
The State of Immunotherapy in Hepatobiliary Cancers

Abstract
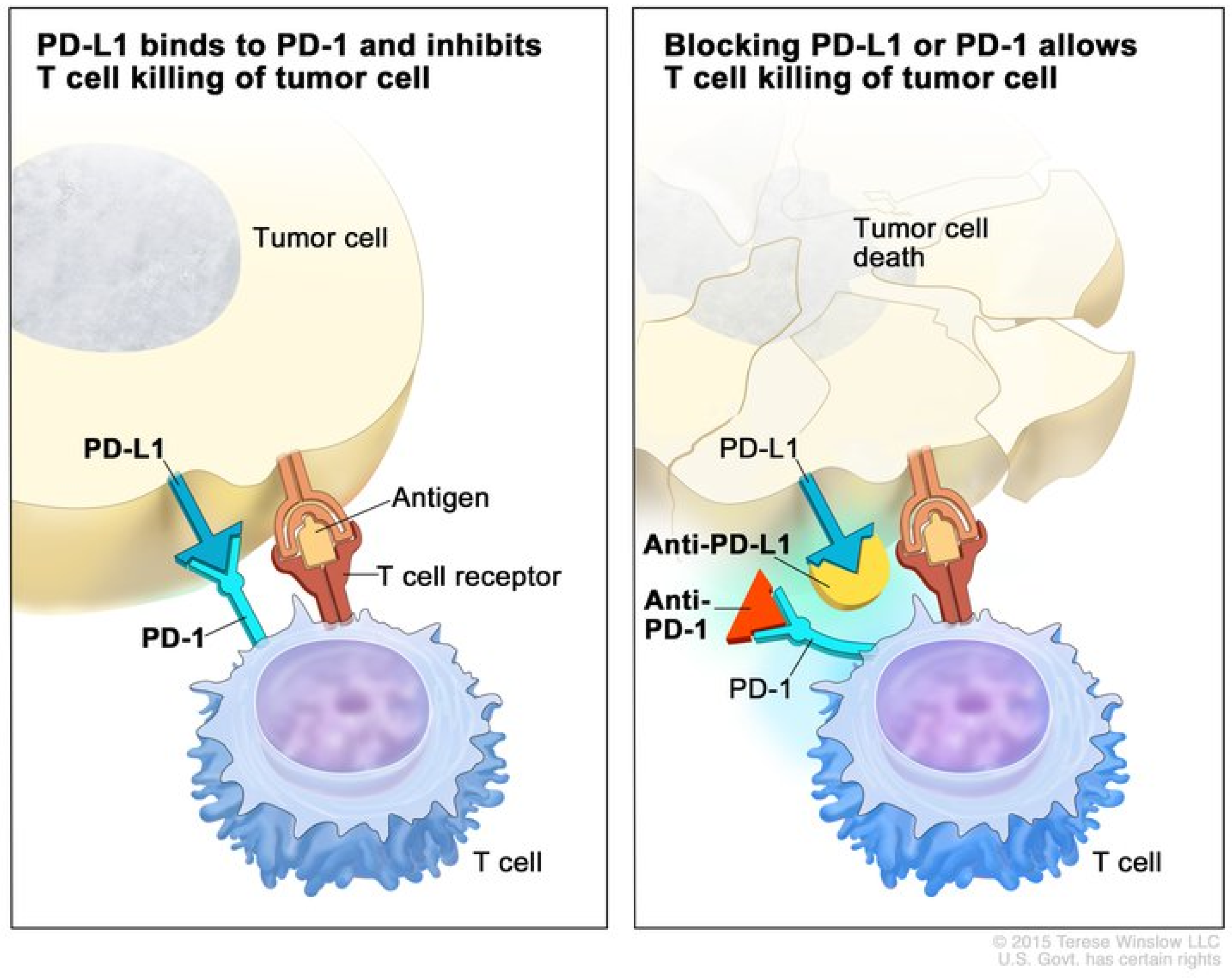
:1. Introduction
2. Hepatocellular Carcinoma
2.1. Current Therapeutic Approach
2.2. The Challenging Tumor Microenvironment of HCC
2.3. Immune Checkpoint Inhibitors
2.4. Future Directions and Novel Approaches
3. Biliary Tract Cancer
3.1. Current Therapeutic Approach
3.2. The Challenging Tumor Microenvironment of Biliary Tract Cancers
3.3. Immune Checkpoint Inhibitors
3.4. Future Directions and Novel Approaches
4. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Seth, R.; Messersmith, H.; Kaur, V.; Kirkwood, J.M.; Kudchadkar, R.; McQuade, J.L.; Provenzano, A.; Swami, U.; Weber, J.; Alluri, K.C.; et al. Systemic Therapy for Melanoma: ASCO Guideline. J. Clin. Oncol. 2020, 38, 3947–3970. [Google Scholar] [CrossRef]
- Wrobel, P.; Ahmed, S. Current status of immunotherapy in metastatic colorectal cancer. Int. J. Colorectal Dis. 2019, 34, 13–25. [Google Scholar] [CrossRef]
- Sangro, B.; Sarobe, P.; Hervás-Stubbs, S.; Melero, I. Advances in immunotherapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 525–534. [Google Scholar] [CrossRef]
- Guo, X.; Shen, W. Latest evidence on immunotherapy for cholangiocarcinoma. Oncol. Lett. 2020, 20, 381. [Google Scholar] [CrossRef]
- Moazzami, B.; Majidzadeh, A.K.; Dooghaie-Moghadam, A.; Eslami, P.; Razavi-Khorasani, N.; Iravani, S.; Khoshdel, A.; Shahi, F.; Dashti, H.; Mehrvar, A.; et al. Cholangiocarcinoma: State of the Art. J. Gastrointest. Cancer 2020, 51, 774–781. [Google Scholar] [CrossRef]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [Green Version]
- Ganne-Carrié, N.; Nahon, P. Hepatocellular carcinoma in the setting of alcohol-related liver disease. J. Hepatol. 2019, 70, 284–293. [Google Scholar] [CrossRef] [Green Version]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273.e1. [Google Scholar] [CrossRef] [Green Version]
- WHO. World Health Organization GLOBOCAN 2020 Database; WHO International Agency for Research on Cancer: New York, NY, USA, 2020. [Google Scholar]
- Jemal, A.; Ward, E.M.; Johnson, C.J.; Cronin, K.A.; Ma, J.; Ryerson, B.; Mariotto, A.; Lake, A.J.; Wilson, R.; Sherman, R.L.; et al. Annual Report to the Nation on the Status of Cancer, 1975–2014, Featuring Survival. J. Natl. Cancer Inst. 2017, 109, djx030. [Google Scholar] [CrossRef]
- Roayaie, S.; Jibara, G.; Tabrizian, P.; Park, J.W.; Yang, J.; Yan, L.; Schwartz, M.; Han, G.; Izzo, F.; Chen, M.; et al. The role of hepatic resection in the treatment of hepatocellular cancer. Hepatology 2015, 62, 440–451. [Google Scholar] [CrossRef]
- Marrero, J.A.; Kulik, L.M.; Sirlin, C.B.; Zhu, A.X.; Finn, R.S.; Abecassis, M.M.; Roberts, L.R.; Heimbach, J.K. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2018, 68, 723–750. [Google Scholar] [CrossRef] [Green Version]
- Mazzaferro, V.; Regalia, E.; Doci, R.; Andreola, S.; Pulvirenti, A.; Bozzetti, F.; Montalto, F.; Ammatuna, M.; Morabito, A.; Gennari, L. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N. Engl. J. Med. 1996, 334, 693–699. [Google Scholar] [CrossRef]
- Ishizawa, T.; Hasegawa, K.; Aoki, T.; Takahashi, M.; Inoue, Y.; Sano, K.; Imamura, H.; Sugawara, Y.; Kokudo, N.; Makuuchi, M. Neither multiple tumors nor portal hypertension are surgical contraindications for hepatocellular carcinoma. Gastroenterology 2008, 134, 1908–1916. [Google Scholar] [CrossRef]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.N.; Fessas, P.; Dominy, K.; Mauri, F.A.; Kaneko, T.; Parcq, P.D.; Khorashad, J.; Toniutto, P.; Goldin, R.D.; Avellini, C.; et al. Qualification of tumour mutational burden by targeted next-generation sequencing as a biomarker in hepatocellular carcinoma. Liver Int. 2021, 41, 192–203. [Google Scholar] [CrossRef]
- Crispe, I.N. The liver as a lymphoid organ. Annu. Rev. Immunol. 2009, 27, 147–163. [Google Scholar] [CrossRef]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Dunham, R.M.; Thapa, M.; Velazquez, V.M.; Elrod, E.J.; Denning, T.L.; Pulendran, B.; Grakoui, A. Hepatic stellate cells preferentially induce Foxp3+ regulatory T cells by production of retinoic acid. J. Immunol. 2013, 190, 2009–2016. [Google Scholar] [CrossRef] [Green Version]
- Ormandy, L.A.; Hillemann, T.; Wedemeyer, H.; Manns, M.P.; Greten, T.F.; Korangy, F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005, 65, 2457–2464. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.C.; Chen, C.H.; Liang, X.; Wang, L.; Gandhi, C.R.; Fung, J.J.; Lu, L.; Qian, S. Inhibition of T-cell responses by hepatic stellate cells via B7-H1-mediated T-cell apoptosis in mice. Hepatology 2004, 40, 1312–1321. [Google Scholar] [CrossRef]
- Sangro, B.; Gomez-Martin, C.; de la Mata, M.; Iñarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.I.; Larrea, E.; Alfaro, C.; Sarobe, P.; et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Greene, J.L.; Brady, W.; Bajorath, J.; Ledbetter, J.A.; Peach, R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1994, 1, 793–801. [Google Scholar] [CrossRef]
- Riley, J.L.; Mao, M.; Kobayashi, S.; Biery, M.; Burchard, J.; Cavet, G.; Gregson, B.P.; June, C.H.; Linsley, P.S. Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 11790–11795. [Google Scholar] [CrossRef] [Green Version]
- Schneider, H.; Downey, J.; Smith, A.; Zinselmeyer, B.H.; Rush, C.; Brewer, J.M.; Wei, B.; Hogg, N.; Garside, P.; Rudd, C.E. Reversal of the TCR stop signal by CTLA-4. Science 2006, 313, 1972–1975. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Blank, C.; Brown, I.; Peterson, A.C.; Spiotto, M.; Iwai, Y.; Honjo, T.; Gajewski, T.F. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004, 64, 1140–1145. [Google Scholar] [CrossRef] [Green Version]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [Green Version]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef]
- Zheng, C.; Zheng, L.; Yoo, J.K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356 e1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab as Second-Line Therapy in Patients with Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate 040. J. Clin. Oncol. 2019, 37, 4012. [Google Scholar] [CrossRef]
- Sangro, B.; Park, J.; Finn, R.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.; Harding, J.; Merle, P.; Rosmorduc, O.; et al. CheckMate 459: Long-term (minimum follow-up 33.6 months) survival outcomes with nivolumab versus sorafenib as first-line treatment in patients with advanced hepatocellular carcinoma. Ann. Oncol. 2020, 31 (Suppl. 3), S241–S242. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, R.K.; Sangro, B.; Harris, W.P.; Ikeda, M.; Okusaka, T.; Kang, Y.-K.; Qin, S.; Tai, W.M.D.; Lim, H.Y.; Yau, T.; et al. Efficacy, tolerability, and biologic activity of a novel regimen of tremelimumab (T) in combination with durvalumab (D) for patients (pts) with advanced hepatocellular carcinoma (aHCC). J. Clin. Oncol. 2020, 38, 4508. [Google Scholar] [CrossRef]
- Yau, T.; Zagonel, V.; Santoro, A.; Acosta-Rivera, M.; Choo, S.P.; Matilla, A.; He, A.R.; Gracián, A.C.; El-Khoueiry, A.B.; Sangro, B.; et al. Nivolumab (NIVO) + ipilimumab (IPI) + cabozantinib (CABO) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate 040. J. Clin. Oncol. 2020, 38, 478. [Google Scholar] [CrossRef]
- van Doorn, D.J.; Takkenberg, R.B.; Klümpen, H.J. Immune Checkpoint Inhibitors in Hepatocellular Carcinoma: An Overview. Pharmaceuticals 2020, 14, 3. [Google Scholar] [CrossRef]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; ElGindi, M.; et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Cubero, E.; Larrubia, J.R. Specific CD8(+) T cell response immunotherapy for hepatocellular carcinoma and viral hepatitis. World J. Gastroenterol. 2016, 22, 6469–6483. [Google Scholar] [CrossRef] [PubMed]
- Li, F.J.; Zhang, Y.; Jin, G.X.; Yao, L.; Wu, D.Q. Expression of LAG-3 is coincident with the impaired effector function of HBV-specific CD8(+) T cell in HCC patients. Immunol. Lett. 2013, 150, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakuishi, K.; Jayaraman, P.; Behar, S.M.; Anderson, A.C.; Kuchroo, V.K. Emerging Tim-3 functions in antimicrobial and tumor immunity. Trends Immunol. 2011, 32, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wu, K.; Tao, K.; Chen, L.; Zheng, Q.; Lu, X.; Liu, J.; Shi, L.; Liu, C.; Wang, G.; et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology 2012, 56, 1342–1351. [Google Scholar] [CrossRef]
- Ganjalikhani Hakemi, M.; Jafarinia, M.; Azizi, M.; Rezaeepoor, M.; Isayev, O.; Bazhin, A.V. The Role of TIM-3 in Hepatocellular Carcinoma: A Promising Target for Immunotherapy? Front. Oncol. 2020, 10, 601661. [Google Scholar] [CrossRef]
- Song, B.; Zhen, S.; Meng, F. T cell inflammation profile after surgical resection may predict tumor recurrence in HBV-related hepatocellular carcinoma. Int. Immunopharmacol. 2016, 41, 35–41. [Google Scholar] [CrossRef]
- Liu, J.F.; Wu, L.; Yang, L.L.; Deng, W.W.; Mao, L.; Wu, H.; Zhang, W.F.; Sun, Z.J. Blockade of TIM3 relieves immunosuppression through reducing regulatory T cells in head and neck cancer. J. Exp. Clin. Cancer Res. 2018, 37, 44. [Google Scholar] [CrossRef] [Green Version]
- Le Naour, J.; Galluzzi, L.; Zitvogel, L.; Kroemer, G.; Vacchelli, E. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology 2020, 9, 1777625. [Google Scholar] [CrossRef] [PubMed]
- van Baren, N.; Van den Eynde, B.J. Tryptophan-degrading enzymes in tumoral immune resistance. Front. Immunol. 2015, 6, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, Z.J.; Yu, S.J.; Heinrich, B.; Ma, C.; Fu, Q.; Sandhu, M.; Agdashian, D.; Zhang, Q.; Korangy, F.; Greten, T.F. Indoleamine 2,3-dioxygenase provides adaptive resistance to immune checkpoint inhibitors in hepatocellular carcinoma. Cancer Immunol. Immunother. 2018, 67, 1305–1315. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.J.; Rosenberg, S.A.; Dudley, M.E.; Yang, J.C.; White, D.E.; Butman, J.A.; Sherry, R.M. Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin. Cancer Res. 2010, 16, 4892–4898. [Google Scholar] [CrossRef] [Green Version]
- Takayama, T.; Sekine, T.; Makuuchi, M.; Yamasaki, S.; Kosuge, T.; Yamamoto, J.; Shimada, K.; Sakamoto, M.; Hirohashi, S.; Ohashi, Y.; et al. Adoptive immunotherapy to lower postsurgical recurrence rates of hepatocellular carcinoma: A randomised trial. Lancet 2000, 356, 802–807. [Google Scholar] [CrossRef]
- Ma, Y.; Xu, Y.C.; Tang, L.; Zhang, Z.; Wang, J.; Wang, H.X. Cytokine-induced killer (CIK) cell therapy for patients with hepatocellular carcinoma: Efficacy and safety. Exp. Hematol. Oncol. 2012, 1, 11. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.Z.; Wang, Q.J.; Dan, J.Q.; Pan, K.; Li, Y.Q.; Zhang, Y.J.; Zhao, J.J.; Weng, D.S.; Tang, Y.; Huang, L.X.; et al. A nomogram for predicting the benefit of adjuvant cytokine-induced killer cell immunotherapy in patients with hepatocellular carcinoma. Sci. Rep. 2015, 5, 9202. [Google Scholar] [CrossRef] [Green Version]
- Pan, K.; Li, Y.Q.; Wang, W.; Xu, L.; Zhang, Y.J.; Zheng, H.X.; Zhao, J.J.; Qiu, H.J.; Weng, D.S.; Li, J.J.; et al. The efficacy of cytokine-induced killer cell infusion as an adjuvant therapy for postoperative hepatocellular carcinoma patients. Ann. Surg. Oncol. 2013, 20, 4305–4311. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, Z.; Liu, Z.; Wei, D.; Wu, X.; Bian, H.; Chen, Z. Adoptive cell transfer therapy for hepatocellular carcinoma. Front. Med. 2019, 13, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sermer, D.; Brentjens, R. CAR T-cell therapy: Full speed ahead. Hematol. Oncol. 2019, 37 (Suppl. 1), 95–100. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Jiang, X.; Chen, S.; Lai, Y.; Wei, X.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; Liang, Q.; et al. Anti-GPC3-CAR T Cells Suppress the Growth of Tumor Cells in Patient-Derived Xenografts of Hepatocellular Carcinoma. Front. Immunol. 2016, 7, 690. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Burga, R.A.; Thorn, M.; Point, G.R.; Guha, P.; Nguyen, C.T.; Licata, L.A.; DeMatteo, R.P.; Ayala, A.; Joseph Espat, N.; Junghans, R.P.; et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol. Immunother. 2015, 64, 817–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizell, M.; Sternby Eilard, M.; Andersson, M.; Andersson, B.; Karlsson-Parra, A.; Suenaert, P. Phase 1 Trial With the Cell-Based Immune Primer Ilixadencel, Alone, and Combined With Sorafenib, in Advanced Hepatocellular Carcinoma. Front. Oncol. 2019, 9, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawada, Y.; Yoshikawa, T.; Nobuoka, D.; Shirakawa, H.; Kuronuma, T.; Motomura, Y.; Mizuno, S.; Ishii, H.; Nakachi, K.; Konishi, M.; et al. Phase I trial of a glypican-3-derived peptide vaccine for advanced hepatocellular carcinoma: Immunologic evidence and potential for improving overall survival. Clin. Cancer Res. 2012, 18, 3686–3696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greten, T.F.; Forner, A.; Korangy, F.; N’Kontchou, G.; Barget, N.; Ayuso, C.; Ormandy, L.A.; Manns, M.P.; Beaugrand, M.; Bruix, J. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer 2010, 10, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Zhu, Y.; Xia, J.; Sawakami, T.; Kokudo, N.; Zhang, N. Status of and prospects for cancer vaccines against hepatocellular carcinoma in clinical trials. Biosci. Trends 2016, 10, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Ang, C.; Klempner, S.J.; Ali, S.M.; Madison, R.; Ross, J.S.; Severson, E.A.; Fabrizio, D.; Goodman, A.; Kurzrock, R.; Suh, J.; et al. Prevalence of established and emerging biomarkers of immune checkpoint inhibitor response in advanced hepatocellular carcinoma. Oncotarget 2019, 10, 4018–4025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loffler, M.W.; Mohr, C.; Bichmann, L.; Freudenmann, L.K.; Walzer, M.; Schroeder, C.M.; Trautwein, N.; Hilke, F.J.; Zinser, R.S.; Muhlenbruch, L.; et al. Multi-omics discovery of exome-derived neoantigens in hepatocellular carcinoma. Genome Med. 2019, 11, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Goetze, T.O. Gallbladder carcinoma: Prognostic factors and therapeutic options. World J. Gastroenterol. 2015, 21, 12211–12217. [Google Scholar] [CrossRef]
- Goetze, T.O.; Paolucci, V. Adequate extent in radical re-resection of incidental gallbladder carcinoma: Analysis of the German Registry. Surg. Endosc. 2010, 24, 2156–2164. [Google Scholar] [CrossRef]
- Ethun, C.G.; Le, N.; Lopez-Aguiar, A.G.; Pawlik, T.M.; Poultsides, G.; Tran, T.; Idrees, K.; Isom, C.A.; Fields, R.C.; Krasnick, B.A.; et al. Pathologic and Prognostic Implications of Incidental versus Nonincidental Gallbladder Cancer: A 10-Institution Study from the United States Extrahepatic Biliary Malignancy Consortium. Am. Surg. 2017, 83, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Gamboa, A.C.; Maithel, S.K. The Landmark Series: Gallbladder Cancer. Ann. Surg. Oncol. 2020, 27, 2846–2858. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.G.; Penn, I.; James, L. Liver transplantation for cholangiocarcinoma: Results in 207 patients. Transplantation 2000, 69, 1633–1637. [Google Scholar] [CrossRef]
- Darwish Murad, S.; Kim, W.R.; Harnois, D.M.; Douglas, D.D.; Burton, J.; Kulik, L.M.; Botha, J.F.; Mezrich, J.D.; Chapman, W.C.; Schwartz, J.J.; et al. Efficacy of neoadjuvant chemoradiation, followed by liver transplantation, for perihilar cholangiocarcinoma at 12 US centers. Gastroenterology 2012, 143, 88–98.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rea, D.J.; Heimbach, J.K.; Rosen, C.B.; Haddock, M.G.; Alberts, S.R.; Kremers, W.K.; Gores, G.J.; Nagorney, D.M. Liver transplantation with neoadjuvant chemoradiation is more effective than resection for hilar cholangiocarcinoma. Ann. Surg. 2005, 242, 451–458; discussion 458–461. [Google Scholar] [CrossRef]
- Ethun, C.G.; Lopez-Aguiar, A.G.; Anderson, D.J.; Adams, A.B.; Fields, R.C.; Doyle, M.B.; Chapman, W.C.; Krasnick, B.A.; Weber, S.M.; Mezrich, J.D.; et al. Transplantation versus Resection for Hilar Cholangiocarcinoma: An Argument for Shifting Treatment Paradigms for Resectable Disease. Ann. Surg. 2018, 267, 797–805. [Google Scholar] [CrossRef]
- Sapisochin, G.; Facciuto, M.; Rubbia-Brandt, L.; Marti, J.; Mehta, N.; Yao, F.Y.; Vibert, E.; Cherqui, D.; Grant, D.R.; Hernandez-Alejandro, R.; et al. Liver transplantation for "very early" intrahepatic cholangiocarcinoma: International retrospective study supporting a prospective assessment. Hepatology 2016, 64, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Mavros, M.N.; Economopoulos, K.P.; Alexiou, V.G.; Pawlik, T.M. Treatment and Prognosis for Patients with Intrahepatic Cholangiocarcinoma: Systematic Review and Meta-analysis. JAMA Surg. 2014, 149, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Squires, M.H.; Cloyd, J.M.; Dillhoff, M.; Schmidt, C.; Pawlik, T.M. Challenges of surgical management of intrahepatic cholangiocarcinoma. Expert. Rev. Gastroenterol. Hepatol. 2018, 12, 671–681. [Google Scholar] [CrossRef]
- Tsilimigras, D.I.; Sahara, K.; Paredes, A.Z.; Moro, A.; Mehta, R.; Moris, D.; Guglielmi, A.; Aldrighetti, L.; Weiss, M.; Bauer, T.W.; et al. Predicting Lymph Node Metastasis in Intrahepatic Cholangiocarcinoma. J. Gastrointest. Surg. 2021, 25, 1156–1163. [Google Scholar] [CrossRef]
- Rahnemai-Azar, A.A.; Pandey, P.; Kamel, I.; Pawlik, T.M. Monitoring outcomes in intrahepatic cholangiocarcinoma patients following hepatic resection. Hepat. Oncol. 2016, 3, 223–239. [Google Scholar] [CrossRef]
- Tsilimigras, D.I.; Sahara, K.; Wu, L.; Moris, D.; Bagante, F.; Guglielmi, A.; Aldrighetti, L.; Weiss, M.; Bauer, T.W.; Alexandrescu, S.; et al. Very Early Recurrence after Liver Resection for Intrahepatic Cholangiocarcinoma: Considering Alternative Treatment Approaches. JAMA Surg. 2020, 155, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.S.; Zhang, X.F.; Weiss, M.; Popescu, I.; Marques, H.P.; Aldrighetti, L.; Maithel, S.K.; Pulitano, C.; Bauer, T.W.; Shen, F.; et al. Recurrence Patterns and Timing Courses Following Curative-Intent Resection for Intrahepatic Cholangiocarcinoma. Ann. Surg. Oncol. 2019, 26, 2549–2557. [Google Scholar] [CrossRef]
- Zhang, X.F.; Beal, E.W.; Bagante, F.; Chakedis, J.; Weiss, M.; Popescu, I.; Marques, H.P.; Aldrighetti, L.; Maithel, S.K.; Pulitano, C.; et al. Early versus late recurrence of intrahepatic cholangiocarcinoma after resection with curative intent. Br. J. Surg. 2018, 105, 848–856. [Google Scholar] [CrossRef]
- Groot Koerkamp, B.; Fong, Y. Outcomes in biliary malignancy. J. Surg. Oncol. 2014, 110, 585–591. [Google Scholar] [CrossRef]
- Strijker, M.; Belkouz, A.; van der Geest, L.G.; van Gulik, T.M.; van Hooft, J.E.; de Meijer, V.E.; Haj Mohammad, N.; de Reuver, P.R.; Verheij, J.; de Vos-Geelen, J.; et al. Treatment and survival of resected and unresected distal cholangiocarcinoma: A nationwide study. Acta Oncol. 2019, 58, 1048–1055. [Google Scholar] [CrossRef]
- Groot Koerkamp, B.; Wiggers, J.K.; Allen, P.J.; Besselink, M.G.; Blumgart, L.H.; Busch, O.R.; Coelen, R.J.; D’Angelica, M.I.; DeMatteo, R.P.; Gouma, D.J.; et al. Recurrence Rate and Pattern of Perihilar Cholangiocarcinoma after Curative Intent Resection. J. Am. Coll. Surg. 2015, 221, 1041–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beal, E.W.; Cloyd, J.M.; Pawlik, T.M. Surgical Treatment of Intrahepatic Cholangiocarcinoma: Current and Emerging Principles. J. Clin. Med. 2020, 10, 104. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fostea, R.M.; Fontana, E.; Torga, G.; Arkenau, H.T. Recent Progress in the Systemic Treatment of Advanced/Metastatic Cholangiocarcinoma. Cancers 2020, 12, 2599. [Google Scholar] [CrossRef] [PubMed]
- Rimassa, L.; Personeni, N.; Aghemo, A.; Lleo, A. The immune milieu of cholangiocarcinoma: From molecular pathogenesis to precision medicine. J. Autoimmun. 2019, 100, 17–26. [Google Scholar] [CrossRef]
- Zatonski, W.A.; Lowenfels, A.B.; Boyle, P.; Maisonneuve, P.; Bueno de Mesquita, H.B.; Ghadirian, P.; Jain, M.; Przewozniak, K.; Baghurst, P.; Moerman, C.J.; et al. Epidemiologic aspects of gallbladder cancer: A case-control study of the SEARCH Program of the International Agency for Research on Cancer. J. Natl. Cancer Inst. 1997, 89, 1132–1138. [Google Scholar] [CrossRef] [Green Version]
- Stephen, A.E.; Berger, D.L. Carcinoma in the porcelain gallbladder: A relationship revisited. Surgery 2001, 129, 699–703. [Google Scholar] [CrossRef]
- Lewis, J.T.; Talwalkar, J.A.; Rosen, C.B.; Smyrk, T.C.; Abraham, S.C. Prevalence and risk factors for gallbladder neoplasia in patients with primary sclerosing cholangitis: Evidence for a metaplasia-dysplasia-carcinoma sequence. Am. J. Surg. Pathol. 2007, 31, 907–913. [Google Scholar] [CrossRef]
- Tyson, G.L.; El-Serag, H.B. Risk factors for cholangiocarcinoma. Hepatology 2011, 54, 173–184. [Google Scholar] [CrossRef]
- Isomoto, H.; Mott, J.L.; Kobayashi, S.; Werneburg, N.W.; Bronk, S.F.; Haan, S.; Gores, G.J. Sustained IL-6/STAT-3 signaling in cholangiocarcinoma cells due to SOCS-3 epigenetic silencing. Gastroenterology 2007, 132, 384–396. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Tadlock, L.; Gores, G.J.; Patel, T. Inhibition of interleukin 6-mediated mitogen-activated protein kinase activation attenuates growth of a cholangiocarcinoma cell line. Hepatology 1999, 30, 1128–1133. [Google Scholar] [CrossRef]
- Subimerb, C.; Pinlaor, S.; Khuntikeo, N.; Leelayuwat, C.; Morris, A.; McGrath, M.S.; Wongkham, S. Tissue invasive macrophage density is correlated with prognosis in cholangiocarcinoma. Mol. Med. Rep. 2010, 3, 597–605. [Google Scholar] [CrossRef] [Green Version]
- Pang, L.; Han, S.; Jiao, Y.; Jiang, S.; He, X.; Li, P. Bu Fei Decoction attenuates the tumor associated macrophage stimulated proliferation, migration, invasion and immunosuppression of non-small cell lung cancer, partially via IL-10 and PD-L1 regulation. Int. J. Oncol. 2017, 51, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.K.; Kumari, N.; Krishnani, N. Molecular pathogenesis of gallbladder cancer: An update. Mutat. Res. 2019, 816–818, 111674. [Google Scholar] [CrossRef]
- Mody, K.; Starr, J.; Saul, M.; Poorman, K.; Weinberg, B.A.; Salem, M.E.; VanderWalde, A.; Shields, A.F. Patterns and genomic correlates of PD-L1 expression in patients with biliary tract cancers. J. Gastrointest. Oncol. 2019, 10, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Peng, C.; Lu, X.; Ding, X.; Zhang, S.; Zou, X.; Zhang, X. Downregulation of FOXP3 inhibits invasion and immune escape in cholangiocarcinoma. Biochem. Biophys. Res. Commun. 2015, 458, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Ghidini, M.; Cascione, L.; Carotenuto, P.; Lampis, A.; Trevisani, F.; Previdi, M.C.; Hahne, J.C.; Said-Huntingford, I.; Raj, M.; Zerbi, A.; et al. Characterisation of the immune-related transcriptome in resected biliary tract cancers. Eur. J. Cancer 2017, 86, 158–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Liu, F.; Zhang, F.; Zhou, W.; Jiang, X.; Yang, Y.; Qu, K.; Wang, Y.; Ma, Q.; Wang, T.; et al. Genomic ERBB2/ERBB3 mutations promote PD-L1-mediated immune escape in gallbladder cancer: A whole-exome sequencing analysis. Gut 2019, 68, 1024–1033. [Google Scholar] [CrossRef]
- Piha-Paul, S.A.; Oh, D.Y.; Ueno, M.; Malka, D.; Chung, H.C.; Nagrial, A.; Kelley, R.K.; Ros, W.; Italiano, A.; Nakagawa, K.; et al. Efficacy and safety of pembrolizumab for the treatment of advanced biliary cancer: Results from the KEYNOTE-158 and KEYNOTE-028 studies. Int. J. Cancer 2020, 147, 2190–2198. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.D.; Chung, V.; Alese, O.B.; El-Rayes, B.F.; Li, D.; Al-Toubah, T.E.; Schell, M.J.; Zhou, J.M.; Mahipal, A.; Kim, B.H.; et al. A Phase 2 Multi-institutional Study of Nivolumab for Patients with Advanced Refractory Biliary Tract Cancer. JAMA Oncol. 2020, 6, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Winkelmann, R.; Schneider, M.; Hartmann, S.; Schnitzbauer, A.A.; Zeuzem, S.; Peveling-Oberhag, J.; Hansmann, M.L.; Walter, D. Microsatellite Instability Occurs Rarely in Patients with Cholangiocarcinoma: A Retrospective Study from a German Tertiary Care Hospital. Int. J. Mol. Sci. 2018, 19, 1421. [Google Scholar] [CrossRef] [Green Version]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Lim, Y.J.; Koh, J.; Kim, K.; Chie, E.K.; Kim, S.; Lee, K.B.; Jang, J.Y.; Kim, S.W.; Oh, D.Y.; Bang, Y.J. Clinical Implications of Cytotoxic T Lymphocyte Antigen-4 Expression on Tumor Cells and Tumor-Infiltrating Lymphocytes in Extrahepatic Bile Duct Cancer Patients Undergoing Surgery Plus Adjuvant Chemoradiotherapy. Target Oncol. 2017, 12, 211–218. [Google Scholar] [CrossRef]
- Zhou, G.; Sprengers, D.; Mancham, S.; Erkens, R.; Boor, P.P.C.; van Beek, A.A.; Doukas, M.; Noordam, L.; Campos Carrascosa, L.; de Ruiter, V.; et al. Reduction of immunosuppressive tumor microenvironment in cholangiocarcinoma by ex vivo targeting immune checkpoint molecules. J. Hepatol. 2019, 71, 753–762. [Google Scholar] [CrossRef]
- Ioka, T.; Ueno, M.; Oh, D.-Y.; Fujiwara, Y.; Chen, J.-S.; Doki, Y.; Mizuno, N.; Park, K.; Asagi, A.; Hayama, M.; et al. Evaluation of safety and tolerability of durvalumab (D) with or without tremelimumab (T) in patients (pts) with biliary tract cancer (BTC). J. Clin. Oncol. 2019, 37, 387. [Google Scholar] [CrossRef]
- Oh, D.-Y.; Lee, K.-H.; Lee, D.-W.; Kim, T.Y.; Bang, J.-H.; Nam, A.-R.; Lee, Y.; Zhang, Q.; Rebelatto, M.; Li, W.; et al. Phase II study assessing tolerability, efficacy, and biomarkers for durvalumab (D) ± tremelimumab (T) and gemcitabine/cisplatin (GemCis) in chemo-naïve advanced biliary tract cancer (aBTC). J. Clin. Oncol. 2020, 38, 4520. [Google Scholar] [CrossRef]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef]
- Higuchi, R.; Yamamoto, M.; Hatori, T.; Shimizu, K.; Imai, K.; Takasaki, K. Intrahepatic cholangiocarcinoma with lymph node metastasis successfully treated by immunotherapy with CD3-activated T cells and dendritic cells after surgery: Report of a case. Surg. Today 2006, 36, 559–562. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Sangsuwannukul, T.; Supimon, K.; Sujjitjoon, J.; Phanthaphol, N.; Chieochansin, T.; Poungvarin, N.; Wongkham, S.; Junking, M.; Yenchitsomanus, P.T. Anti-tumour effect of the fourth-generation chimeric antigen receptor T cells targeting CD133 against cholangiocarcinoma cells. Int. Immunopharmacol. 2020, 89, 107069. [Google Scholar] [CrossRef]
- Guo, Y.; Feng, K.; Liu, Y.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Jia, H.; Han, W. Phase I Study of Chimeric Antigen Receptor-Modified T Cells in Patients with EGFR-Positive Advanced Biliary Tract Cancers. Clin. Cancer Res. 2018, 24, 1277–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaida, M.; Morita-Hoshi, Y.; Soeda, A.; Wakeda, T.; Yamaki, Y.; Kojima, Y.; Ueno, H.; Kondo, S.; Morizane, C.; Ikeda, M.; et al. Phase 1 trial of Wilms tumor 1 (WT1) peptide vaccine and gemcitabine combination therapy in patients with advanced pancreatic or biliary tract cancer. J. Immunother. 2011, 34, 92–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Ueno, T.; Kawaoka, T.; Hazama, S.; Fukui, M.; Suehiro, Y.; Hamanaka, Y.; Ikematsu, Y.; Imai, K.; Oka, M.; et al. MUC1 peptide vaccination in patients with advanced pancreas or biliary tract cancer. Anticancer Res. 2005, 25, 3575–3579. [Google Scholar]
- Aruga, A.; Takeshita, N.; Kotera, Y.; Okuyama, R.; Matsushita, N.; Ohta, T.; Takeda, K.; Yamamoto, M. Phase I clinical trial of multiple-peptide vaccination for patients with advanced biliary tract cancer. J. Transl. Med. 2014, 12, 61. [Google Scholar] [CrossRef] [Green Version]
- Aruga, A.; Takeshita, N.; Kotera, Y.; Okuyama, R.; Matsushita, N.; Ohta, T.; Takeda, K.; Yamamoto, M. Long-term Vaccination with Multiple Peptides Derived from Cancer-Testis Antigens Can Maintain a Specific T-cell Response and Achieve Disease Stability in Advanced Biliary Tract Cancer. Clin. Cancer Res. 2013, 19, 2224–2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, K.; Kotera, Y.; Aruga, A.; Takeshita, N.; Takasaki, K.; Yamamoto, M. Clinical utilization of postoperative dendritic cell vaccine plus activated T-cell transfer in patients with intrahepatic cholangiocarcinoma. J. Hepatobiliary Pancreat. Sci. 2012, 19, 171–178. [Google Scholar] [CrossRef]
- Fukuda, K.; Abei, M.; Ugai, H.; Seo, E.; Wakayama, M.; Murata, T.; Todoroki, T.; Tanaka, N.; Hamada, H.; Yokoyama, K.K. E1A, E1B double-restricted adenovirus for oncolytic gene therapy of gallbladder cancer. Cancer Res. 2003, 63, 4434–4440. [Google Scholar]
- Weng, M.; Gong, W.; Ma, M.; Chu, B.; Qin, Y.; Zhang, M.; Lun, X.; McFadden, G.; Forsyth, P.; Yang, Y.; et al. Targeting gallbladder cancer: Oncolytic virotherapy with myxoma virus is enhanced by rapamycin in vitro and further improved by hyaluronan in vivo. Mol. Cancer 2014, 13, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, K.; Todo, T.; Chijiiwa, K.; Tanaka, M. Therapeutic efficacy of G207, a conditionally replicating herpes simplex virus type 1 mutant, for gallbladder carcinoma in immunocompetent hamsters. Mol. Ther. 2001, 3, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Pugalenthi, A.; Mojica, K.; Ady, J.W.; Johnsen, C.; Love, D.; Chen, N.G.; Aguilar, R.J.; Szalay, A.A.; Fong, Y. Recombinant vaccinia virus GLV-1h68 is a promising oncolytic vector in the treatment of cholangiocarcinoma. Cancer Gene Ther. 2015, 22, 591–596. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Trial Identifier | Phase | Status | Immunotherapy | Co-Treatments | Participants |
|---|---|---|---|---|---|
| NCT04740307 | II | Recruiting | Combination immune checkpoint inhibitors (CTLA-4 with PD-1) | Lenvatinib | 110 |
| NCT02821754 | II | Recruiting | Combination immune checkpoint inhibitors (CTLA-4 with PD-1) | Transarterial catheter chemoembolization, radiofrequency ablation, cryoablation | 90 |
| NCT04785287 | I/II | Recruiting | Combination immune checkpoint inhibitors (CTLA-4 with PD-1) | Stereotactic Body Radiation Therapy | 80 |
| NCT04430452 | II | Not yet recruiting | Combination immune checkpoint inhibitors (CTLA-4 with PD-1) | Hypofractionated Radiation Therapy | 30 |
| NCT03638141 | II | Recruiting | Combination immune checkpoint inhibitors (CTLA-4 with PD-1) | Drug-eluting bead transarterial chemoembolization | 30 |
| NCT03222076 | II | Active not recruiting | Combination immune checkpoint inhibitors (CTLA-4 with PD-1) | N/A | 30 |
| NCT03652077 | I | Active, not recruiting | Immune checkpoint inhibitor (TIM-3) | N/A | 40 |
| NCT03680508 | II | Recruiting | Combination immune checkpoint inhibitors (TIM-3 with PD-1) | N/A | 42 |
| NCT04658147 | I | Recruiting | Combination immune checkpoint inhibitors (LAG-3 with PD-1) | N/A | 20 |
| NCT03695250 | I/II | Active, not recruiting | Immune checkpoint inhibitor (PD-1) with IDO1 inhibitor | N/A | 8 |
| NCT04728321 | II | Recruiting | Bispecific antibody against CTLA-4 and PD-1 | lenvatinib | 75 |
| NCT04444167 | I/II | Recruiting | Bispecific antibody against CTLA-4 and PD-1 | Lenvatinib | 30 |
| NCT04601610 | I/II | Not yet recruiting | Bispecific antibody against CTLA-4 and PD-L1 | Ningetinib Tosylate (tyrosine kinase inhibitor) | 70 |
| NCT03849469 | I | Recruiting | Bispecific antibody against CTLA-4 and LAG-3, combination bispecific antibody and immune checkpoint inhibitor (PD-1) | N/A | 242 |
| NCT03980288 | I | Recruiting | GPC3 CAR-T cell | Fludarabine, cyclophosphamide | 36 |
| NCT04121273 | I | Recruiting | GPC3 CAR-T cell | N/A | 20 |
| NCT03884751 | I | Recruiting | GPC3 CAR-T cells | N/A | 15 |
| NCT02905188 | I | Recruiting | GPC3 CAR-T cells | Cytoxan, Fludarabine | 14 |
| NCT04951141 | I | Recruiting | GPC3 CAR-T cells | N/A | 10 |
| NCT03302403 | N/A | Active, not recruiting | GPC3 CAR-T cell | Fludarabine, Cyclophosphamide | 18 |
| NCT04506983 | 1 | Not yet recruiting | GPC3-CAR-T cells | N/A | 12 |
| NCT03198546 | I | Recruiting | GPC3 and/or TGFβ targeting CAR-T cells | N/A | 30 |
| NCT03638206 | I/II | Recruiting | DR5 CAR-T cells | N/A | 73 |
| NCT03941626 | I/II | Recruiting | DR5 CAR-T/TCR-T cells | N/A | 50 |
| NCT03993743 | I | Recruiting | CD147 CAR-T cells | N/A | 34 |
| NCT04550663 | I | Not yet recruiting | NKG2dLs CAR-T cells | N/A | 10 |
| NCT03441100 | 1 | Recruiting | MAGEA1 TCR engineered T cells | Cyclophosphamide, fludarabine, Interleukin-2 | 15 |
| NCT04502082 | I/II | Recruiting | Alpha fetoprotein peptide/HLA-A2 complex TCR engineered T cells | N/A | 50 |
| NCT04634357 | I/II | Not yet recruiting | Alpha fetoprotein peptide/HLA-A2 complex TCR engineered T cells | N/A | 25 |
| NCT04518774 | I | Recruiting | expanded allogeneic gamma-delta T cells | N/A | 8 |
| NCT03836352 | II | Recruiting | Induction of survivin-specific cytotoxic T lymphocytes with immune checkpoint inhibitors (PD-1) | Cyclophosphamide | 184 |
| NCT04417764 | I | Recruiting | PD-1 knockout engineered T cells | Transarterial catheter chemoembolization | 10 |
| NCT04317248 | II | Recruiting | Autologous dendritic cell vaccine | Cyclophosphamide | 600 |
| NCT04912765 | II | Recruiting | Neoantigen Dendritic Cell Vaccine with immune checkpoint inhibitor (PD-1) | N/A | 60 |
| NCT04147078 | I | Recruiting | Autologous dendritic cells vaccine | N/A | 80 |
| NCT03228667 | II | Active, not recruiting | PD-L1 targeting high-affinity natural killer with immune checkpoint inhibitors (PD-1 or PD-L1) | N-803 (interleukin-15 superagonist) | 145 |
| NCT03311334 | I/II | Recruiting | Peptide vaccine against WT1 with immune checkpoint inhibitors (PD-1) | N/A | 104 |
| NCT04246671 | I/II | Recruiting | Peptide vaccine against HER-2/neu | N/A | 45 |
| NCT02432963 | I | Active, not recruiting | Peptide vaccine against P53 with immune checkpoint inhibitors (PD-1) | N/A | 19 |
| NCT04251117 | I/II | Recruiting | Personalized neoantigen DNA vaccine with immune checkpoint inhibitor (PD-1) | Plasmid-encoded interleukin 12 | 24 |
| NCT04248569 | I | Recruiting | Peptide vaccine against DNAJB1-PRKACA fusion kinase with combination immune checkpoint inhibitors (CTLA-4 with PD-1) | N/A | 12 |
| NCT03071094 | I/II | Active, not recruiting | Oncolytic vaccine with immune checkpoint inhibitor (PD-1) | N/A | 30 |
| NCT04665362 | I | Not yet recruiting | Oncolytic vaccine with immune checkpoint inhibitor (PD-1) | Apatinib | 10 |
| NCT04665362 | I | Not yet recruiting | Oncolytic virus with immune checkpoint inhibitor (PD-1) | Apatinib | 10 |
| Trial Identifier | Phase | Status | Immunotherapy | Co-Treatments | Participants |
|---|---|---|---|---|---|
| NCT02834013 | II | Recruiting | Combination immune checkpoint inhibitors (CTLA-4 with PD-1) | N/A | 818 |
| NCT03473574 | II | Active, not recruiting | Combination immune checkpoint inhibitors (CTLA-4 with PD-1) | Gemcitabine, Cisplatin | 128 |
| NCT02821754 | II | Recruiting | Combination immune checkpoint inhibitors (CTLA-4 with PD-1) | Transarterial catheter chemoembolization, radiofrequency ablation, cryoablation | 90 |
| NCT04634058 | II | Not yet recruiting | Combination immune checkpoint inhibitors (CTLA-4 with PD-1) | N/A | 40 |
| NCT03058289 | I/II | Recruiting | Combination immune checkpoint inhibitors (CTLA-4 with PD-1) | Novel chemotherapy (INT230-6) | 180 |
| NCT03849469 | I | Recruiting | Bispecific antibody against CTLA-4 and LAG-3, combination bispecific antibody and immune checkpoint inhibitor (PD-1) | N/A | 242 |
| NCT04641871 | I | Active, not recruiting | Combination immune checkpoint inhibitors (PD-1 with LAG-3 or TIM-3) | N/A | 200 |
| NCT04672434 | I | Recruiting | Combination immune checkpoint inhibitors (PD-1 with CD73) | N/A | 100 |
| NCT03872947 | I | Recruiting | Monoclonal antibody against undisclosed tumor-associated antigen with immune checkpoint inhibitors (PD-1 or CTLA-4) | Imiquimod Cream, Irinotecan, Leucovorin, 5-FU, Gemcitabine, Cisplatin, Carboplatin, Ramucirumab, Paclitaxel | 75 |
| NCT03801083 | II | Recruiting | TIL adoptive cell transfer | N/A | 59 |
| NCT03633773 | I/II | Recruiting | MUC-1 CAR-T cell | N/A | 9 |
| NCT04951141 | I | Recruiting | GPC3 CAR-T cells | N/A | 10 |
| NCT01868490 | I/II | Recruiting | CIK adoptive cell transfer | N/A | 13 |
| NCT03942328 | I | Recruiting | Autologous dendritic cells | External Beam Radiation Therapy, Pneumococcal 13-valent Conjugate Vaccine | 26 |
| NCT04853017 | I/II | Recruiting | Peptide vaccine against KRAS mutations | N/A | 159 |
| Trial Identifier | Phase | Status | Immunotherapy | Co-Treatments | Participants |
|---|---|---|---|---|---|
| NCT02834013 | II | Recruiting | Combination immune checkpoint inhibitors (CTLA-4 with PD-1) | N/A | 818 |
| NCT03473574 | II | Active, not recruiting | Combination immune checkpoint inhibitors (CTLA-4 with PD-1) | Gemcitabine, Cisplatin | 128 |
| NCT02821754 | II | Recruiting | Combination immune checkpoint inhibitors (CTLA-4 with PD-1) | Transarterial catheter chemoembolization, radiofrequency ablation, cryoablation | 90 |
| NCT04641871 | I | Active, not recruiting | Combination immune checkpoint inhibitors (PD-1 with LAG-3 or TIM-3) | N/A | 200 |
| NCT04672434 | I | Recruiting | Combination immune checkpoint inhibitors (PD-1 with CD73) | N/A | 100 |
| NCT03801083 | II | Recruiting | TIL adoptive cell transfer | N/A | 59 |
| NCT04426669 | I/II | Recruiting | CRISPR gene editing in TIL adoptive cell transfer | Aldesleukin, Cyclophosphamide, Fludarabine | 20 |
| NCT04853017 | I/II | Recruiting | Peptide vaccine against KRAS mutations | N/A | 159 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilyas, F.Z.; Beane, J.D.; Pawlik, T.M. The State of Immunotherapy in Hepatobiliary Cancers. Cells 2021, 10, 2096. https://doi.org/10.3390/cells10082096
Ilyas FZ, Beane JD, Pawlik TM. The State of Immunotherapy in Hepatobiliary Cancers. Cells. 2021; 10(8):2096. https://doi.org/10.3390/cells10082096
Chicago/Turabian StyleIlyas, Farhan Z., Joal D. Beane, and Timothy M. Pawlik. 2021. "The State of Immunotherapy in Hepatobiliary Cancers" Cells 10, no. 8: 2096. https://doi.org/10.3390/cells10082096