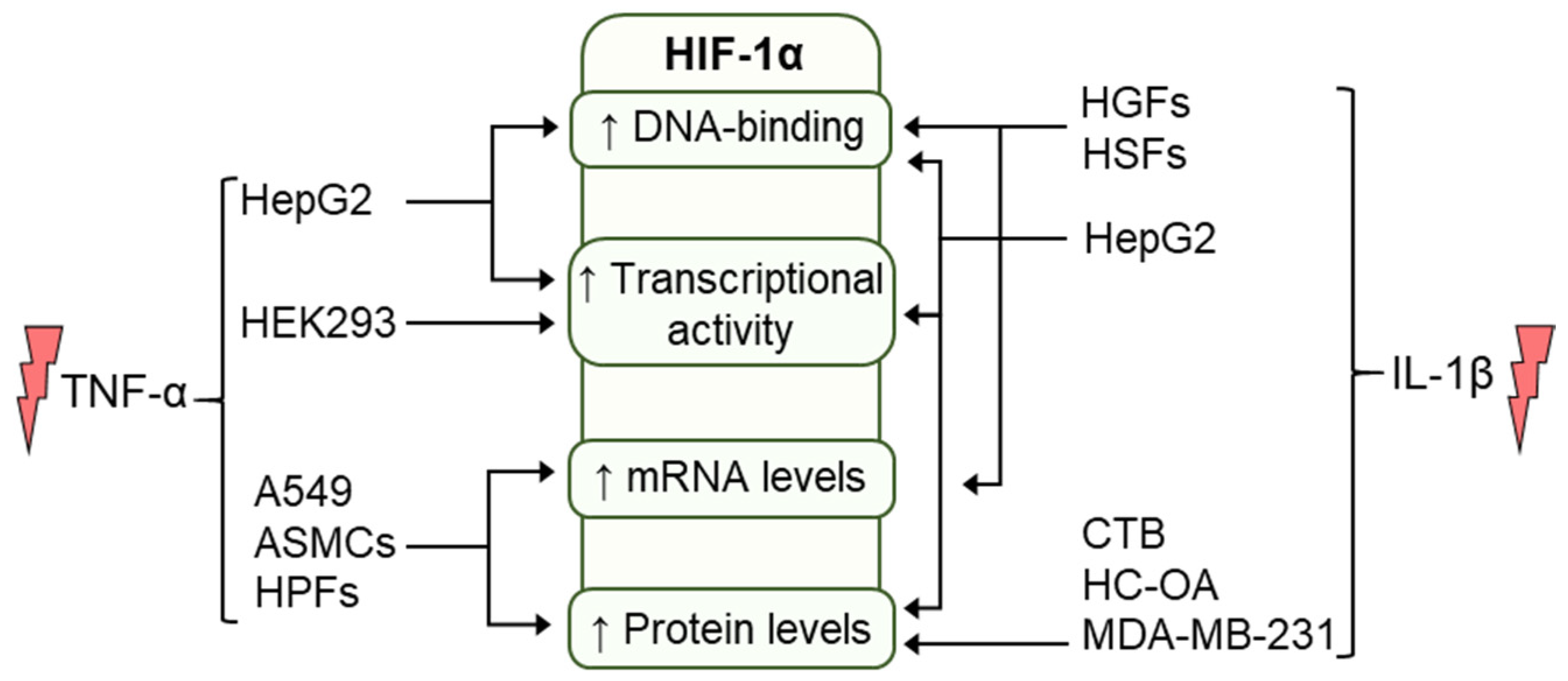
5. Regulation of HIF by Cytokines
5.1. Regulation of HIF by TNF-α
5.2. Regulation of HIF by IL-1β
6. HIF and Cytokines Crosstalk in Cancer and Inflammation
7. Conclusions
Funding
Conflicts of Interest
References
- Taylor, C.T.; Pouyssegur, J. Oxygen, hypoxia, and stress. Ann. N. Y. Acad. Sci. 2007, 1113, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factor 1: Oxygen homeostasis and disease pathophysiology. Trends Mol. Med. 2001, 7, 345–350. [Google Scholar] [CrossRef]
- Grocott, M.P.; Martin, D.S.; Levett, D.Z.; McMorrow, R.; Windsor, J.; Montgomery, H.E.; Group, C.X.E.R. Arterial blood gases and oxygen content in climbers on Mount Everest. N. Engl. J. Med. 2009, 360, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Cummins, E.P.; Doherty, G.A.; Taylor, C.T. Hydroxylases as therapeutic targets in inflammatory bowel disease. Lab. Investig. 2013, 93, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeley, T.P.; Mann, G.E. Defining Physiological Normoxia for Improved Translation of Cell Physiology to Animal Models and Humans. Physiol. Rev. 2019, 99, 161–234. [Google Scholar] [CrossRef] [Green Version]
- Taylor, C.T.; Colgan, S.P. Hypoxia and gastrointestinal disease. J. Mol. Med. 2007, 85, 1295–1300. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [Green Version]
- Sluimer, J.C.; Daemen, M.J. Novel concepts in atherogenesis: Angiogenesis and hypoxia in atherosclerosis. J. Pathol. 2009, 218, 7–29. [Google Scholar] [CrossRef]
- Brown, J.M.; Giaccia, A.J. The unique physiology of solid tumors: Opportunities (and problems) for cancer therapy. Cancer Res. 1998, 58, 1408–1416. [Google Scholar]
- Karhausen, J.; Furuta, G.T.; Tomaszewski, J.E.; Johnson, R.S.; Colgan, S.P.; Haase, V.H. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J. Clin. Investig. 2004, 114, 1098–1106. [Google Scholar] [CrossRef]
- Brown, E.; Rowan, C.; Strowitzki, M.J.; Fagundes, R.R.; Faber, K.N.; Güntsch, A.; Halligan, D.N.; Kugler, J.; Jones, F.; Lee, C.T.; et al. Mucosal inflammation downregulates PHD1 expression promoting a barrier-protective HIF-1α response in ulcerative colitis patients. FASEB J. 2020, 34, 3732–3742. [Google Scholar] [CrossRef] [Green Version]
- Imtiyaz, H.Z.; Williams, E.P.; Hickey, M.M.; Patel, S.A.; Durham, A.C.; Yuan, L.J.; Hammond, R.; Gimotty, P.A.; Keith, B.; Simon, M.C. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J. Clin. Investig. 2010, 120, 2699–2714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, K.P.; Ganju, L. Influence of high altitude exposure on the immune system: A review. Immunol. Investig. 2010, 39, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, G.; Tschöp, M.; Fischer, R.; Bidlingmaier, C.; Riepl, R.; Tschöp, K.; Hautmann, H.; Endres, S.; Toepfer, M. High altitude increases circulating interleukin-6, interleukin-1 receptor antagonist and C-reactive protein. Cytokine 2000, 12, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jiang, H.; Duan, J.; Chen, J.; Wang, Q.; Liu, X. Exploration of Acute Phase Proteins and Inflammatory Cytokines in Early Stage Diagnosis of Acute Mountain Sickness. High Alt. Med. Biol. 2018, 19, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Lundeberg, J.; Feiner, J.R.; Schober, A.; Sall, J.W.; Eilers, H.; Bickler, P.E. Increased Cytokines at High Altitude: Lack of Effect of Ibuprofen on Acute Mountain Sickness, Physiological Variables, or Cytokine Levels. High Alt. Med. Biol. 2018, 19, 249–258. [Google Scholar] [CrossRef]
- Taylor, C.T.; McElwain, J.C. Ancient atmospheres and the evolution of oxygen sensing via the hypoxia-inducible factor in metazoans. Physiology 2010, 25, 272–279. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem. J. 2007, 405, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Kaelin, W.G.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solaini, G.; Baracca, A.; Lenaz, G.; Sgarbi, G. Hypoxia and mitochondrial oxidative metabolism. Biochim. Biophys. Acta 2010, 1797, 1171–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaelin, W.G. Proline hydroxylation and gene expression. Annu. Rev. Biochem. 2005, 74, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Minamishima, Y.A.; Moslehi, J.; Bardeesy, N.; Cullen, D.; Bronson, R.T.; Kaelin, W.G. Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood 2008, 111, 3236–3244. [Google Scholar] [CrossRef] [Green Version]
- Colgan, S.P.; Taylor, C.T. Hypoxia: An alarm signal during intestinal inflammation. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 281–287. [Google Scholar] [CrossRef] [Green Version]
- Serocki, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. miRNAs regulate the HIF switch during hypoxia: A novel therapeutic target. Angiogenesis 2018, 21, 183–202. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.T.; Lin, J.F.; Li, T.; Li, J.J.; Xu, R.H.; Ju, H.Q. LncRNA-mediated posttranslational modifications and reprogramming of energy metabolism in cancer. Cancer Commun. 2021, 41, 109–120. [Google Scholar] [CrossRef]
- Filippopoulou, C.; Simos, G.; Chachami, G. The Role of Sumoylation in the Response to Hypoxia: An Overview. Cells 2020, 9, 2359. [Google Scholar] [CrossRef]
- Hon, W.C.; Wilson, M.I.; Harlos, K.; Claridge, T.D.; Schofield, C.J.; Pugh, C.W.; Maxwell, P.H.; Ratcliffe, P.J.; Stuart, D.I.; Jones, E.Y. Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature 2002, 417, 975–978. [Google Scholar] [CrossRef]
- Hewitson, K.S.; McNeill, L.A.; Riordan, M.V.; Tian, Y.M.; Bullock, A.N.; Welford, R.W.; Elkins, J.M.; Oldham, N.J.; Bhattacharya, S.; Gleadle, J.M.; et al. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J. Biol. Chem. 2002, 277, 26351–26355. [Google Scholar] [CrossRef] [Green Version]
- Hagen, T.; Taylor, C.T.; Lam, F.; Moncada, S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: Effect on HIF1alpha. Science 2003, 302, 1975–1978. [Google Scholar] [CrossRef]
- Semenza, G.L.; Jiang, B.H.; Leung, S.W.; Passantino, R.; Concordet, J.P.; Maire, P.; Giallongo, A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef] [Green Version]
- Rocha, S. Gene regulation under low oxygen: Holding your breath for transcription. Trends Biochem. Sci. 2007, 32, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.L.; Luo, C.W.; Dai, Z.K.; Shaw, K.P.; Chai, C.Y.; Wu, C.C. Hypoxia-inducible factor-1α, vascular endothelial growth factor, inducible nitric oxide synthase, and endothelin-1 expression correlates with angiogenesis in congenital heart disease. Kaohsiung. J. Med. Sci. 2016, 32, 348–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Aguila, H.L.; Parikh, N.S.; Li, X.; Lamothe, K.; Duan, L.J.; Takeda, H.; Lee, F.S.; Fong, G.H. Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood 2008, 111, 3229–3235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, M.M.; Linde, N.S.; Wynter, A.; Metzger, M.; Wong, C.; Langsetmo, I.; Lin, A.; Smith, R.; Rodgers, G.P.; Donahue, R.E.; et al. HIF prolyl hydroxylase inhibition results in endogenous erythropoietin induction, erythrocytosis, and modest fetal hemoglobin expression in rhesus macaques. Blood 2007, 110, 2140–2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colgan, S.P.; Furuta, G.T.; Taylor, C.T. Hypoxia and Innate Immunity: Keeping Up with the HIFsters. Annu. Rev. Immunol. 2020, 38, 341–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, S.F.; Tambuwala, M.M.; Bruning, U.; Schaible, B.; Scholz, C.C.; Byrne, A.; O’Connor, A.; Gallagher, W.M.; Lenihan, C.R.; Garvey, J.F.; et al. An intact canonical NF-κB pathway is required for inflammatory gene expression in response to hypoxia. J. Immunol. 2011, 186, 1091–1096. [Google Scholar] [CrossRef] [Green Version]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Taylor, C.T.; Doherty, G.; Fallon, P.G.; Cummins, E.P. Hypoxia-dependent regulation of inflammatory pathways in immune cells. J. Clin. Investig. 2016, 126, 3716–3724. [Google Scholar] [CrossRef] [Green Version]
- Hu, F.; Liu, H.; Xu, L.; Li, Y.; Liu, X.; Shi, L.; Su, Y.; Qiu, X.; Zhang, X.; Yang, Y.; et al. Hypoxia-inducible factor-1α perpetuates synovial fibroblast interactions with T cells and B cells in rheumatoid arthritis. Eur. J. Immunol. 2016, 46, 742–751. [Google Scholar] [CrossRef] [Green Version]
- Flück, K.; Breves, G.; Fandrey, J.; Winning, S. Hypoxia-inducible factor 1 in dendritic cells is crucial for the activation of protective regulatory T cells in murine colitis. Mucosal Immunol. 2016, 9, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Shah, Y.M.; Ito, S.; Morimura, K.; Chen, C.; Yim, S.H.; Haase, V.H.; Gonzalez, F.J. Hypoxia-inducible factor augments experimental colitis through an MIF-dependent inflammatory signaling cascade. Gastroenterology 2008, 134, 2036–2048.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, X.; Ramakrishnan, S.; Anderson, E.; Taylor, M.; Zimmermann, E.M.; Spence, J.R.; Huang, S.; Greenson, J.K.; Shah, Y.M. Endothelial PAS domain protein 1 activates the inflammatory response in the intestinal epithelium to promote colitis in mice. Gastroenterology 2013, 145, 831–841. [Google Scholar] [CrossRef] [Green Version]
- Mole, D.R.; Schlemminger, I.; McNeill, L.A.; Hewitson, K.S.; Pugh, C.W.; Ratcliffe, P.J.; Schofield, C.J. 2-oxoglutarate analogue inhibitors of HIF prolyl hydroxylase. Bioorg. Med. Chem. Lett. 2003, 13, 2677–2680. [Google Scholar] [CrossRef]
- Cummins, E.P.; Seeballuck, F.; Keely, S.J.; Mangan, N.E.; Callanan, J.J.; Fallon, P.G.; Taylor, C.T. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology 2008, 134, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Furusho, H.; Hirota, K.; Sasaki, H. Activation of hypoxia-inducible factor 1 attenuates periapical inflammation and bone loss. Int. J. Oral Sci. 2018, 10, 12. [Google Scholar] [CrossRef]
- Shang, L.; Kang, W.; Li, S.; Ge, S. Prolyl hydroxylase inhibitor DMOG suppressed inflammatory cytokine production in human gingival fibroblasts stimulated with Fusobacterium nucleatum. Clin. Oral Investig. 2019, 23, 3123–3132. [Google Scholar] [CrossRef]
- Taylor, C.T.; Colgan, S.P. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat. Rev. Immunol. 2017, 17, 774. [Google Scholar] [CrossRef]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Josephs, S.F.; Ichim, T.E.; Prince, S.M.; Kesari, S.; Marincola, F.M.; Escobedo, A.R.; Jafri, A. Unleashing endogenous TNF-alpha as a cancer immunotherapeutic. J. Transl. Med. 2018, 16, 242. [Google Scholar] [CrossRef] [PubMed]
- Hellwig-Bürgel, T.; Rutkowski, K.; Metzen, E.; Fandrey, J.; Jelkmann, W. Interleukin-1beta and tumor necrosis factor-alpha stimulate DNA binding of hypoxia-inducible factor-1. Blood 1999, 94, 1561–1567. [Google Scholar] [CrossRef]
- Stiehl, D.P.; Jelkmann, W.; Wenger, R.H.; Hellwig-Bürgel, T. Normoxic induction of the hypoxia-inducible factor 1alpha by insulin and interleukin-1beta involves the phosphatidylinositol 3-kinase pathway. FEBS Lett. 2002, 512, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.W.; Lee, S.J.; Kim, J.C. TNF-α upregulates HIF-1α expression in pterygium fibroblasts and enhances their susceptibility to VEGF independent of hypoxia. Exp. Eye Res. 2017, 164, 74–81. [Google Scholar] [CrossRef]
- Zhou, J.; Schmid, T.; Brüne, B. Tumor necrosis factor-alpha causes accumulation of a ubiquitinated form of hypoxia inducible factor-1alpha through a nuclear factor-kappaB-dependent pathway. Mol. Biol. Cell 2003, 14, 2216–2225. [Google Scholar] [CrossRef]
- Görlach, A.; Bonello, S. The cross-talk between NF-kappaB and HIF-1: Further evidence for a significant liaison. Biochem. J. 2008, 412, e17–e19. [Google Scholar] [CrossRef]
- van Uden, P.; Kenneth, N.S.; Rocha, S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem. J. 2008, 412, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Tsapournioti, S.; Mylonis, I.; Hatziefthimiou, A.; Ioannou, M.G.; Stamatiou, R.; Koukoulis, G.K.; Simos, G.; Molyvdas, P.A.; Paraskeva, E. TNFα induces expression of HIF-1α mRNA and protein but inhibits hypoxic stimulation of HIF-1 transcriptional activity in airway smooth muscle cells. J. Cell. Physiol. 2013, 228, 1745–1753. [Google Scholar] [CrossRef]
- Goryo, K.; Torii, S.; Yasumoto, K.; Sogawa, K. Tumour necrosis factor-α suppresses the hypoxic response by NF-κB-dependent induction of inhibitory PAS domain protein in PC12 cells. J. Biochem. 2011, 150, 311–318. [Google Scholar] [CrossRef]
- Augstein, A.; Poitz, D.M.; Braun-Dullaeus, R.C.; Strasser, R.H.; Schmeisser, A. Cell-specific and hypoxia-dependent regulation of human HIF-3α: Inhibition of the expression of HIF target genes in vascular cells. Cell. Mol. Life Sci. 2011, 68, 2627–2642. [Google Scholar] [CrossRef] [PubMed]
- Albina, J.E.; Mastrofrancesco, B.; Vessella, J.A.; Louis, C.A.; Henry, W.L.; Reichner, J.S. HIF-1 expression in healing wounds: HIF-1alpha induction in primary inflammatory cells by TNF-alpha. Am. J. Physiol. Cell Physiol. 2001, 281, C1971–C1977. [Google Scholar] [CrossRef]
- Basic, V.T.; Jacobsen, A.; Sirsjö, A.; Abdel-Halim, S.M. TNF stimulation induces VHL overexpression and impairs angiogenic potential in skeletal muscle myocytes. Int. J. Mol. Med. 2014, 34, 228–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Chen, X.; He, Y.; Tian, Y.; Xu, L.; Ma, Y.; Hu, P.; Su, K.; Luo, Z.; Wei, L.; et al. TNF-α inhibits xenograft tumor formation by A549 lung cancer cells in nude mice via the HIF-1α/VASP signaling pathway. Oncol. Rep. 2019, 41, 2418–2430. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, N.; Kurata, M.; Yamamoto, T.; Morikawa, S.; Masumoto, J. The role of interleukin-1 in general pathology. Inflamm. Regen. 2019, 39, 12. [Google Scholar] [CrossRef] [Green Version]
- Naldini, A.; Filippi, I.; Miglietta, D.; Moschetta, M.; Giavazzi, R.; Carraro, F. Interleukin-1β regulates the migratory potential of MDAMB231 breast cancer cells through the hypoxia-inducible factor-1α. Eur. J. Cancer 2010, 46, 3400–3408. [Google Scholar] [CrossRef]
- Qian, D.; Lin, H.Y.; Wang, H.M.; Zhang, X.; Liu, D.L.; Li, Q.L.; Zhu, C. Normoxic induction of the hypoxic-inducible factor-1 alpha by interleukin-1 beta involves the extracellular signal-regulated kinase 1/2 pathway in normal human cytotrophoblast cells. Biol. Reprod. 2004, 70, 1822–1827. [Google Scholar] [CrossRef] [Green Version]
- Thornton, R.D.; Lane, P.; Borghaei, R.C.; Pease, E.A.; Caro, J.; Mochan, E. Interleukin 1 induces hypoxia-inducible factor 1 in human gingival and synovial fibroblasts. Biochem. J. 2000, 350 Pt 1, 307–312. [Google Scholar] [CrossRef]
- Sartori-Cintra, A.R.; Mara, C.S.; Argolo, D.L.; Coimbra, I.B. Regulation of hypoxia-inducible factor-1α (HIF-1α) expression by interleukin-1β (IL-1 β), insulin-like growth factors I (IGF-I) and II (IGF-II) in human osteoarthritic chondrocytes. Clinics 2012, 67, 35–40. [Google Scholar] [CrossRef]
- Maxwell, P.J.; Gallagher, R.; Seaton, A.; Wilson, C.; Scullin, P.; Pettigrew, J.; Stratford, I.J.; Williams, K.J.; Johnston, P.G.; Waugh, D.J. HIF-1 and NF-kappaB-mediated upregulation of CXCR1 and CXCR2 expression promotes cell survival in hypoxic prostate cancer cells. Oncogene 2007, 26, 7333–7345. [Google Scholar] [CrossRef] [Green Version]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Wicinski, J.; Cervera, N.; Finetti, P.; Hur, M.H.; Diebel, M.E.; Monville, F.; Dutcher, J.; et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009, 69, 1302–1313. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.R.; Ohlmeyer, M. Protein phosphatase 2A as a therapeutic target in inflammation and neurodegeneration. Pharmacol. Ther. 2019, 201, 181–201. [Google Scholar] [CrossRef]
- Berra, E.; Pagès, G.; Pouysségur, J. MAP kinases and hypoxia in the control of VEGF expression. Cancer Metastasis Rev. 2000, 19, 139–145. [Google Scholar] [CrossRef]
- Michiels, C.; Minet, E.; Michel, G.; Mottet, D.; Piret, J.P.; Raes, M. HIF-1 and AP-1 cooperate to increase gene expression in hypoxia: Role of MAP kinases. IUBMB Life 2001, 52, 49–53. [Google Scholar] [CrossRef]
- Koshida, Y.; Koizumi, W.; Sasabe, M.; Katoh, Y.; Okayasu, I. Association of Helicobacter pylori-dependent gastritis with gastric carcinomas in young Japanese patients: Histopathological comparison of diffuse and intestinal type cancer cases. Histopathology 2000, 37, 124–130. [Google Scholar] [CrossRef]
- Yanagisawa, N.; Mikami, T.; Koike, M.; Okayasu, I. Enhanced cell kinetics, p53 accumulation and high p21WAF1 expression in chronic cholecystitis: Comparison with background mucosa of gallbladder carcinomas. Histopathology 2000, 36, 54–61. [Google Scholar] [CrossRef]
- Okayasu, I.; Fujiwara, M.; Hara, Y.; Tanaka, Y.; Rose, N.R. Association of chronic lymphocytic thyroiditis and thyroid papillary carcinoma. A study of surgical cases among Japanese, and white and African Americans. Cancer 1995, 76, 2312–2318. [Google Scholar] [CrossRef]
- Sarra, M.; Pallone, F.; Macdonald, T.T.; Monteleone, G. IL-23/IL-17 axis in IBD. Inflamm. Bowel Dis. 2010, 16, 1808–1813. [Google Scholar] [CrossRef]
- Kobayashi, T.; Okamoto, S.; Hisamatsu, T.; Kamada, N.; Chinen, H.; Saito, R.; Kitazume, M.T.; Nakazawa, A.; Sugita, A.; Koganei, K.; et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn’s disease. Gut 2008, 57, 1682–1689. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, S.S.; Li, X.; Miranti, C.K. The host microenvironment influences prostate cancer invasion, systemic spread, bone colonization, and osteoblastic metastasis. Front. Oncol. 2014, 4, 364. [Google Scholar] [CrossRef] [Green Version]
- Serviss, J.T.; Johnsson, P.; Grandér, D. An emerging role for long non-coding RNAs in cancer metastasis. Front. Genet. 2014, 5, 234. [Google Scholar] [CrossRef] [Green Version]
- Inácio Pinto, N.; Carnier, J.; Oyama, L.M.; Otoch, J.P.; Alcântara, P.S.; Tokeshi, F.; Nascimento, C.M. Cancer as a Proinflammatory Environment: Metastasis and Cachexia. Mediators Inflamm. 2015, 2015, 791060. [Google Scholar] [CrossRef] [Green Version]
- Mucaj, V.; Shay, J.E.; Simon, M.C. Effects of hypoxia and HIFs on cancer metabolism. Int. J. Hematol. 2012, 95, 464–470. [Google Scholar] [CrossRef] [Green Version]
- Nasr, Z.; Pelletier, J. Tumor progression and metastasis: Role of translational deregulation. Anticancer Res. 2012, 32, 3077–3084. [Google Scholar]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Hursting, S.D.; Hursting, M.J. Growth signals, inflammation, and vascular perturbations: Mechanistic links between obesity, metabolic syndrome, and cancer. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1766–1770. [Google Scholar] [CrossRef] [Green Version]
- Bates, R.C.; Mercurio, A.M. Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol. Biol. Cell 2003, 14, 1790–1800. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, D.E.; Ferris, M.; Nguyen, H.; Abboud, E.; Brody, A.R. TNF-alpha induces TGF-beta1 expression in lung fibroblasts at the transcriptional level via AP-1 activation. J. Cell. Mol. Med. 2009, 13, 1866–1876. [Google Scholar] [CrossRef] [PubMed]
- Tafani, M.; Pucci, B.; Russo, A.; Schito, L.; Pellegrini, L.; Perrone, G.A.; Villanova, L.; Salvatori, L.; Ravenna, L.; Petrangeli, E.; et al. Modulators of HIF1α and NFkB in Cancer Treatment: Is it a Rational Approach for Controlling Malignant Progression? Front. Pharmacol. 2013, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toffoli, S.; Michiels, C. Intermittent hypoxia is a key regulator of cancer cell and endothelial cell interplay in tumours. FEBS J. 2008, 275, 2991–3002. [Google Scholar] [CrossRef]
- Tafani, M.; Di Vito, M.; Frati, A.; Pellegrini, L.; De Santis, E.; Sette, G.; Eramo, A.; Sale, P.; Mari, E.; Santoro, A.; et al. Pro-inflammatory gene expression in solid glioblastoma microenvironment and in hypoxic stem cells from human glioblastoma. J. Neuroinflamm. 2011, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.Z.; Guan, W.P.; Maeda, T.; Makino, N. Different levels of hypoxia regulate telomere length and telomerase activity. Aging Clin. Exp. Res. 2012, 24, 213–217. [Google Scholar] [CrossRef]
- Zhu, P.; Ning, Y.; Yao, L.; Chen, M.; Xu, C. The proliferation, apoptosis, invasion of endothelial-like epithelial ovarian cancer cells induced by hypoxia. J. Exp. Clin. Cancer Res. 2010, 29, 124. [Google Scholar] [CrossRef] [Green Version]
- Suswam, E.A.; Nabors, L.B.; Huang, Y.; Yang, X.; King, P.H. IL-1beta induces stabilization of IL-8 mRNA in malignant breast cancer cells via the 3′ untranslated region: Involvement of divergent RNA-binding factors HuR, KSRP and TIAR. Int. J. Cancer 2005, 113, 911–919. [Google Scholar] [CrossRef]
- Chen, D.R.; Lu, D.Y.; Lin, H.Y.; Yeh, W.L. Mesenchymal stem cell-induced doxorubicin resistance in triple negative breast cancer. BioMed Res. Int. 2014, 2014, 532161. [Google Scholar] [CrossRef]
- Jin, K.; Pandey, N.B.; Popel, A.S. Crosstalk between stromal components and tumor cells of TNBC via secreted factors enhances tumor growth and metastasis. Oncotarget 2017, 8, 60210–60222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, D.; Sinha, A.; Bogan, D.; Davies, J.; Koziol, J.; ElShamy, W.M. A niche that triggers aggressiveness within BRCA1-IRIS overexpressing triple negative tumors is supported by reciprocal interactions with the microenvironment. Oncotarget 2017, 8, 103182–103206. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, P.; Gilkes, D.M.; Takano, N.; Semenza, G.L. Hypoxia-inducible factor-dependent signaling between triple-negative breast cancer cells and mesenchymal stem cells promotes macrophage recruitment. Proc. Natl. Acad. Sci. USA 2014, 111, E2120–E2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemik, O.; Kemik, A.S.; Begenik, H.; Erdur, F.M.; Emre, H.; Sumer, A.; Purisa, S.; Tuzun, S.; Kotan, C. The relationship among acute-phase responce proteins, cytokines, and hormones in various gastrointestinal cancer types patients with cachectic. Hum. Exp. Toxicol. 2012, 31, 117–125. [Google Scholar] [CrossRef]
- Kim, S.; Keku, T.O.; Martin, C.; Galanko, J.; Woosley, J.T.; Schroeder, J.C.; Satia, J.A.; Halabi, S.; Sandler, R.S. Circulating levels of inflammatory cytokines and risk of colorectal adenomas. Cancer Res. 2008, 68, 323–328. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; Group, C.T. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369. [Google Scholar] [CrossRef] [Green Version]
- Isambert, N.; Hervieu, A.; Rébé, C.; Hennequin, A.; Borg, C.; Zanetta, S.; Chevriaux, A.; Richard, C.; Derangère, V.; Limagne, E.; et al. Fluorouracil and bevacizumab plus anakinra for patients with metastatic colorectal cancer refractory to standard therapies (IRAFU): A single-arm phase 2 study. Oncoimmunology 2018, 7, e1474319. [Google Scholar] [CrossRef]
- Hamaguchi, T.; Wakabayashi, H.; Matsumine, A.; Sudo, A.; Uchida, A. TNF inhibitor suppresses bone metastasis in a breast cancer cell line. Biochem. Biophys. Res. Commun. 2011, 407, 525–530. [Google Scholar] [CrossRef]
- Holen, I.; Lefley, D.V.; Francis, S.E.; Rennicks, S.; Bradbury, S.; Coleman, R.E.; Ottewell, P. IL-1 drives breast cancer growth and bone metastasis in vivo. Oncotarget 2016, 7, 75571–75584. [Google Scholar] [CrossRef] [Green Version]
- Braegger, C.P.; Nicholls, S.; Murch, S.H.; Stephens, S.; MacDonald, T.T. Tumour necrosis factor alpha in stool as a marker of intestinal inflammation. Lancet 1992, 339, 89–91. [Google Scholar] [CrossRef]
- Komatsu, M.; Kobayashi, D.; Saito, K.; Furuya, D.; Yagihashi, A.; Araake, H.; Tsuji, N.; Sakamaki, S.; Niitsu, Y.; Watanabe, N. Tumor necrosis factor-alpha in serum of patients with inflammatory bowel disease as measured by a highly sensitive immuno-PCR. Clin. Chem. 2001, 47, 1297–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruder, B.; Atreya, R.; Becker, C. Tumour Necrosis Factor Alpha in Intestinal Homeostasis and Gut Related Diseases. Int. J. Mol. Sci. 2019, 20, 1887. [Google Scholar] [CrossRef] [Green Version]
- Cummins, E.P.; Crean, D. Hypoxia and inflammatory bowel disease. Microbes Infect. 2017, 19, 210–221. [Google Scholar] [CrossRef]
- van Dullemen, H.M.; van Deventer, S.J.; Hommes, D.W.; Bijl, H.A.; Jansen, J.; Tytgat, G.N.; Woody, J. Treatment of Crohn’s disease with anti-tumor necrosis factor chimeric monoclonal antibody (cA2). Gastroenterology 1995, 109, 129–135. [Google Scholar] [CrossRef]
- Adegbola, S.O.; Sahnan, K.; Warusavitarne, J.; Hart, A.; Tozer, P. Anti-TNF Therapy in Crohn’s Disease. Int. J. Mol. Sci. 2018, 19, 2244. [Google Scholar] [CrossRef] [Green Version]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Danese, S.; Colombel, J.F.; Peyrin-Biroulet, L.; Rutgeerts, P.; Reinisch, W. Review article: The role of anti-TNF in the management of ulcerative colitis—Past, present and future. Aliment. Pharmacol. Ther. 2013, 37, 855–866. [Google Scholar] [CrossRef] [Green Version]
- Lim, W.C.; Wang, Y.; MacDonald, J.K.; Hanauer, S. Aminosalicylates for induction of remission or response in Crohn’s disease. Cochrane Database Syst. Rev. 2016, 7, CD008870. [Google Scholar] [CrossRef]
- Ben-Horin, S.; Kopylov, U.; Chowers, Y. Optimizing anti-TNF treatments in inflammatory bowel disease. Autoimmun. Rev. 2014, 13, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Hindryckx, P.; De Vos, M.; Jacques, P.; Ferdinande, L.; Peeters, H.; Olievier, K.; Bogaert, S.; Brinkman, B.; Vandenabeele, P.; Elewaut, D.; et al. Hydroxylase inhibition abrogates TNF-alpha-induced intestinal epithelial damage by hypoxia-inducible factor-1-dependent repression of FADD. J. Immunol. 2010, 185, 6306–6316. [Google Scholar] [CrossRef] [Green Version]
- Marchbank, T.; Mahmood, A.; Harten, S.; Maxwell, P.H.; Playford, R.J. Dimethyloxalyglycine stimulates the early stages of gastrointestinal repair processes through VEGF-dependent mechanisms. Lab. Investig. 2011, 91, 1684–1694. [Google Scholar] [CrossRef]
- Hams, E.; Saunders, S.P.; Cummins, E.P.; O’Connor, A.; Tambuwala, M.T.; Gallagher, W.M.; Byrne, A.; Campos-Torres, A.; Moynagh, P.M.; Jobin, C.; et al. The hydroxylase inhibitor dimethyloxallyl glycine attenuates endotoxic shock via alternative activation of macrophages and IL-10 production by B1 cells. Shock 2011, 36, 295–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaible, B.; McClean, S.; Selfridge, A.; Broquet, A.; Asehnoune, K.; Taylor, C.T.; Schaffer, K. Hypoxia modulates infection of epithelial cells by Pseudomonas aeruginosa. PLoS ONE 2013, 8, e56491. [Google Scholar] [CrossRef] [Green Version]
- Van Welden, S.; Selfridge, A.C.; Hindryckx, P. Intestinal hypoxia and hypoxia-induced signalling as therapeutic targets for IBD. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 596–611. [Google Scholar] [CrossRef] [PubMed]
- McDonnel, M. Single Ascending Dose Study to Assess the Safety, Tolerability, PK and PD Effects in Male Volunteers (Identification No. NCT02914262). Available online: https://clinicaltrials.gov/ct2/show/NCT02914262 (accessed on 10 July 2021).
- Levesque, B.; Meadows, K.T.; Buch, A.; Flynn, M.; Peters, K.; Olson, A.; Shen, J.; Sergeeva, M.; Slee, D.; Van Biene, C.; et al. P006 GB004, a novel gut-targeted prolyl hydroxylase inhibitor for inflammatory bowel disease: First-in-human, multiple-dose study in healthy subjects with gut biopsies. Inflamm. Bowel Dis. 2020, 26, S9–S10. [Google Scholar] [CrossRef]
- Sun, M.; He, C.; Wu, W.; Zhou, G.; Liu, F.; Cong, Y.; Liu, Z. Hypoxia inducible factor-1α-induced interleukin-33 expression in intestinal epithelia contributes to mucosal homeostasis in inflammatory bowel disease. Clin. Exp. Immunol. 2017, 187, 428–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malkov, M.I.; Lee, C.T.; Taylor, C.T. Regulation of the Hypoxia-Inducible Factor (HIF) by Pro-Inflammatory Cytokines. Cells 2021, 10, 2340. https://doi.org/10.3390/cells10092340
Malkov MI, Lee CT, Taylor CT. Regulation of the Hypoxia-Inducible Factor (HIF) by Pro-Inflammatory Cytokines. Cells. 2021; 10(9):2340. https://doi.org/10.3390/cells10092340
Chicago/Turabian StyleMalkov, Mykyta I., Chee Teik Lee, and Cormac T. Taylor. 2021. "Regulation of the Hypoxia-Inducible Factor (HIF) by Pro-Inflammatory Cytokines" Cells 10, no. 9: 2340. https://doi.org/10.3390/cells10092340