
Role of MicroRNA in Inflammatory Bowel Disease: Clinical Evidence and the Development of Preclinical Animal Models

 , ,
, ,
Abstract
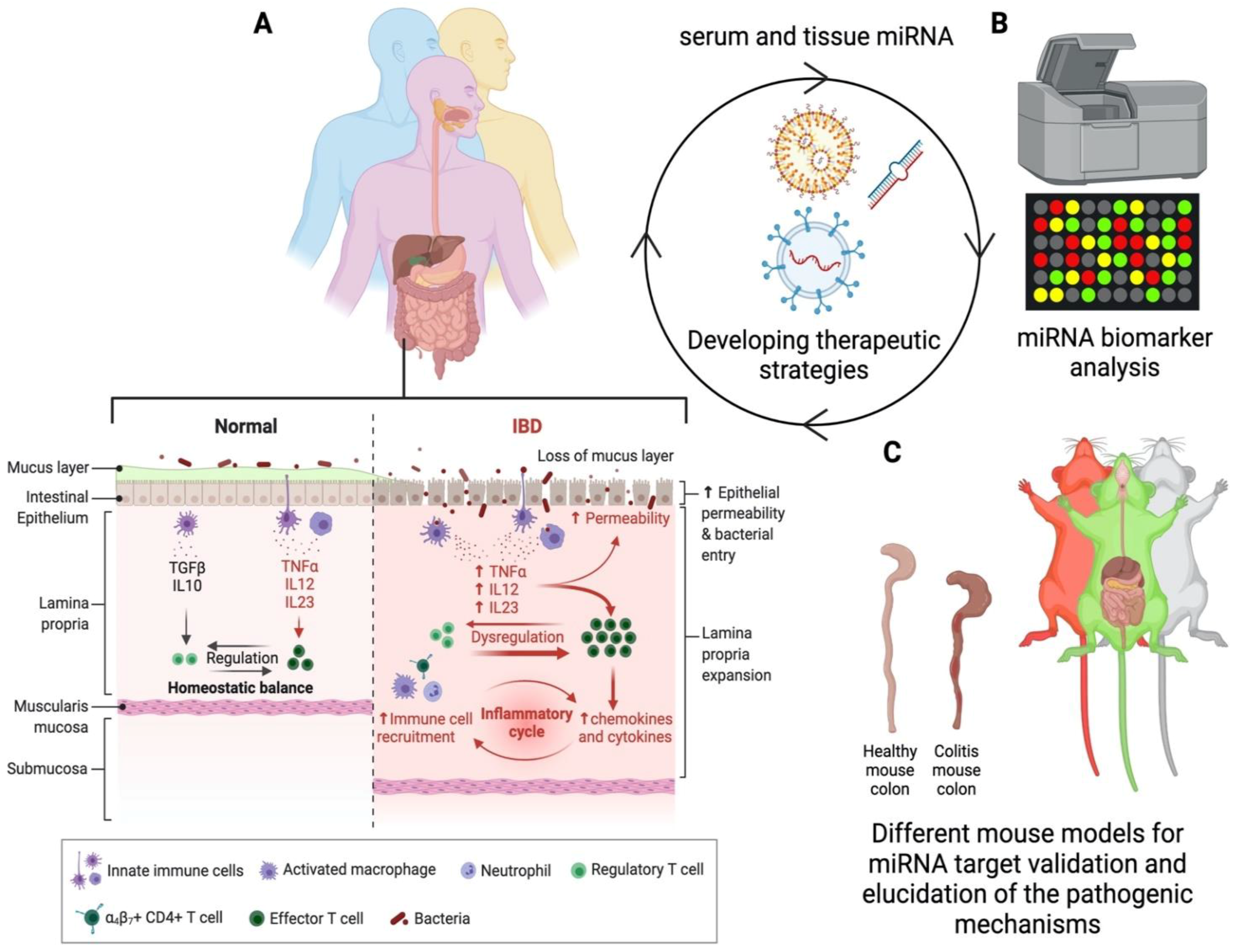
:1. Introduction
2. MicroRNA and IBD
2.1. Synthesis and Physiological Significance of miRNA
2.2. Detection of miRNA
2.3. Therapeutic Potential of miRNA
2.4. Limitations in Current Methodologies in miRNA Research
3. Developing Novel Mouse Models of IBD
3.1. Humanized Mouse Models
3.2. Genetic Diversity and IBD
3.3. Importance of Choosing the Right Mouse Model(s) to Study and Validate miRNA as Therapeutic Targets
4. MicroRNA as a Therapeutic Target in IBD
5. Exosome-Aided Communication
6. Host–Microbiota Interactions Can Impact IBD Via Modulating miRNA
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baumgart, D.C.; Sandborn, W.J. Inflammatory bowel disease: Clinical aspects and established and evolving therapies. Lancet 2007, 369, 1641–1657. [Google Scholar] [CrossRef]
- Rodríguez, L.A.G.; Ruigómez, A.; Panés, J. Acute gastroenteritis is followed by an increased risk of inflammatory bowel disease. Gastroenterology 2006, 130, 1588–1594. [Google Scholar] [CrossRef]
- Abraham, C.; Cho, J.H. Inflammatory bowel disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.; McGovern, D.P.; Hui, K.Y.; Lee, J.; Schumm, P.; Sharma, Y.; Anderson, C.A.; et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Torres, J.; Mehandru, S.; Colombel, J.F.; Peyrin-Biroulet, L. Crohn’s disease. Lancet 2017, 389, 1741–1755. [Google Scholar] [CrossRef]
- Ordás, I.; Eckmann, L.; Talamini, M.; Baumgart, D.C.; Sandborn, W.J. Ulcerative colitis. Lancet 2012, 380, 1606–1619. [Google Scholar] [CrossRef] [Green Version]
- Adams, S.M.; Bornemann, P.H. Ulcerative colitis. Am. Fam. Physician 2013, 87, 699–705. [Google Scholar] [PubMed]
- Roberti, R.; Iannone, L.F.; Palleria, C.; De Sarro, C.; Spagnuolo, R.; Barbieri, M.A.; Vero, A.; Manti, A.; Pisana, V.; Fries, W.; et al. Safety profiles of biologic agents for inflammatory bowel diseases: A prospective pharmacovigilance study in Southern Italy. Curr. Med. Res. Opin. 2020, 36, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Freeman, K.; Connock, M.; Auguste, P.; Taylor-Phillips, S.; Mistry, H.; Shyangdan, D.; Court, R.; Arasaradnam, R.; Sutcliffe, P.; Clarke, A. Clinical effectiveness and cost-effectiveness of use of therapeutic monitoring of tumour necrosis factor alpha (TNF-α) inhibitors [LISA-TRACKER® enzyme-linked immunosorbent assay (ELISA) kits, TNF-α-Blocker ELISA kits and Promonitor® ELISA kits] versus standard care in patients with Crohn’s disease: Systematic reviews and economic modelling. Health Technol. Assess. 2016, 20, 1–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, R.; Tappenden, P.; Ren, S.; James, M.M.-S.; Harvey, R.; Basarir, H.; Stevens, J.; Carroll, C.; Cantrell, A.; Lobo, A.; et al. Infliximab, adalimumab and golimumab for treating moderately to severely active ulcerative colitis after the failure of conventional therapy (including a review of TA140 and TA262): Clinical effectiveness systematic review and economic model. Health Technol. Assess. 2016, 20, 1–326. [Google Scholar] [CrossRef] [PubMed]
- Dahlhamer, J.M.; Zammitti, E.P.; Ward, B.W.; Wheaton, A.G.; Croft, J.B. Prevalence of Inflammatory Bowel Disease Among Adults Aged ≥18 Years—United States, 2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 1166–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, G.C.; Chong, C.A.; Chong, R.Y. National estimates of the burden of inflammatory bowel disease among racial and ethnic groups in the United States. J. Crohn’s Coliti 2014, 8, 288–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.; Panaccione, R.; Ghosh, S.; Barkema, H.; et al. Increasing Incidence and Prevalence of the Inflammatory Bowel Diseases with Time, Based on Systematic Review. Gastroenterology 2012, 142, 46–54.e42. [Google Scholar] [CrossRef] [Green Version]
- Dalal, S.R.; Kwon, J.H. The Role of MicroRNA in Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2010, 6, 714–722. [Google Scholar]
- Chapman, C.G.; Pekow, J. The emerging role of miRNAs in inflammatory bowel disease: A review. Ther. Adv. Gastroenterol. 2014, 8, 4–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalla, R.; Ventham, N.T.; Kennedy, N.; Quintana, J.F.; Nimmo, E.R.; Buck, A.; Satsangi, J. MicroRNAs: New players in IBD. Gut 2014, 64, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-Coding RNAs and their Integrated Networks. J. Integr. Bioinform. 2019, 16. [Google Scholar] [CrossRef]
- Uchida, S.; Adams, J. Physiological roles of non-coding RNAs. Am. J. Physiol. Physiol. 2019, 317, C1–C2. [Google Scholar] [CrossRef]
- Bhartiya, D.; Scaria, V. Genomic variations in non-coding RNAs: Structure, function and regulation. Genomics 2016, 107, 59–68. [Google Scholar] [CrossRef]
- Dai, X.; Kaushik, A.C.; Zhang, J. The Emerging Role of Major Regulatory RNAs in Cancer Control. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Friedman, R.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2008, 19, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Yu, X.; Hu, S.; Yu, J. A Brief Review on the Mechanisms of miRNA Regulation. Genom. Proteom. Bioinform. 2009, 7, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Du, T.; Zamore, P.D. Beginning to understand microRNA function. Cell Res. 2007, 17, 661–663. [Google Scholar] [CrossRef]
- Roden, C.; Gaillard, J.; Kanoria, S.; Rennie, W.; Barish, S.; Cheng, J.; Pan, W.; Liu, J.; Cotsapas, C.; Ding, Y. Novel determinants of mammalian primary microRNA processing revealed by systematic evaluation of hairpin-containing transcripts and human genetic variation. Genome Res. 2017, 27, 374–384. [Google Scholar] [CrossRef] [Green Version]
- Shibata, C.; Otsuka, M.; Kishikawa, T.; Yoshikawa, T.; Ohno, M.; Takata, A.; Koike, K. Current status of miRNA-targeting therapeutics and preclinical studies against gastroenterological carcinoma. Mol. Cell. Ther. 2013, 1, 5. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Zhang, P.-Y.; Li, P.; Xie, J.-W.; Wang, J.-B.; Lin, J.-X.; Chen, Q.-Y.; Cao, L.-L.; Huang, C.-M.; Zheng, C.-H. Circular RNA hsa_circ_0001368 suppresses the progression of gastric cancer by regulating miR-6506–5p/FOXO3 axis. Biochem. Biophys. Res. Commun. 2019, 512, 29–33. [Google Scholar] [CrossRef]
- Chuang, A.Y.; Chuang, J.C.; Zhai, Z.; Wu, F.; Kwon, J.H. NOD2 expression is regulated by microRNAs in colonic epithelial HCT116 cells. Inflamm. Bowel Dis. 2014, 20, 126–135. [Google Scholar] [CrossRef] [Green Version]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Davidson-Moncada, J.; Papavasiliou, F.N.; Tam, W. MiRNAs of the Immune System: Roles in Inflammation and Cancer. Ann. N. Y. Acad. Sci. 2010, 1183, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Ambros, V.; Bartel, B.; Bartel, D.P.; Burge, C.B.; Carrington, J.; Chen, X.; Dreyfuss, G.; Eddy, S.; Griffiths-Jones, S.; Marshall, M.; et al. A uniform system for microRNA annotation. RNA 2003, 9, 277–279. [Google Scholar] [CrossRef] [Green Version]
- Metias, S.M.; Lianidou, E.; Yousef, G.M. MicroRNAs in clinical oncology: At the crossroads between promises and problems. J. Clin. Pathol. 2009, 62, 771–776. [Google Scholar] [CrossRef]
- Davison, T.S.; Johnson, C.D.; Andruss, B.F. Analyzing MicroRNA Expression Using Microarrays. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2006; pp. 14–34. [Google Scholar]
- Mohammadi, A.; Kelly, O.B.; Smith, M.; Kabakchiev, B.; Silverberg, M.S. Differential miRNA Expression in Ileal and Colonic Tissues Reveals an Altered Immunoregulatory Molecular Profile in Individuals with Crohn’s Disease versus Healthy Subjects. J. Crohn’s Coliti 2019, 13, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zikusoka, M.; Trindade, A.; Dassopoulos, T.; Harris, M.L.; Bayless, T.M.; Brant, S.R.; Chakravarti, S.; Kwon, J.H. MicroRNAs Are Differentially Expressed in Ulcerative Colitis and Alter Expression of Macrophage Inflammatory Peptide-2α. Gastroenterology 2008, 135, 1624–1635.e24. [Google Scholar] [CrossRef] [PubMed]
- Takagi, T.; Naito, Y.; Mizushima, K.; Hirata, I.; Yagi, N.; Tomatsuri, N.; Ando, T.; Oyamada, Y.; Isozaki, Y.; Hongo, H.; et al. Increased expression of microRNA in the inflamed colonic mucosa of patients with active ulcerative colitis. J. Gastroenterol. Hepatol. 2010, 25, S129–S133. [Google Scholar] [CrossRef] [PubMed]
- Lennox, K.A.; Behlke, M.A. Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther. 2011, 18, 1111–1120. [Google Scholar] [CrossRef] [Green Version]
- Ebert, M.S.; Sharp, P.A. MicroRNA sponges: Progress and possibilities. RNA 2010, 16, 2043–2050. [Google Scholar] [CrossRef] [Green Version]
- Baumann, V.; Winkler, J. miRNA-based therapies: Strategies and delivery platforms for oligonucleotide and non-oligonucleotide agents. Future Med. Chem. 2014, 6, 1967–1984. [Google Scholar] [CrossRef] [Green Version]
- Mognato, M.; Celotti, L. MicroRNAs Used in Combination with Anti-Cancer Treatments Can Enhance Therapy Efficacy. Mini-Rev. Med. Chem. 2015, 15, 1052–1062. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2007, 36, D154–D158. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Kwon, J.E.; Cho, M.-L. Immunological pathogenesis of inflammatory bowel disease. Intest. Res. 2018, 16, 26–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiesler, P.; Fuss, I.J.; Strober, W. Experimental Models of Inflammatory Bowel Diseases. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 154–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boismenu, R.; Chen, Y. Insights from mouse models of colitis. J. Leukoc. Biol. 2000, 67, 267–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichele, D.D.; Kharbanda, K.K. Dextran sodium sulfate colitis murine model: An indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J. Gastroenterol. 2017, 23, 6016–6029. [Google Scholar] [CrossRef]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104, 15.25.1–15.25.14. [Google Scholar] [CrossRef]
- Alex, P.; Zachos, N.C.; Nguyen, T.; Gonzales, L.; Chen, T.E.; Conklin, L.S.; Centola, M.; Li, X. Distinct Cytokine Patterns Identified from Multiplex Profiles of Murine DSS and TNBS-Induced Colitis. Inflamm. Bowel Dis. 2009, 15, 341–352. [Google Scholar] [CrossRef]
- Lee, J.; Park, E.J.; Yuki, Y.; Ahmad, S.; Mizuguchi, K.; Ishii, K.; Shimaoka, M.; Kiyono, H. Profiles of microRNA networks in intestinal epithelial cells in a mouse model of colitis. Sci. Rep. 2015, 5, 18174. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, J.S.; Montufar-Solis, D.; Vigneswaran, N.; Klein, J.R. Selective upregulation of microRNA expression in peripheral blood leukocytes in IL-10−/− mice precedes expression in the colon. J. Immunol. 2011, 187, 5834–5841. [Google Scholar] [CrossRef] [Green Version]
- Antoniou, E.; Margonis, G.A.; Angelou, A.; Pikouli, A.; Argiri, P.; Karavokyros, I.; Papalois, A.; Pikoulis, E. The TNBS-induced colitis animal model: An overview. Ann. Med. Surg. 2016, 11, 9–15. [Google Scholar] [CrossRef]
- Wu, F.; Dong, F.; Arendovich, N.; Zhang, J.; Huang, Y.; Kwon, J.H. Divergent Influence of MicroRNA-21 Deletion on Murine Colitis Phenotypes. Inflamm. Bowel Dis. 2014, 20, 1972–1985. [Google Scholar] [CrossRef]
- Meroni, E.; Stakenborg, N.; Gomez-Pinilla, P.J.; De Hertogh, G.; Goverse, G.; Matteoli, G.; Verheijden, S.; Boeckxstaens, G.E. Functional characterization of oxazolone-induced colitis and survival improvement by vagus nerve stimulation. PLoS ONE 2018, 13, e0197487. [Google Scholar] [CrossRef]
- Waldner, M.J.; Neurath, M.F. Chemically induced mouse models of colitis. Curr. Protoc. Pharmacol. 2009, 46, 5–55. [Google Scholar] [CrossRef] [PubMed]
- Pizarro, T.T.; Pastorelli, L.; Bamias, G.; Garg, R.R.; Reuter, B.K.; Mercado, J.R.; Marcello, C.; Arseneau, K.O.; Ley, K.; Cominelli, F.; et al. The SAMP1/YitFc Mouse Strain: A Spontaneous Model of Crohn’s Disease-Like Ileitis. Inflamm. Bowel Dis. 2011, 17, 2566–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cominelli, F.; Arseneau, K.O.; Rodriguez-Palacios, A.; Pizarro, T.T. Uncovering Pathogenic Mechanisms of Inflammatory Bowel Disease Using Mouse Models of Crohn’s Disease-Like Ileitis: What is the Right Model? Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 19–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundberg, J.P.; Elson, C.O.; Bedigian, H.; Birkenmeier, E.H. Spontaneous, heritable colitis in a new substrain of C3H/HeJ mice. Gastroenterology 1994, 107, 1726–1735. [Google Scholar] [CrossRef]
- Elson, C.O.; Cong, Y.; Sundberg, J. The C3H/HeJBir Mouse Model: A High Susceptibility Phenotype for Colitis. Int. Rev. Immunol. 2000, 19, 63–75. [Google Scholar] [CrossRef]
- Duijvis, N.W.; Moerland, P.D.; Kunne, C.; Slaman, M.M.W.; Van Dooren, F.H.; Vogels, E.W.; De Jonge, W.J.; Meijer, S.; Fluiter, K.; Velde, A.A.T. Inhibition of miR-142-5P ameliorates disease in mouse models of experimental colitis. PLoS ONE 2017, 12, e0185097. [Google Scholar] [CrossRef] [Green Version]
- Keubler, L.M.; Buettner, M.; Häger, C.; Bleich, A. A Multihit Model: Colitis Lessons from the Interleukin-10-deficient Mouse. Inflamm. Bowel Dis. 2015, 21, 1967–1975. [Google Scholar] [CrossRef]
- Ostanin, D.V.; Bao, J.; Koboziev, I.; Gray, L.; Robinson-Jackson, S.A.; Kosloski-Davidson, M.; Price, V.H.; Grisham, M.B. T cell transfer model of chronic colitis: Concepts, considerations, and tricks of the trade. Am. J. Physiol. Liver Physiol. 2009, 296, G135–G146. [Google Scholar] [CrossRef] [Green Version]
- Eri, R.; McGuckin, M.; Wadley, R. T Cell Transfer Model of Colitis: A Great Tool to Assess the Contribution of T Cells in Chronic Intestinal Inflammation. In Leucocytes: Methods and Protocols; Humana Press: Totowa, NJ, USA, 2011; pp. 261–275. [Google Scholar]
- Hosur, V.; Skelly, D.A.; Francis, C.; Low, B.E.; Kohar, V.; Burzenski, L.M.; Amiji, M.A.; Shultz, L.D.; Wiles, M.V. Improved mouse models and advanced genetic and genomic technologies for the study of neutrophils. Drug Discov. Today 2020, 25, 1013–1025. [Google Scholar] [CrossRef]
- Shultz, L.D.; Brehm, M.; Garcia-Martinez, J.V.; Greiner, D.L. Humanized mice for immune system investigation: Progress, promise and challenges. Nat. Rev. Immunol. 2012, 12, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.-E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized Mouse Models of Clinical Disease. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 187–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudreau, J.; Liu, X.-R.; Zhao, Z.; Zhang, A.; Shultz, L.D.; Greiner, D.L.; Dupont, B.; Hsu, K.C. Cell-Extrinsic MHC Class I Molecule Engagement Augments Human NK Cell Education Programmed by Cell-Intrinsic MHC Class I. Immunity 2016, 45, 280–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.; Pearson, T.; Friberg, H.; Shultz, L.D.; Greiner, D.L.; Rothman, A.L.; Mathew, A. Dengue virus infection and virus-specific HLA-A2 restricted immune responses in humanized NOD-scid IL2rgammanull mice. PLoS ONE 2009, 4, e7251. [Google Scholar] [CrossRef] [PubMed]
- Johanna, I.; Hernández-López, P.; Heijhuurs, S.; Bongiovanni, L.; De Bruin, A.; Beringer, D.; Van Dooremalen, S.; Shultz, L.D.; Ishikawa, F.; Sebestyen, Z.; et al. TEG011 persistence averts extramedullary tumor growth without exerting off-target toxicity against healthy tissues in a humanized HLA-A*24:02 transgenic mice. J. Leukoc. Biol. 2020, 107, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Saito, Y.; Najima, Y.; Tanaka, S.; Ochi, T.; Tomizawa, M.; Doi, T.; Sone, A.; Suzuki, N.; Fujiwara, H.; et al. Generation of functional human T-cell subsets with HLA-restricted immune responses in HLA class I expressing NOD/SCID/IL2r gamma(null) humanized mice. Proc. Natl. Acad. Sci. USA 2010, 107, 13022–13027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goettel, J.A.; Gandhi, R.; Kenison, J.; Yeste, A.; Murugaiyan, G.; Sambanthamoorthy, S.; Griffith, A.E.; Patel, B.; Shouval, D.S.; Weiner, H.L.; et al. AHR Activation Is Protective against Colitis Driven by T Cells in Humanized Mice. Cell Rep. 2016, 17, 1318–1329. [Google Scholar] [CrossRef] [Green Version]
- Jodeleit, H.; Caesar, J.; Aguilera, C.V.; Sterz, S.; Holdt, L.; Beigel, F.; Stallhofer, J.; Breiteneicher, S.; Bartnik, E.; Siebeck, M.; et al. The Combination of Patient Profiling and Preclinical Studies in a Mouse Model Based on NOD/Scid IL2Rγ null Mice Reconstituted with Peripheral Blood Mononuclear Cells from Patients with Ulcerative Colitis May Lead to Stratification of Patients for Treatment with Adalimumab. Inflamm. Bowel Dis. 2019, 26, 557–569. [Google Scholar] [CrossRef]
- Seyhan, A.A. Lost in translation: The valley of death across preclinical and clinical divide—Identification of problems and overcoming obstacles. Transl. Med. Commun. 2019, 4, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Farmer, M.A.; Sundberg, J.P.; Bristol, I.J.; Churchill, G.A.; Li, R.; Elson, C.O.; Leiter, E.H. A major quantitative trait locus on chromosome 3 controls colitis severity in IL-10-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 13820–13825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mähler, M.; Most, C.; Schmidtke, S.; Sundberg, J.P.; Li, R.; Hedrich, H.J.; Churchill, G.A. Genetics of colitis susceptibility in IL-10-deficient mice: Backcross versus F2 results contrasted by principal component analysis. Genomics 2002, 80, 274–282. [Google Scholar] [CrossRef]
- Lahue, K.G.; Lara, M.K.; Linton, A.A.; Lavoie, B.; Fang, Q.; McGill, M.M.; Crothers, J.W.; Teuscher, C.; Mawe, G.M.; Tyler, A.L.; et al. Identification of novel loci controlling inflammatory bowel disease susceptibility utilizing the genetic diversity of wild-derived mice. Genes Immun. 2020, 21, 311–325. [Google Scholar] [CrossRef]
- Doetschman, T. Influence of Genetic Background on Genetically Engineered Mouse Phenotypes. Methods Mol. Biol. 2009, 530, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Sittig, L.; Carbonetto, P.; Engel, K.A.; Krauss, K.; Barrios-Camacho, C.M.; Palmer, A.A. Genetic Background Limits Generalizability of Genotype-Phenotype Relationships. Neuron 2016, 91, 1253–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saul, M.; Philip, V.; Reinholdt, L.G.; Chesler, E.J. High-Diversity Mouse Populations for Complex Traits. Trends Genet. 2019, 35, 501–514. [Google Scholar] [CrossRef] [Green Version]
- Rogala, A.R.; Morgan, A.P.; Christensen, A.M.; Gooch, T.J.; Bell, T.A.; Miller, D.R.; Godfrey, V.L.; De Villena, F.P.-M. The Collaborative Cross as a Resource for Modeling Human Disease: CC011/Unc, a New Mouse Model for Spontaneous Colitis. Mamm. Genome 2014, 25, 95–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubenthal, M.; Franke, A.; Lipinski, S.; Juzenas, S. MicroRNAs and Inflammatory Bowel Disease. In Molecular Genetics of Inflammatory Bowel Disease; Hedin, C., Rioux, J.D., D’Amato, M., Eds.; Springer: Cham, Switzerland, 2019; pp. 203–230. [Google Scholar]
- Schaefer, J.S.; Attumi, T.; Opekun, A.R.; Abraham, B.; Hou, J.; Shelby, H.; Graham, D.Y.; Streckfus, C.; Klein, J.R. MicroRNA signatures differentiate Crohn’s disease from ulcerative colitis. BMC Immunol. 2015, 16, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhang, S.; Dassopoulos, T.; Harris, M.L.; Bayless, T.M.; Meltzer, S.J.; Steven, R.B.; Kwon, J.H. Identification of MicroRNAs Associated with Ileal and Colonic Crohn’s Disease. Inflamm. Bowel Dis. 2010, 16, 1729–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peck, B.C.E.; Weiser, M.; Lee, S.E.; Gipson, G.R.; Iyer, V.B.; Sartor, R.B.; Herfarth, H.H.; Long, M.D.; Hansen, J.J.; Isaacs, K.L.; et al. MicroRNAs Classify Different Disease Behavior Phenotypes of Crohn’s Disease and May Have Prognostic Utility. Inflamm. Bowel Dis. 2015, 21, 2178–2187. [Google Scholar] [CrossRef] [Green Version]
- Fasseu, M.; Treton, X.; Guichard, C.; Pedruzzi, E.; Cazals-Hatem, D.; Richard, C.; Aparicio, T.; Daniel, F.; Soulé, J.-C.; Moreau, R.; et al. Identification of Restricted Subsets of Mature microRNA Abnormally Expressed in Inactive Colonic Mucosa of Patients with Inflammatory Bowel Disease. PLoS ONE 2010, 5, e13160. [Google Scholar] [CrossRef]
- Vijay-Kumar, M.; Chassaing, B.; Kumar, M.; Baker, M.; Singh, V. Mammalian gut immunity. Biomed. J. 2014, 37, 246. [Google Scholar] [CrossRef]
- Thoo, L.; Noti, M.; Krebs, P. Keep calm: The intestinal barrier at the interface of peace and war. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iborra, M.; Bernuzzi, F.; Correale, C.; Vetrano, S.; Fiorino, G.; Beltrán, B.; Marabita, F.; Locati, M.; Spinelli, A.; Nos, P.; et al. Identification of serum and tissue micro-RNA expression profiles in different stages of inflammatory bowel disease. Clin. Exp. Immunol. 2013, 173, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Nijhuis AFau–Kumagai, T.; Kumagai TFau–Lindsay, J.; Lindsay JFau–Silver, A.; Silver, A. Defects in the Adherens Junction Complex (E-Cadherin/ B-Catenin) in Inflammatory Bowel Disease; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- López-Posadas, R.; Stürzl, M.; Atreya, I.; Neurath, M.F.; Britzen-Laurent, N. Interplay of GTPases and Cytoskeleton in Cellular Barrier Defects during Gut Inflammation. Front. Immunol. 2017, 8, 1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, A.A.; Uppada, S.; Achkar, I.; Hashem, S.; Yadav, S.K.; Shanmugakonar, M.; Al-Naemi, H.A.; Haris, M.; Uddin, S. Tight Junction Proteins and Signaling Pathways in Cancer and Inflammation: A Functional Crosstalk. Front. Physiol. 2019, 9, 1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cichon, C.; Sabharwal, H.; Rüter, C.; Schmidt, M.A. MicroRNAs regulate tight junction proteins and modulate epithelial/endothelial barrier functions. Tissue Barriers 2014, 2, e944446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landy, J.; Ronde, E.; English, N.; Clark, S.K.; Hart, A.L.; Knight, S.C.; Ciclitira, P.J.; Al-Hassi, H.O. Tight junctions in inflammatory bowel diseases and inflammatory bowel disease associated colorectal cancer. World J. Gastroenterol. 2016, 22, 3117–3126. [Google Scholar] [CrossRef]
- Aihara, E.; Engevik, K.A.; Montrose, M.H. Trefoil Factor Peptides and Gastrointestinal Function. Annu. Rev. Physiol. 2017, 79, 357–380. [Google Scholar] [CrossRef] [Green Version]
- Aamann, L.; Vestergaard, E.M.; Grønbæk, H. Trefoil factors in inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 3223–3230. [Google Scholar] [CrossRef]
- Sun, Y.; Zhu, Y.; Wang, L.; Mao, X.; Peng, X.; Peng, Y. Recombinant Adenovirus-Mediated Intestinal Trefoil Factor Gene Therapy for Burn-Induced Intestinal Mucosal Injury. PLoS ONE 2013, 8, e62429. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto-Furusho, J.K.; Mendivil, E.J.; Fonseca-Camarillo, G. Differential expression of occludin in patients with ulcerative colitis and healthy controls. Inflamm. Bowel Dis. 2012, 18, E1999. [Google Scholar] [CrossRef]
- Ye, D.; Guo, S.; Al–Sadi, R.; Ma, T.Y. MicroRNA Regulation of Intestinal Epithelial Tight Junction Permeability. Gastroenterology 2011, 141, 1323–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayburgh, D.; Barrett, T.; Tang, Y.; Meddings, J.B.; Van Eldik, L.J.; Watterson, D.; Clarke, L.L.; Mrsny, R.J.; Turner, J.R. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J. Clin. Investig. 2005, 115, 2702–2715. [Google Scholar] [CrossRef] [PubMed]
- Al-Sadi, R.; Ye, D.; Said, H.M.; Ma, T.Y. IL-1β-Induced Increase in Intestinal Epithelial Tight Junction Permeability Is Mediated by MEKK-1 Activation of Canonical NF-κB Pathway. Am. J. Pathol. 2010, 177, 2310–2322. [Google Scholar] [CrossRef]
- Al-Sadi, R.; Ye, D.; Dokladny, K.; Ma, T.Y. Mechanism of IL-1β-Induced Increase in Intestinal Epithelial Tight Junction Permeability. J. Immunol. 2008, 180, 5653–5661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Sadi, R.; Guo, S.; Ye, D.; Dokladny, K.; Alhmoud, T.; Ereifej, L.; Said, H.M.; Ma, T.Y. Mechanism of IL-1β Modulation of Intestinal Epithelial Barrier Involves p38 Kinase and Activating Transcription Factor-2 Activation. J. Immunol. 2013, 190, 6596–6606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawat, M.; Nighot, M.; Al-Sadi, R.; Gupta, Y.; Viszwapriya, D.; Yochum, G.; Koltun, W.; Ma, T.Y. IL1B Increases Intestinal Tight Junction Permeability by Up-regulation of MIR200C-3p, Which Degrades Occludin mRNA. Gastroenterology 2020, 159, 1375–1389. [Google Scholar] [CrossRef]
- Shi, C.; Liang, Y.; Yang, J.; Xia, Y.; Chen, H.; Han, H.; Yang, Y.; Wu, W.; Gao, R.; Qin, H. MicroRNA-21 Knockout Improve the Survival Rate in DSS Induced Fatal Colitis through Protecting against Inflammation and Tissue Injury. PLoS ONE 2013, 8, e66814. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Costinean, S.; Croce, C.M.; Brasier, A.R.; Merwat, S.; Larson, S.A.; Basra, S.; Verne, G.N. MicroRNA 29 targets nuclear factor-kappaB-repressing factor and Claudin 1 to increase intestinal permeability. Gastroenterology 2015, 148, 158–169. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chao, K.; Ng, S.C.; Bai, A.H.; Yu, Q.; Yu, J.; Li, M.; Cui, Y.; Chen, M.; Hu, J.-F.; et al. Pro-inflammatory miR-223 mediates the cross-talk between the IL23 pathway and the intestinal barrier in inflammatory bowel disease. Genome Biol. 2016, 17, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhu, L.; Chen, P.; Wang, Y.; Yang, G.; Zhou, G.; Li, L.; Feng, R.; Qiu, Y.; Han, J.; et al. MALAT1 Maintains the Intestinal Mucosal Homeostasis in Crohn’s Disease via the miR-146b-5p-CLDN11/NUMB Pathway. J. Crohn’s Coliti 2021. [Google Scholar] [CrossRef]
- Guo, J.; Yang, L.-J.; Sun, M.; Xu, L.-F. Inhibiting microRNA-7 Expression Exhibited a Protective Effect on Intestinal Mucosal Injury in TNBS-Induced Inflammatory Bowel Disease Animal Model. Inflammation 2019, 42, 2267–2277. [Google Scholar] [CrossRef]
- Chen, Y.; Shan, T.; Qu, H.; Chen, Y.; Wang, N.; Xia, J. Inhibition of miR-16 Ameliorates Inflammatory Bowel Disease by Modulating Bcl-2 in Mouse Models. J. Surg. Res. 2020, 253, 185–192. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, J.; Li, Y.; Zhao, R.; Du, S.; Lv, C.; Wu, W.; Liu, R.; Sheng, X.; Song, Y.; et al. MicroRNA-31 Reduces Inflammatory Signaling and Promotes Regeneration in Colon Epithelium, and Delivery of Mimics in Microspheres Reduces Colitis in Mice. Gastroenterology 2019, 156, 2281–2296.e6. [Google Scholar] [CrossRef]
- Garo, L.P.; Ajay, A.K.; Fujiwara, M.; Gabriely, G.; Raheja, R.; Kuhn, C.; Kenyon, B.; Skillin, N.; Kadowaki-Saga, R.; Saxena, S.; et al. MicroRNA-146a limits tumorigenic inflammation in colorectal cancer. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Polytarchou, C.; Hommes, D.W.; Palumbo, T.; Hatziapostolou, M.; Koutsioumpa, M.; Koukos, G.; van der Meulen-de Jong, A.E.; Oikonomopoulos, A.; van Deen, W.K.; Vorvis, C.; et al. MicroRNA214 Is Associated with Progression of Ulcerative Colitis, and Inhibition Reduces Development of Colitis and Colitis-Associated Cancer in Mice. Gastroenterology 2015, 149, 981–992.e11. [Google Scholar] [CrossRef] [Green Version]
- Neudecker, V.; Haneklaus, M.; Jensen, O.; Khailova, L.; Masterson, J.C.; Tye, H.; Biette, K.; Jedlicka, P.; Brodsky, K.S.; Gerich, M.E.; et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J. Exp. Med. 2017, 214, 1737–1752. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Zhang, H.; Hao, Y.; Xu, F.; Yang, J.; Zhang, R.; Lu, G.; Zheng, Z.; Cui, M.; Qi, C.-F.; et al. Reprogramming macrophage orientation by microRNA 146b targeting transcription factor IRF5. eBioMedicine 2016, 14, 83–96. [Google Scholar] [CrossRef] [Green Version]
- Deng, F.; He, S.; Cui, S.; Shi, Y.; Tan, Y.; Li, Z.; Huang, C.; Liu, D.; Zhi, F.; Peng, L. A Molecular Targeted Immunotherapeutic Strategy for Ulcerative Colitis via Dual-targeting Nanoparticles Delivering miR-146b to Intestinal Macrophages. J. Crohn’s Coliti 2018, 13, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wang, Q.; Yang, W.; Yang, Q.; Pei, Y.; Zhang, W. MiR-98-5p expression inhibits polarization of macrophages to an M2 phenotype by targeting Trib1 in inflammatory bowel disease. Acta Biochim. Pol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, N.; Cui, L.; Li, Y.; Cao, Z.; Wu, X.; Wang, Q.; Zhang, B.; Ma, C.; Cheng, Y. Long Non-coding RNA MEG3 Alleviated Ulcerative Colitis Through Upregulating miR-98-5p-Sponged IL-10. Inflammation 2021, 44, 1049–1059. [Google Scholar] [CrossRef]
- Singh, U.P.; Murphy, A.E.; Enos, R.; Shamran, H.; Singh, N.P.; Guan, H.; Hegde, V.L.; Fan, D.; Price, R.L.; Taub, D.D.; et al. miR-155 deficiency protects mice from experimental colitis by reducing T helper type 1/type 17 responses. Immunology 2014, 143, 478–489. [Google Scholar] [CrossRef]
- Zhu, F.; Li, H.; Liu, Y.; Tan, C.; Liu, X.; Fan, H.; Wu, H.; Dong, Y.; Yu, T.; Chu, S.; et al. miR-155 antagomir protect against DSS-induced colitis in mice through regulating Th17/Treg cell balance by Jarid2/Wnt/Ī²-catenin. Biomed. Pharmacother. 2020, 126, 109909. [Google Scholar] [CrossRef]
- Shi, T.; Xie, Y.; Fu, Y.; Zhou, Q.; Ma, Z.; Ma, J.; Huang, Z.; Zhang, J.; Chen, J. The signaling axis of microRNA-31/interleukin-25 regulates Th1/Th17-mediated inflammation response in colitis. Mucosal Immunol. 2017, 10, 983–995. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Shi, Y.; Wu, R.; Sun, M.; Fang, L.; Wu, W.; Liu, C.; Tang, M.; Li, Z.; Wang, P.; et al. miR-301a promotes intestinal mucosal inflammation through induction of IL-17A and TNF-α in IBD. Gut 2015, 65, 1938–1950. [Google Scholar] [CrossRef]
- Shi, Y.; Dai, S.; Qiu, C.; Wang, T.; Zhou, Y.; Xue, C.; Yao, J.; Xu, Y. MicroRNA-219a-5p suppresses intestinal inflammation through inhibiting Th1/Th17-mediated immune responses in inflammatory bowel disease. Mucosal Immunol. 2019, 13, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Yu, J.; Wang, J.; Xue, B.; He, N.; Tian, Y.; Yang, L.; Wang, Y.; Wang, Y.; Tang, Q. DNA Methylation of miR-122 Aggravates Oxidative Stress in Colitis Targeting SELENBP1 Partially by p65NF-κB Signaling. Oxidative Med. Cell. Longev. 2019, 2019, 5294105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, K.; Law, I.K.M.; Padua, D.; Sideri, A.; Huang, V.; Kevil, C.G.; Iliopoulos, D.; Pothoulakis, C. MicroRNA-31-3p Is Involved in Substance P (SP)-Associated Inflammation in Human Colonic Epithelial Cells and Experimental Colitis. Am. J. Pathol. 2018, 188, 586–599. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Ma, Y.; Shi, C.; Chen, H.; Zhang, H.; Chen, N.; Zhang, P.; Wang, F.; Yang, J.; Yang, J.; et al. Overexpression of miR-21 in patients with ulcerative colitis impairs intestinal epithelial barrier function through targeting the Rho GTPase RhoB. Biochem. Biophys. Res. Commun. 2013, 434, 746–752. [Google Scholar] [CrossRef]
- Tsukita, S.; Tanaka, H.; Tamura, A. The Claudins: From Tight Junctions to Biological Systems. Trends Biochem. Sci. 2019, 44, 141–152. [Google Scholar] [CrossRef]
- McGovern, D.; Powrie, F. The IL23 axis plays a key role in the pathogenesis of IBD. Gut 2007, 56, 1333–1336. [Google Scholar] [CrossRef] [Green Version]
- Moschen, A.R.; Tilg, H.; Raine, T. IL-12, IL-23 and IL-17 in IBD: Immunobiology and therapeutic targeting. Nat. Rev. Gastroenterol. Hepatol. 2018, 16, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. IL-23 in inflammatory bowel diseases and colon cancer. Cytokine Growth Factor Rev. 2018, 45, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Soroosh, A.; Rankin, C.R.; Polytarchou, C.; Lokhandwala, Z.A.; Patel, A.; Chang, L.; Pothoulakis, C.; Iliopoulos, D.; Padua, D.M. miR-24 Is Elevated in Ulcerative Colitis Patients and Regulates Intestinal Epithelial Barrier Function. Am. J. Pathol. 2019, 189, 1763–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.-I.; Yi, E.-J.; Kim, Y.-D.; Lee, A.R.; Chung, J.; Ha, H.C.; Cho, J.M.; Kim, S.-R.; Ko, H.-J.; Cheon, J.-H.; et al. Local Stabilization of Hypoxia-Inducible Factor-1Ī± Controls Intestinal Inflammation via Enhanced Gut Barrier Function and Immune Regulation. Front. Immunol. 2021, 11, 609689. [Google Scholar] [CrossRef]
- Halligan, D.N.; Khan, M.N.; Brown, E.; Rowan, C.R.; Coulter, I.S.; Doherty, G.A.; Tambuwala, M.; Taylor, C.T. Hypoxia-inducible factor hydroxylase inhibition enhances the protective effects of cyclosporine in colitis. Am. J. Physiol. Liver Physiol. 2019, 317, G90–G97. [Google Scholar] [CrossRef]
- Lou, C.; Li, Y. Functional role of microRNA-135a in colitis. J. Inflamm. 2018, 15, 7. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Xu, L.; Teng, X.; Sun, M. MicroRNA-7-5p regulates the proliferation and migration of intestinal epithelial cells by targeting trefoil factor 3 via inhibiting the phosphoinositide 3-kinase/Akt signalling pathway. Int. J. Mol. Med. 2017, 40, 1435–1443. [Google Scholar] [CrossRef]
- Guo, J.; Sun, M.; Teng, X.; Xu, L. MicroRNA-7-5p regulates the expression of TFF3 in inflammatory bowel disease. Mol. Med. Rep. 2017, 16, 1200–1206. [Google Scholar] [CrossRef] [Green Version]
- Tian, R.; Wang, R.-L.; Xie, H.; Jin, W.; Yu, K.-L. Overexpressed miRNA-155 dysregulates intestinal epithelial apical junctional complex in severe acute pancreatitis. World J. Gastroenterol. 2013, 19, 8282–8291. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, F.; He, D.-K.; Fan, X.-M.; Shen, J. MicroRNA-21 is upregulated during intestinal barrier dysfunction induced by ischemia reperfusion. Kaohsiung J. Med. Sci. 2018, 34, 556–563. [Google Scholar] [CrossRef]
- Ma, F.; Zhang, X.; Yin, K.-J. MicroRNAs in central nervous system diseases: A prospective role in regulating blood-brain barrier integrity. Exp. Neurol. 2019, 323, 113094. [Google Scholar] [CrossRef]
- Ahluwalia, B.; Moraes, L.; Magnusson, M.; Öhman, L. Immunopathogenesis of inflammatory bowel disease and mechanisms of biological therapies. Scand. J. Gastroenterol. 2018, 53, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-R. Dysregulation of mucosal immune response in pathogenesis of inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 3255–3264. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Taganov, K.D.; Boldin, M.P.; Chang, K.-J.; Baltimore, D. NF-B-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [Green Version]
- Béres, N.J.; Szabó, D.; Kocsis, D.; Szűcs, D.; Kiss, Z.; Müller, K.E.; Lendvai, G.A.; Kiss, A.; Arato, A.; Sziksz, E.; et al. Role of Altered Expression of miR-146a, miR-155, and miR-122 in Pediatric Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2016, 22, 327–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzola, A.; González, R.; Gámez-Belmonte, R.; Ocón, B.; Aranda, C.J.; Martínez-Moya, P.; López-Posadas, R.; Hernández-Chirlaque, C.; De Medina, F.S.; Martínez-Augustin, O. miR-146a regulates the crosstalk between intestinal epithelial cells, microbial components and inflammatory stimuli. Sci. Rep. 2018, 8, 17350. [Google Scholar] [CrossRef] [PubMed]
- Boldin, M.P.; Taganov, K.D.; Rao, D.; Yang, L.; Zhao, J.L.; Kalwani, M.; Garcia-Flores, Y.; Luong, M.; Devrekanli, A.; Xu, J.; et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 2011, 208, 1189–1201. [Google Scholar] [CrossRef]
- Lu, L.-F.; Boldin, M.; Chaudhry, A.; Lin, L.-L.; Taganov, K.D.; Hanada, T.; Yoshimura, A.; Baltimore, D.; Rudensky, A.Y. Function of miR-146a in Controlling Treg Cell-Mediated Regulation of Th1 Responses. Cell 2010, 142, 914–929. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Boldin, M.; Yu, Y.; Liu, C.S.; Ea, C.-K.; Ramakrishnan, P.; Taganov, K.D.; Zhao, J.L.; Baltimore, D. miR-146a controls the resolution of T cell responses in mice. J. Exp. Med. 2012, 209, 1655–1670. [Google Scholar] [CrossRef] [Green Version]
- Tourkochristou, E.; Aggeletopoulou, I.; Konstantakis, C.; Triantos, C. Role of NLRP3 inflammasome in inflammatory bowel diseases. World J. Gastroenterol. 2019, 25, 4796–4804. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, F.; Rieger, A.; Schildberg, F.A.; Knolle, P.A.; Schmid-Burgk, J.; Hornung, V. NLRP3 Inflammasome Activity Is Negatively Controlled by miR-223. J. Immunol. 2012, 189, 4175–4181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnnidis, J.B.; Harris, M.H.; Wheeler, R.T.; Stehling-Sun, S.; Lam, M.H.; Kirak, O.; Brummelkamp, T.R.; Fleming, M.; Camargo, F.D. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 2008, 451, 1125–1129. [Google Scholar] [CrossRef]
- Roffel, M.P.; Bracke, K.R.; Heijink, I.H.; Maes, T. miR-223: A Key Regulator in the Innate Immune Response in Asthma and COPD. Front. Med. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-Y. MicroRNA-223: A double-edged sword in rheumatoid arthritis. Rheumatol. Int. 2013, 34, 285–286. [Google Scholar] [CrossRef]
- Yan, L.-N.; Zhang, X.; Xu, F.; Fan, Y.-Y.; Ge, B.; Guo, H.; Li, Z.-L. Four-microRNA signature for detection of type 2 diabetes. World J. Clin. Cases 2020, 8, 1923–1931. [Google Scholar] [CrossRef]
- Sánchez-Ceinos, J.; Rangel-Zuñiga, O.A.; Clemente-Postigo, M.; Podadera-Herreros, A.; Camargo, A.; Alcalá-Diaz, J.F.; Guzmán-Ruiz, R.; López-Miranda, J.; Malagón, M.M. miR-223-3p as a potential biomarker and player for adipose tissue dysfunction preceding type 2 diabetes onset. Mol. Ther. Nucleic Acids 2021, 23, 1035–1052. [Google Scholar] [CrossRef]
- Krausgruber, T.; Blazek, K.; Smallie, T.; Alzabin, S.; Lockstone, H.; Sahgal, N.; Hussell, T.; Feldmann, M.; Udalova, I. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 2011, 12, 231–238. [Google Scholar] [CrossRef]
- Weiss, M.; Blazek, K.; Byrne, A.J.; Perocheau, D.P.; Udalova, I.A. IRF5 is a specific marker of inflammatory macrophages in vivo. Mediat. Inflamm. 2013, 245804. [Google Scholar] [CrossRef] [Green Version]
- Kühn, R.; Löhler, J.; Rennick, D.M.; Rajewsky, K.; Muller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274. [Google Scholar] [CrossRef]
- Chaudhuri, A.A.; So, A.Y.-L.; Sinha, N.; Gibson, W.S.J.; Taganov, K.D.; O’Connell, R.M.; Baltimore, D. MicroRNA-125b Potentiates Macrophage Activation. J. Immunol. 2011, 187, 5062–5068. [Google Scholar] [CrossRef]
- Chaudhuri, A.; So, A.S.; Baltimore, D.; O’connell, R.M. Regulation of macrophage activation using miR-125b. WO2012177565, 2014. [Google Scholar]
- Sugimoto, K. Role of STAT3 in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 5110–5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koukos, G.; Polytarchou, C.; Kaplan, J.L.; Morley–Fletcher, A.; Gras-Miralles, B.; Kokkotou, E.; Baril–Dore, M.; Pothoulakis, C.; Winter, H.S.; Iliopoulos, D. MicroRNA-124 Regulates STAT3 Expression and Is Down-regulated in Colon Tissues of Pediatric Patients with Ulcerative Colitis. Gastroenterology 2013, 145, 842–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.-T.; Wang, J.; Lu, W.; Cao, Y.; Cai, W. Downregulated expression of microRNA-124 in pediatric intestinal failure patients modulates macrophages activation by inhibiting STAT3 and AChE. Cell Death Dis. 2016, 7, e2521. [Google Scholar] [CrossRef]
- Sun, Y.; Qin, Z.; Li, Q.; Wan, J.-J.; Cheng, M.-H.; Wang, P.-Y.; Su, D.-F.; Yu, J.-G.; Liu, X. MicroRNA-124 negatively regulates LPS-induced TNF-α production in mouse macrophages by decreasing protein stability. Acta Pharmacol. Sin. 2016, 37, 889–897. [Google Scholar] [CrossRef] [Green Version]
- Vermeire, S.; Hébuterne, X.; Napora, P.; Wisniewska-Jarosinska, M.; Kiss, G.; Bourreille, A.; Przemysław, Z.; Nitcheu, J.; Gineste, P.; Steens, J.-M.; et al. OP21 ABX464 is safe and efficacious in a proof-of-concept study in ulcerative colitis patients. J. Crohn’s Coliti 2019, 13, S014–S015. [Google Scholar] [CrossRef]
- Tazi, J.; Begon-Pescia, C.; Campos, N.; Apolit, C.; Garcel, A.; Scherrer, D. Specific and selective induction of miR-124 in immune cells by the quinoline ABX464: A transformative therapy for inflammatory diseases. Drug Discov. Today 2020, 26, 1030–1039. [Google Scholar] [CrossRef]
- Chebli, K.; Papon, L.; Paul, C.; Garcel, A.; Campos, N.; Scherrer, D.; Ehrlich, H.J.; Hahne, M.; Tazi, J. The Anti-Hiv Candidate Abx464 Dampens Intestinal Inflammation by Triggering Il-22 Production in Activated Macrophages. Sci. Rep. 2017, 7, 4860. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Lian, B.; Shang, Y.; Li, C.; Meng, Q. miR-135a Protects Dextran Sodium Sulfate-Induced Inflammation in Human Colorectal Cell Lines by Activating STAT3 Signal. Cell. Physiol. Biochem. 2018, 51, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Kanno, T.; Nakayamada, S.; Hirahara, K.; Sciume, G.; Muljo, S.; Kuchen, S.; Casellas, R.; Wei, L.; Kanno, Y.; et al. TGF-β and retinoic acid induce the microRNA miR-10a, which targets Bcl-6 and constrains the plasticity of helper T cells. Nat. Immunol. 2012, 13, 587–595. [Google Scholar] [CrossRef]
- Xue, X.; Feng, T.; Yao, S.; Wolf, K.; Liu, C.-G.; Liu, X.; Elson, C.O.; Cong, Y. Microbiota Downregulates Dendritic Cell Expression of miR-10a, Which Targets IL-12/IL-23p40. J. Immunol. 2011, 187, 5879–5886. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; He, C.; Liu, C.; Cao, A.T.; Xue, X.; Evans-Marin, H.L.; Sun, M.; Fang, L.; Yao, S.; Pinchuk, I.V.; et al. miR-10a inhibits dendritic cell activation and Th1/Th17 cell immune responses in IBD. Gut 2014, 64, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Zhang, D.; Wan, X.; Bai, Y.; Yuan, C.; Wang, T.; Yuan, D.; Zhang, C.; Liu, C. Chlorogenic Acid Suppresses miR-155 and Ameliorates Ulcerative Colitis through the NF-ĪŗB/NLRP3 Inflammasome Pathway. Mol. Nutr. Food Res. 2020, 64, 2000452. [Google Scholar] [CrossRef]
- Seddiki, N.; Brezar, V.; Ruffin, N.; Lévy, Y.; Swaminathan, S. Role of miR-155 in the regulation of lymphocyte immune function and disease. Immunology 2014, 142, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.-B.; Luo, M.-M.; Chen, Z.-Y.; He, B.-H. The Function and Role of the Th17/Treg Cell Balance in Inflammatory Bowel Disease. J. Immunol. Res. 2020, 2020, 1–8. [Google Scholar] [CrossRef]
- Su, J.; Chen, T.; Ji, X.-Y.; Liu, C.; Yadav, P.K.; Wu, R.; Yang, P.; Liu, Z. IL-25 Downregulates Th1/Th17 Immune Response in an IL-10–Dependent Manner in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2013, 19, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Janssen, H.L.A.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; Van Der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV Infection by Targeting MicroRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [Green Version]
- Gebert, L.F.R.; Rebhan, M.A.E.; Crivelli, S.E.M.; Denzler, R.; Stoffel, M.; Hall, J. Miravirsen (SPC3649) can inhibit the biogenesis of miR-122. Nucleic Acids Res. 2013, 42, 609–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wang, C.; Liu, Y.; Tang, L.; Zheng, M.; Xu, C.; Song, J.; Meng, X. miR-122 targets NOD2 to decrease intestinal epithelial cell injury in Crohn’s disease. Biochem. Biophys. Res. Commun. 2013, 438, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nature 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Meng, W.; He, C.; Hao, Y.; Wang, L.; Li, L.; Zhu, G. Prospects and challenges of extracellular vesicle-based drug delivery system: Considering cell source. Drug Deliv. 2020, 27, 585–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, F.; Quarto, R.; Tasso, R. Extracellular Vesicles as Natural, Safe and Efficient Drug Delivery Systems. Pharmaceutics 2019, 11, 557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wani, S.; Law, I.K.M.; Pothoulakis, C. Role and mechanisms of exosomal miRNAs in IBD pathophysiology. Am. J. Physiol. Liver Physiol. 2020, 319, G646–G654. [Google Scholar] [CrossRef]
- Mitsuhashi, S.; Feldbrügge, L.; Csizmadia, E.; Mitsuhashi, M.; Robson, S.C.; Moss, A.C. Luminal Extracellular Vesicles (EVs) in Inflammatory Bowel Disease (IBD) Exhibit Proinflammatory Effects on Epithelial Cells and Macrophages. Inflamm. Bowel Dis. 2016, 22, 1587–1595. [Google Scholar] [CrossRef]
- Wu, H.; Fan, H.; Shou, Z.; Xu, M.; Chen, Q.; Ai, C.; Dong, Y.; Liu, Y.; Nan, Z.; Wang, Y.; et al. Extracellular vesicles containing miR-146a attenuate experimental colitis by targeting TRAF6 and IRAK1. Int. Immunopharmacol. 2019, 68, 204–212. [Google Scholar] [CrossRef]
- Gong, L.; Xiao, J.; Yi, J.; Xiao, J.; Lu, F.; Liu, X. Immunomodulatory Effect of Serum Exosomes from Crohn Disease on Macrophages via Let-7b-5p/TLR4 Signaling. Inflamm. Bowel Dis. 2021. [Google Scholar] [CrossRef]
- Bubier, J.A.; Chesler, E.J.; Weinstock, G.M. Host genetic control of gut microbiome composition. Mamm. Genome 2021, 32, 1–19. [Google Scholar] [CrossRef]
- Singh, N.; Shirdel, E.A.; Waldron, L.; Zhang, R.-H.; Jurisica, I.; Comelli, E.M. The Murine Caecal MicroRNA Signature Depends on the Presence of the Endogenous Microbiota. Int. J. Biol. Sci. 2012, 8, 171–186. [Google Scholar] [CrossRef] [Green Version]
- Filip, A.T.; Balacescu, O.; Marian, C.; Anghel, A. Microbiota Small RNAs in Inflammatory Bowel Disease. J. Gastrointest. Liver Dis. 2016, 25, 509–516. [Google Scholar] [CrossRef]
- Rojas-Feria, M.; Romero-García, T.; Caballero-Rico, J.F.; Ramírez, H.P.; Avilés-Recio, M.; Castro-Fernandez, M.; Porcuna, N.C.; Romero-Gómez, M.; García, F.; Grande, L.; et al. Modulation of faecal metagenome in Crohn’s disease: Role of microRNAs as biomarkers. World J. Gastroenterol. 2018, 24, 5223–5233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Deeke, S.A.; Ning, Z.; Starr, A.E.; Butcher, J.; Li, J.; Mayne, J.; Cheng, K.; Liao, B.; Li, L.; et al. Metaproteomics reveals associations between microbiome and intestinal extracellular vesicle proteins in pediatric inflammatory bowel disease. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Larabi, A.; Dalmasso, G.; Delmas, J.; Barnich, N.; Nguyen, H.T.T. Exosomes transfer miRNAs from cell-to-cell to inhibit autophagy during infection with Crohn’s disease-associated adherent-invasive E. coli. Gut Microbes 2020, 11, 1677–1694. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Group | Subgroup | Model Features | Similarities with Human IBD | References |
|---|---|---|---|---|
| Chemical agents | Dextran Sodium Sulfate (DSS) Colitis Model. |
|
| [43,44,45,46,47,48,49] |
| Chemical agents | Trinitrobenzenesulfonic acid (TNBS). |
|
| [43,47,49,50,51] |
| Chemical agents | Oxazolone |
|
| [43,52,53] |
| Spontaneous colitis | SAMP1/YitFcsJ mouse strain |
|
| [54,55] |
| Spontaneous colitis | C3H/HeJBirLtJ mouse strain |
|
| [56,57] |
| Gene knockout (KO) | B6.129P2-Il10tm1Cgn/J (IL-10 KO) |
|
| [43,49,58,59] |
| Reconstitution of immunodeficient mice with CD4+ T-cells | Adoptive transfer colitis |
|
| [43,58,60,61] |
| Outcome | miRNA | Model | Treatment | Dose and Regimen | Ref |
|---|---|---|---|---|---|
| Occludin upregulation | miR-122a | Intestinal Perfusion Model | Anti-miR-122a | Dose: 25 nM, complexed with Lipofectamine. | [97] |
| miR-200c-3p | 3% DSS | Antagomir-200c-3p | Dose: 800 mg/day, starting two days before DSS treatment and continued for seven days of DSS course; oral gavage. | [101] | |
| miR-21 | miR-21 KO mice, 3.5% DSS | miR-21 deletion showed less susceptibility to DSS induced colitis | N/A | [102] | |
| Claudin CLDN1 upregulation | miR-29a and b | miR-29 KO mice, TNBS | Mice tolerated TNBS induced barrier disruption | N/A | [103] |
| Claudin CLDN8 upregulation | miR-223 | TNBS | Antagomir-223 | Dose: 7.5 mg/kg, prepared as 3 mg/mL in PBS, dosed for three successive days 24 h after TNBS administration; IP administration. | [104] |
| Claudin CLDN11 upregulation | miR-146b-5p | TNBS | antagomir-146b-5p | Not specified | [105] |
| Trefoil factor family 3 (TFF3) upregulation | miR-7 | TNBS | Antagomir-7 | Dose: 100 nmol/kg, tail vein injection, 2 h after TNBS perfusion. | [106] |
| Bcl-2 upregulation | miR-16 | 3% DSS | Anti-miR-16 | Dose: 5 mg/kg IP administration, twice a week, for the two weeks of 3% DSS administration. | [107] |
| Epithelial Regeneration by WNT and Hippo signaling | miR-31 | 3.5% DSS | oxidized konjac glucomannan (OKGM)-PS-miR-31 microspheres | Dose: 3.15 µg, enema. Preventative mode: once per day for 7 d, 2 d gap, 5 d DSS treatment. Therapeutic mode: 5 d DSS treatment, once per day for 7 d; microsphere enema. | [108] |
| NF-kB pathway dampening | miR-146a | 3% DSS | miR-146a mimics | Dose: 5 mg/kg, IP administration. | [109] |
| miR-214 | DSS | AntagomiR-214 | Dose: 12 mg/kg, 2.5 mg/mL diluted in PBS, 4 doses, every 2 days after DSS treatment regime; intracolonic administration. | [110] | |
| IL10RA activation | miR-142-5p | CD4 + CD45RO + hitransfer | AntagomiR-142-5p | 5 mg/kg, IP, 5 consecutive days starting at 5–10% weight loss; IP | [58] |
| NLRP3 inhibition | miR-223 | 3% DSS | miR-223 mimic | Dose: 50 µg, nanoparticle emulsion- DOPC, squalene oil, PS-20 and an antioxidant, on days 1 and 3 after DSS. | [111] |
| Macrophage polarization | miR-146b | IL-10 KO | miR-146b mimics | Dose: 10 mg/kg, twice a week; IP administration | [112] |
| miR-146b | 3% DSS | miR-146b mimic | Dose: 20 µg/kg, encapsulated in mannose modified trimethyl chitosan nanoparticles; oral administration | [113] | |
| miR-98-5p | 4% DSS | Antagomir-98-5p | Caudal vein administration | [114] | |
| miR-98-5p | TNBS | pcDNA3.1-MEG3 mediated reduction in miR-98-5p | Injected, complexed with Lipofectamine 2000 | [115] | |
| Inhibition of Th1/Th17 mediated inflammatory response | miR-155 | miR-155 KO, 1% DSS | Lower levels of Th17 upon DSS induced colitis observed | N/A | [116] |
| miR-155 | 3% DSS | AntagomiR-155 | Dose: 80 mg/kg, from the 5th day of the DSS cycle, for three consecutive days, IP administration | [117] | |
| miR-31 | TNBS | Antimir-31 | Dose: 5 mg/kg, 12 h after TNBS treatment, complexed with PEI; intracolonic administration | [118] | |
| miR-31 | IL10 KO | Antimir-31 | Dose: 5 mg/kg, weekly for IL10 KO mice, complexed with PEI; intracolonic administration | [118] | |
| miR-301a | TNBS | Anti-miR-301a | Dose: 3 optical density, intracolonic administration daily, starting at the day of TNBS induction till 5 d. | [119] | |
| miR-219a-5p | TNBS | Pre-miR-219a-5p | Dose: 5 mg/kg, complexed with PEI, 4 consecutive days, 12 h after TNBS administration | [120] | |
| Oxidative stress and SBP1 downregulation | miR-122 | TNBS | Pre-miR-122 | Dose: 5 mg/kg, 12 h after TNBS treatment | [121] |
| RhoA reduction | miR-31-3p | 2% DSS | AgomiR-31-3p | Dose: 80 µg; days 1, 3 and 5 of DSS treatment, intracolonic administration | [122] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suri, K.; Bubier, J.A.; Wiles, M.V.; Shultz, L.D.; Amiji, M.M.; Hosur, V. Role of MicroRNA in Inflammatory Bowel Disease: Clinical Evidence and the Development of Preclinical Animal Models. Cells 2021, 10, 2204. https://doi.org/10.3390/cells10092204
Suri K, Bubier JA, Wiles MV, Shultz LD, Amiji MM, Hosur V. Role of MicroRNA in Inflammatory Bowel Disease: Clinical Evidence and the Development of Preclinical Animal Models. Cells. 2021; 10(9):2204. https://doi.org/10.3390/cells10092204
Chicago/Turabian StyleSuri, Kanika, Jason A. Bubier, Michael V. Wiles, Leonard D. Shultz, Mansoor M. Amiji, and Vishnu Hosur. 2021. "Role of MicroRNA in Inflammatory Bowel Disease: Clinical Evidence and the Development of Preclinical Animal Models" Cells 10, no. 9: 2204. https://doi.org/10.3390/cells10092204