
Interference of LPS H. pylori with IL-33-Driven Regeneration of Caviae porcellus Primary Gastric Epithelial Cells and Fibroblasts

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
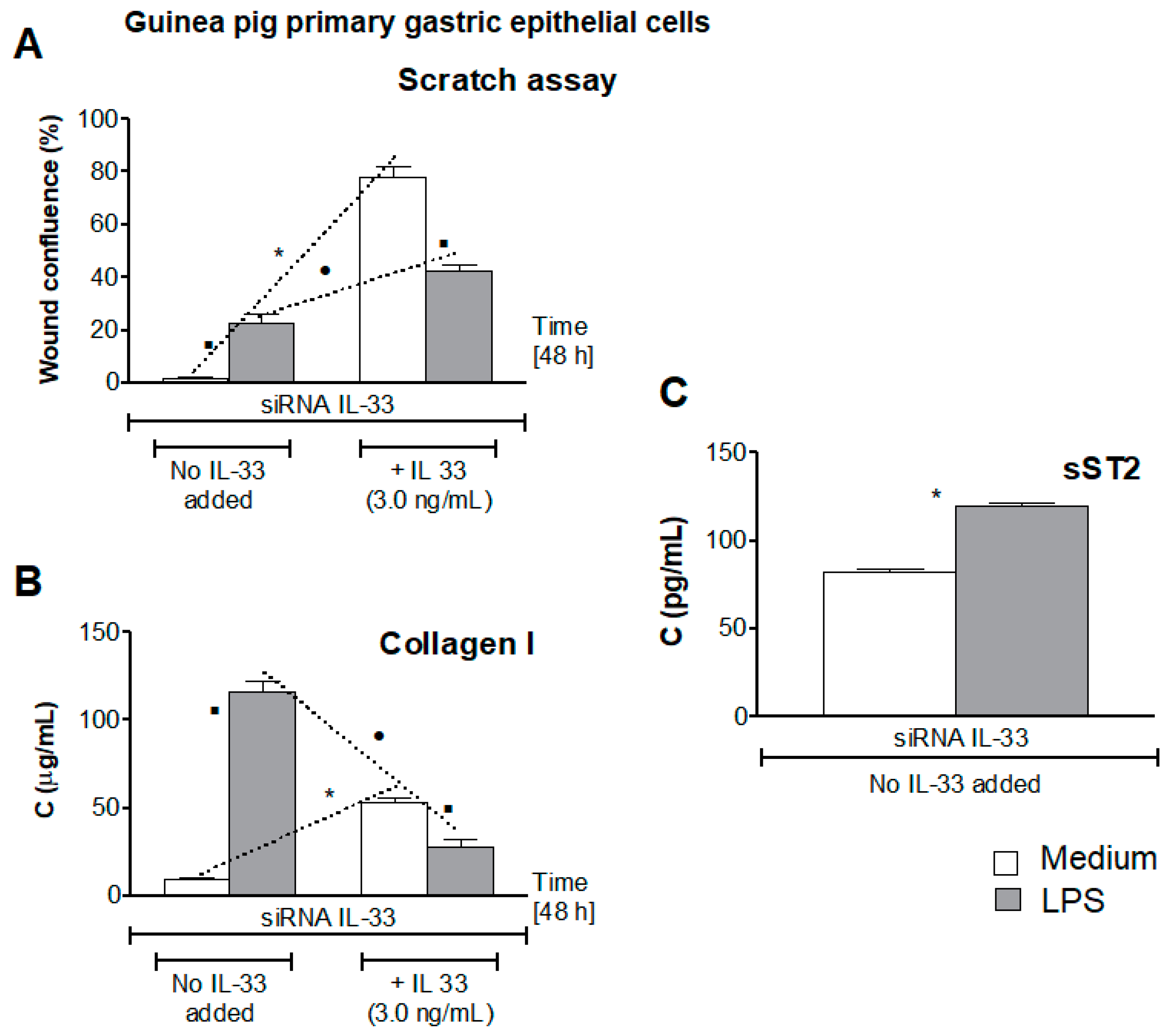{kind=link}
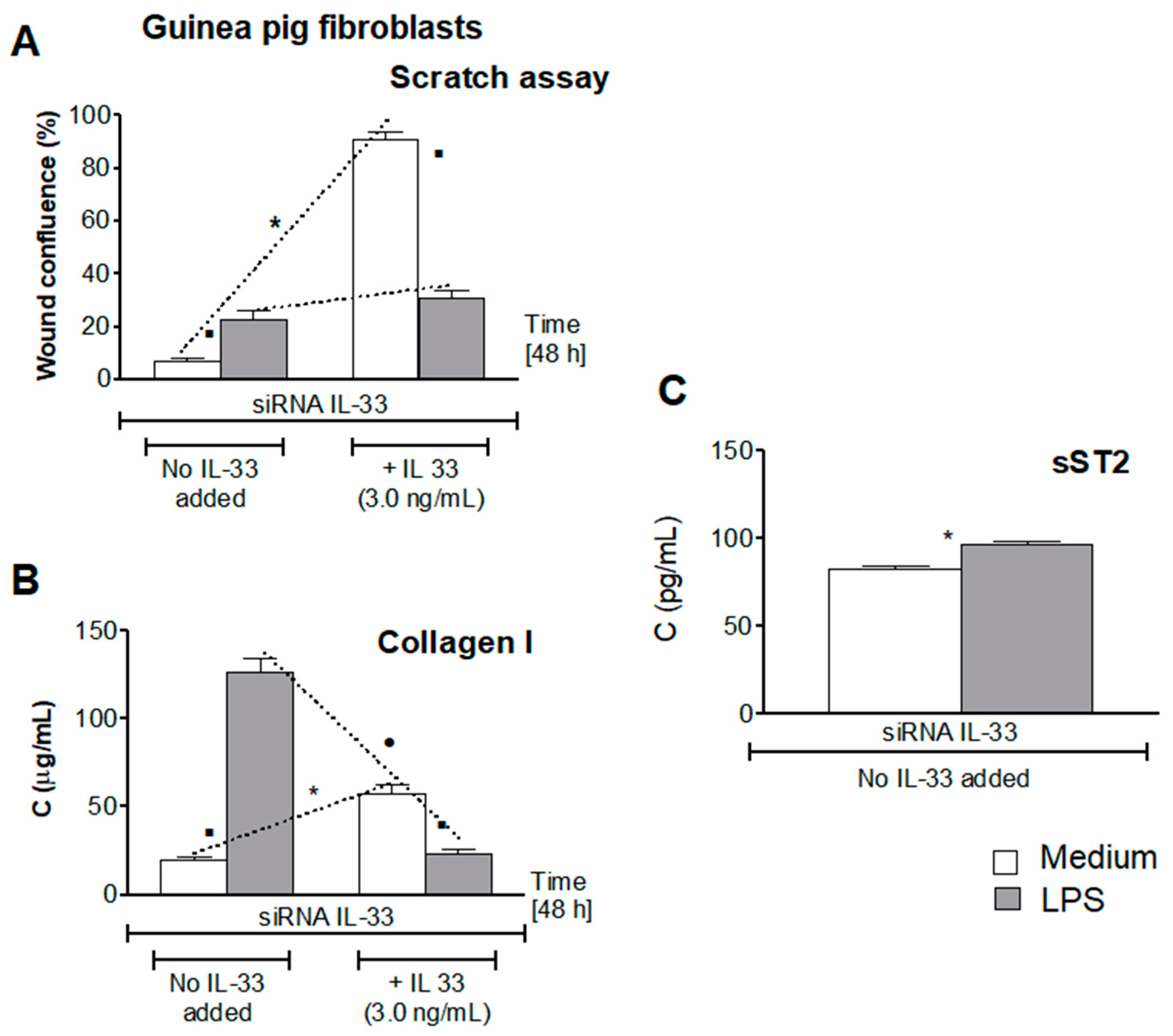{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Bacterial Stimuli
2.3. IL-33 siRNA Silencing
2.4. Lipid Oxidation—4HNE Production
2.5. Erk Activation/Phosphorylation (pErk)
2.6. Cell Apoptosis
2.7. Cell Migration—Wound Healing Assay
2.8. Collagen I Assay
2.9. ELISA for ST2
2.10. Statistical Analysis
3. Results and Discussion
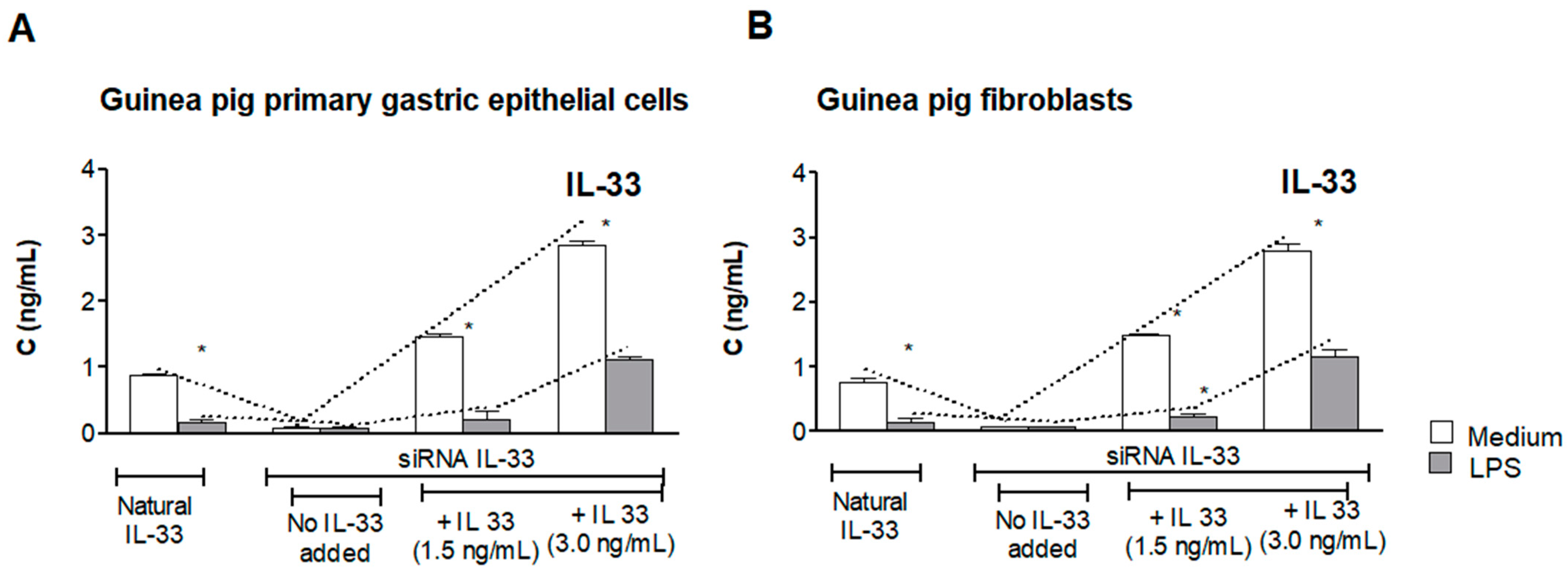
3.1. Assessment of Natural IL-33 or Exogenous IL-33 in Cell Cultures Carried Out in the Presence or Absence of LPS of H. pylori
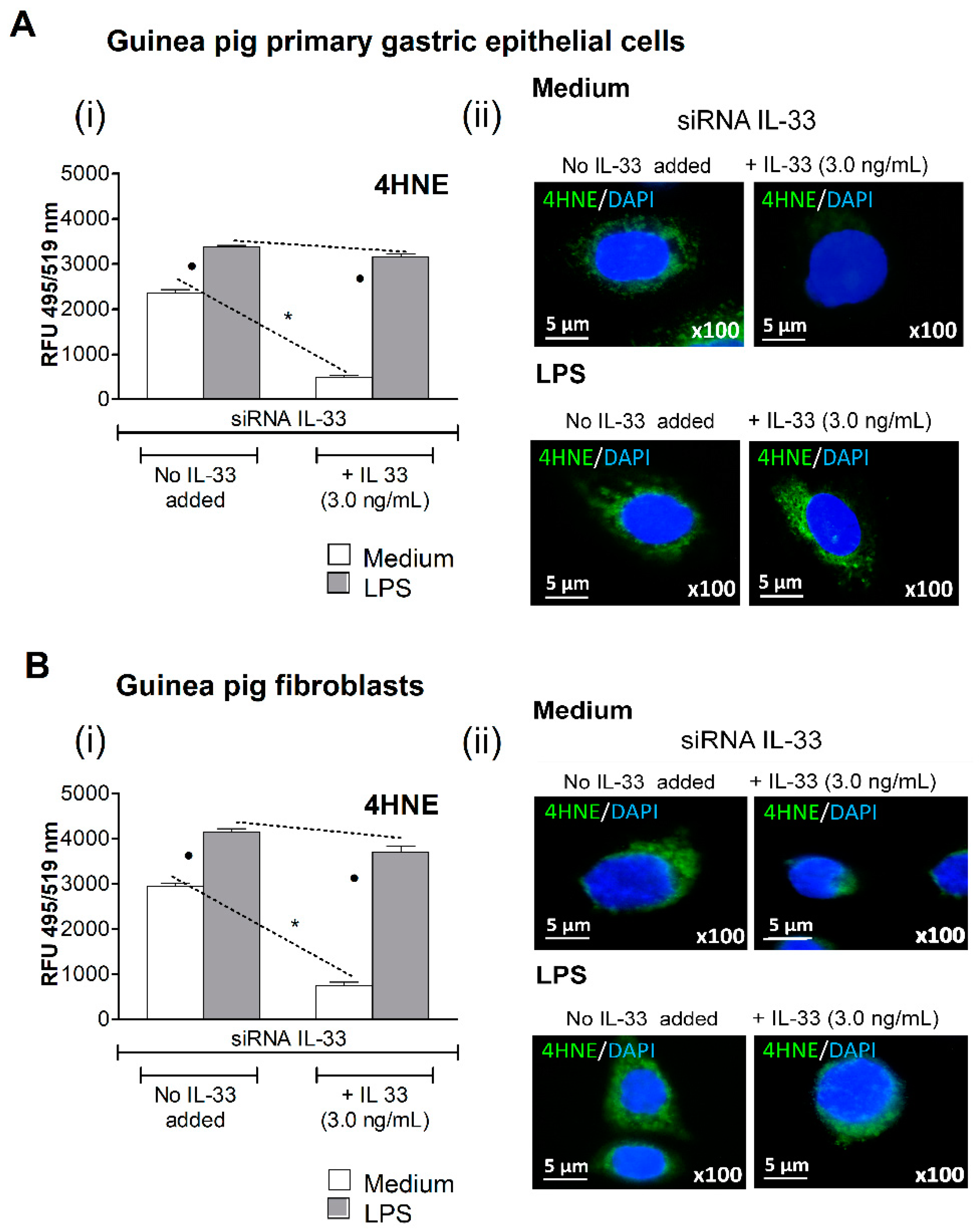
3.2. Oxidative Stress Induced in Cell Cultures Exposed to H. pylori LPS
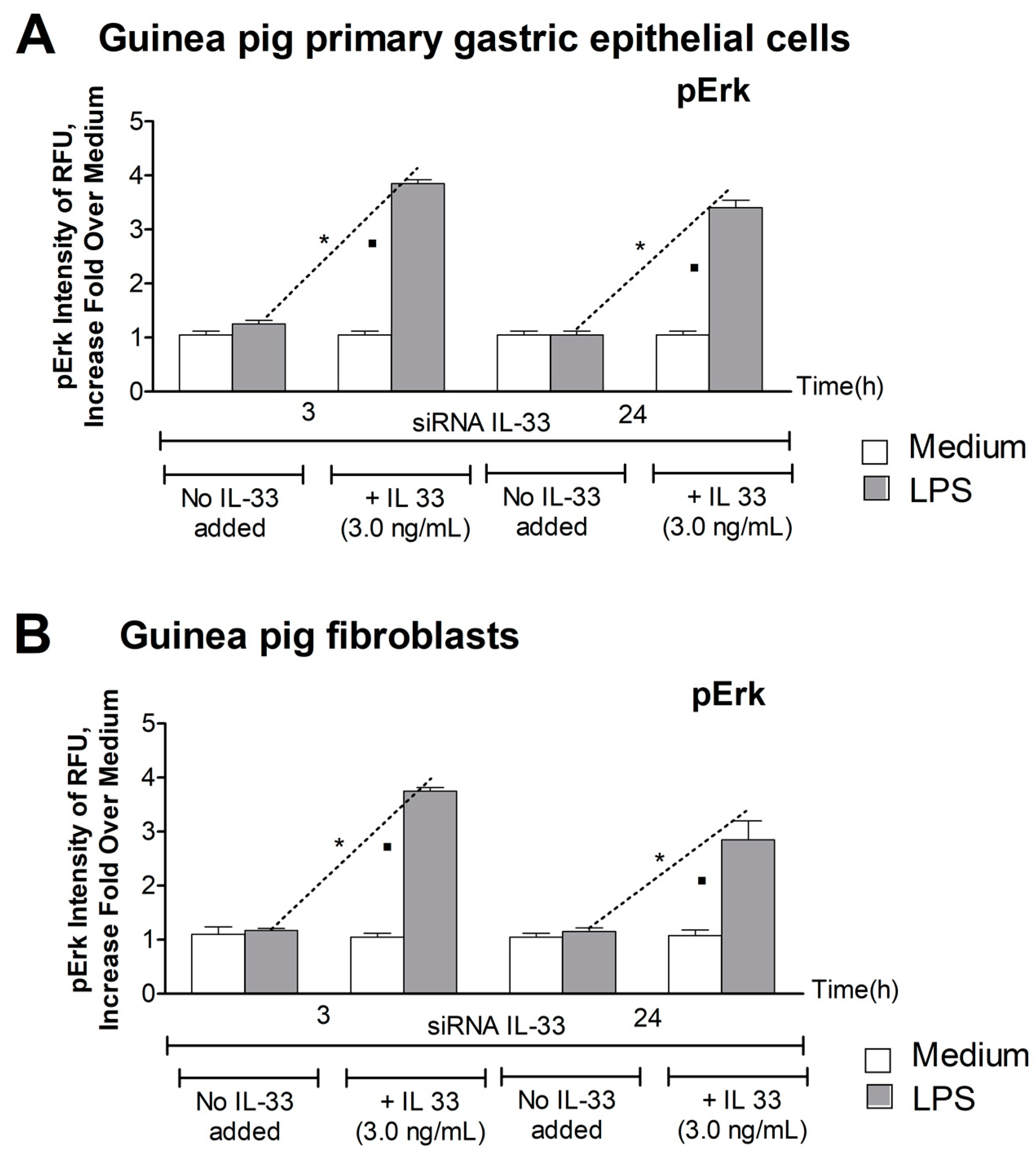
3.3. Level of pErk
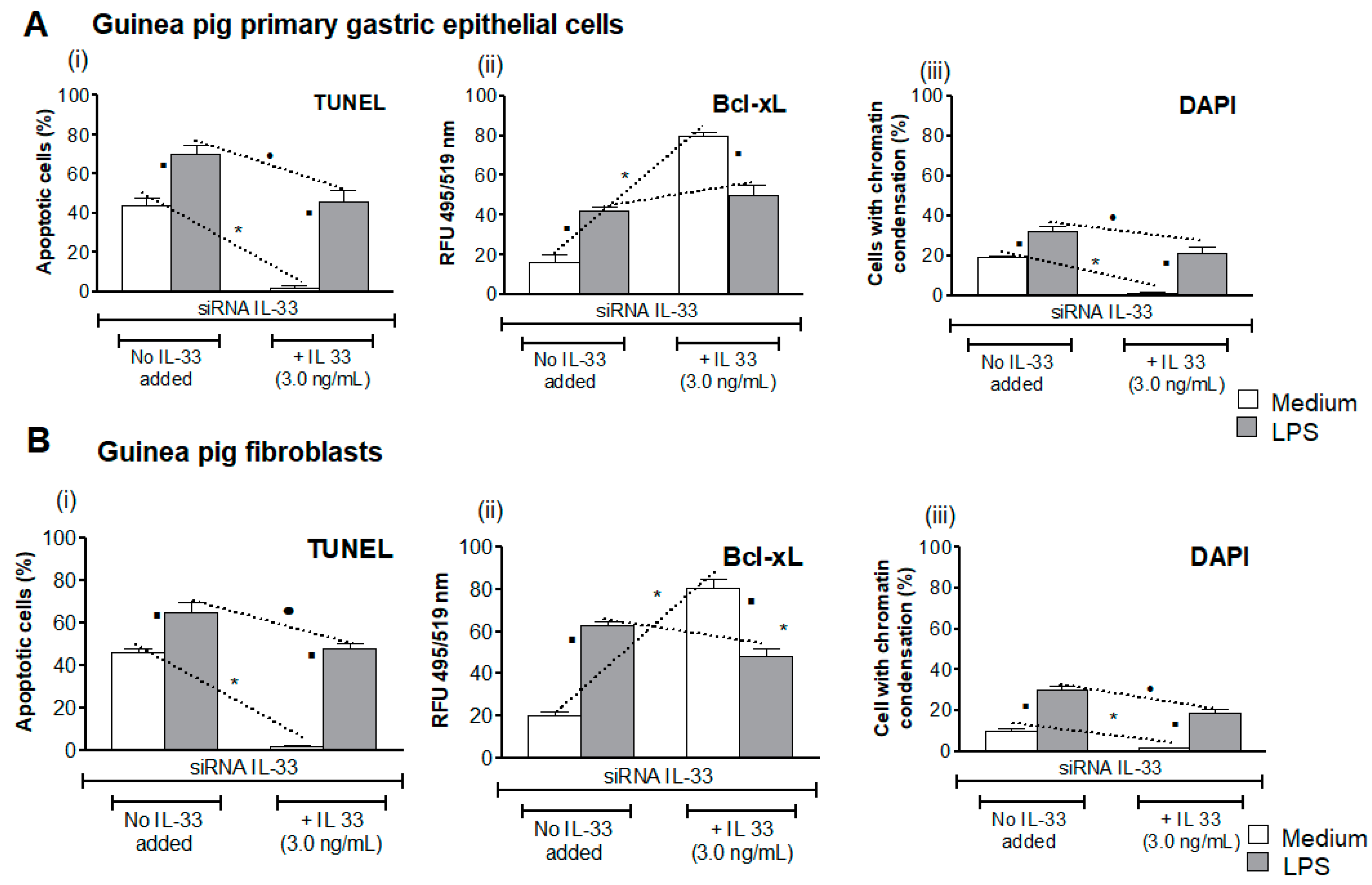
3.4. Influence of LPS H. pylori on IL-33-Driven Downregulation of Cell Apoptosis
3.5. Influence of LPS H. pylori on IL-33-Driven Cell Migration
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Warren, J.R.; Marshall, B. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1983, 1, 1273–1275. [Google Scholar]
- Posselt, G.; Backert, S.; Wessler, S. The functional interplay of H. pylori factors with gastric epithelial cells induces a multi-step process in pathogenesis. Cell Commun. Signal. 2013, 11, 77. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Murata-Kamiya, N.; Yanagiya, K.; Suda, W.; Hattori, M.; Kanda, H.; Bingo, A.; Fujii, Y.; Maeda, S.; Koike, K.; et al. Mutual reinforcement of inflammation and carcinogenesis by the H. pylori CagA oncoprotein. Sci. Rep. 2015, 5, 10024. [Google Scholar] [CrossRef] [Green Version]
- Chmiela, M.; Karwowska, Z.; Gonciarz, W.; Allushi, B.; Stączek, P. Host pathogen interactions in Helicobacter pylori related gastric cancer. World J. Gastroenterol. 2017, 23, 1521–1540. [Google Scholar] [CrossRef]
- Fiorentino, M.; Ding, H.; Blanchard, T.G.; Czinn, S.; Sztein, M.B.; Fasano, A. Helicobacter pylori-induced disruption of monolayer permeability and proinflammatory cytokine secretion in polarized human gastric epithelial cells. Infect. Immun. 2013, 81, 876–883. [Google Scholar] [CrossRef] [Green Version]
- Mnich, E.; Kowalewicz-Kulbat, M.; Sicińska, P.; Hinc, K.; Obuchowski, M.; Gajewski, A.; Moran, A.P.; Chmiela, M. Impact of Helicobacter pylori on the healing process of the gastric barrier. World J. Gastroenterol. 2016, 22, 7536–7558. [Google Scholar] [CrossRef]
- Viala, J.; Chaput, C.; Boneca, I.G.; Cardona, A.; Girardin, S.E.; Moran, A.P.; Athman, R.; Mémet, S.; Huerre, M.R.; Coyle, A.J.; et al. Nod 1 responds to peptidoglycan delivered by the H. pylori cag pathogenicity island. Nat. Immunol. 2004, 5, 1166–1174. [Google Scholar] [CrossRef]
- Backert, S.; Kwok, T.; Schmid, M.; Selbach, M.; Moese, S.; Peek, R.M., Jr.; König, W.; Meyer, T.F.; Jungblut, P.R. Subproteomes of soluble and structure-bound H. pylori proteins analysed by two-dimensional gel electrophoresis and mass spectrometry. Proteomics 2005, 5, 1311–3345. [Google Scholar] [CrossRef]
- Cover, T.L.; Blanke, S.R.H. H. pylori VacA, a paradigm for toxin multifunctionality. Nat. Rev. Microbiol. 2005, 3, 320–332. [Google Scholar] [CrossRef]
- Hoy, B.; Löwer, M.; Weydig, C.; Carra, G.; Tegtmeyer, N.; Geppert, T.; Schröder, P.; Sewald, N.; Backert, S.; Schneider, G.; et al. pylori Htr is a new secreted virulence factor that cleaves E-cadherin to disrupt intercellular adhesion. EMBO Rep. 2010, 11, 798–804. [Google Scholar] [CrossRef] [Green Version]
- Yahiro, K.; Satoh, M.; Nakano, M.; Hisatsune, J.; Isomoto, H.; Sap, J.; Suzuki, H.; Nomura, F.; Noda, M.; Moss, J.; et al. Low-density lipoprotein receptor-related protein-1 (LRP-1) mediates autophagy and apoptosis caused by H. pylori VacA. J. Biol. Chem. 2012, 287, 31104–31115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muotiala, A.; Helander, I.M.; Pyhälä, L.; Kosunen, T.U.; Moran, A.P. Low biological activity of Helicobacter pylori lipopolysaccharide. Infect. Immun. 1992, 60, 1714–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, A.P.; Aspinall, G.O. Unique structural and biological features of Helicobacter pylori lipopolysaccharides. Prog. Clin. Biol. Res. 1998, 397, 37–49. [Google Scholar] [PubMed]
- Appelmelk, B.J.; Simoons-Smit, I.; Negrini, R.; Moran, A.P.; Aspinall, G.O.; Forte, J.G.; De Vries, T.; Quan, H.; Verboom, T.; Maaskant, J.J.; et al. Potential role of molecular mimicry between Helicobacter pylori lipopolysaccharide and host Lewis blood group antigens in autoimmunity. Infect. Immun. 1996, 64, 2031–2040. [Google Scholar] [CrossRef] [Green Version]
- Chmiela, M.; Miszczyk, E.; Rudnicka, K. Structural modifications of Helicobacter pylori lipopolysaccharide: An idea for how to live in peace. World J. Gastroenterol. 2014, 20, 9882–9897. [Google Scholar] [CrossRef]
- Grebowska, A.; Moran, A.P.; Matusiak, A.; Bak-Romaniszyn, L.; Czkwianianc, E.; Rechciński, T.; Walencka, M.; Płaneta-Małecka, I.; Rudnicka, W.; Chmiela, M. Anti-phagocytic activity of Helicobacter pylori lipopolysaccharide (LPS)-possible modulation of the innate immune response to these bacteria. Pol. J. Microbiol. 2008, 57, 185–192. [Google Scholar]
- Paziak-Domańska, B.; Chmiela, M.; Jarosińska, A.; Rudnicka, W. Potential role of CagA in the inhibition of T cell reactivity in Helicobacter pylori infections. Cell Immunol. 2000, 202, 136–139. [Google Scholar] [CrossRef]
- Grebowska, A.; Moran, A.P.; Bielanski, W.; Matusiak, A.; Rechcinski, T.; Rudnicka, K.; Baranowska, A.; Rudnicka, W.; Chmiela, M. Helicobacter pylori lipopolysaccharide activity in human peripheral blood mononuclear leukocyte cultures. J. Physiol. Pharmacol. 2010, 61, 437–442. [Google Scholar]
- Rudnicka, K.; Miszczyk, E.; Matusiak, A.; Walencka, M.; Moran, A.P.; Rudnicka, W.; Chmiela, M. Helicobacter pylori-driven modulation of NK cell expansion, intracellular cytokine expression and cytotoxic activity. Innate Immun. 2015, 21, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Miszczyk, E.; Walencka, M.; Rudnicka, K.; Matusiak, A.; Rudnicka, W.; Chmiela, M. Antigen-specific lymphocyte proliferation as a marker of immune response in guinea pigs with sustained Helicobacter pylori infection. Acta Biochim. Pol. 2014, 61, 295–303. [Google Scholar] [CrossRef]
- Mnich, E.; Gajewski, A.; Rudnicka, K.; Gonciarz, W.; Stawerski, P.; Hinc, K.; Obuchowski, M.; Chmiela, M. Immunoregulation of antigen presenting and secretory functions of monocytic cells by Helicobacter pylori antigens in relation to impairment of lymphocyte expansion. Acta Biochim. Pol. 2015, 62, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Reyes, V.E.; Peniche, A.G. Helicobacter pylori deregulates T and B cell signaling to trigger immune evasion. Curr. Top Microbiol. Immunol. 2019, 421, 229–265. [Google Scholar]
- Miszczyk, E.; Rudnicka, K.; Moran, A.P.; Fol, M.; Kowalewicz-Kulbat, M.; Druszczyńska, M.; Matusiak, A.; Walencka, M.; Rudnicka, W.; Chmiela, M. Interaction of Helicobacter pylori with C-type lectin dendritic cell-specific ICAM grabbing nonintegrin. J. Biomed. Biotechnol. 2012, 2012, 206463. [Google Scholar] [CrossRef] [Green Version]
- Tsay, F.W.; Hsu, P.I.H. H. pylori infection and extra-gastroduodenal diseases. J. Biomed. Sci. 2018, 25, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol 2008, 8, 279–298. [Google Scholar] [CrossRef]
- Shi, Y.; Evans, J.E.; Rock, K.L. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 2003, 425, 516–521. [Google Scholar] [CrossRef]
- Le, H.; Kim, W.; Kim, J.; Cho, H.R.; Kwon, B. Interleukin-33: A mediator of inflammation targeting hematopoietic stem and progenitor cells and their progenies. Front. Immunol. 2013, 4, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.M. Role of IL-33 in inflammation and disease. J. Inflamm. 2011, 8, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastorelli, L.; De Salvo Vecchi, M.; Pizarro, T.T. The role of IL-33 in gut mucosal inflammation. Mediat. Inflamm. 2013, 2013, 608187. [Google Scholar] [CrossRef]
- Caselli, C. Inflammation in cardiac disease: Focus on interleukin-33/ST2 pathway. Inflamm. Cell Signal. 2014, 1, 118–151. [Google Scholar] [CrossRef]
- Buzelli, J.N.; Chalinor, H.V.; Pavlic, D.; Sutton, P.; Menheniott, T.R.; Giraud, A.S.; Judd, L.M. IL-33 is a stomach alarmin that initiates a skewed Th2 response to injury and infection. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 203–221. [Google Scholar] [CrossRef] [Green Version]
- Gonciarz, W.; Krupa, A.; Chmiela, M. Proregenerative activity of IL-33 in gastric tissue cells undergoing Helicobacter pylori-induced apoptosis. Int. J. Mol. Sci. 2020, 21, 1801. [Google Scholar] [CrossRef] [Green Version]
- Kakkar, R.; Lee, R.T. The IL-33/ST2 pathway: Therapeutic target and novel biomarker. Nat. Rev. Drug Discov. 2008, 7, 827–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual-Figal, D.A.; Janizzi, J.L. The biology of ST2: The international ST2 consensus panel. Am. J. Cardiol. 2015, 115, 3B–7B. [Google Scholar] [CrossRef]
- Gao, Q.; Li, Y.; Li, M. The potential role of IL-33/ST2 signaling in fibrotic diseases. J. Leuk. Biol. 2015, 98, 15–22. [Google Scholar] [CrossRef] [PubMed]
- López-Casado, M.A.; Lorite, P.; Palomeque, T.; Torres, M.I. Potential role of the IL-33/ST2 axis in celiac disease. Cell Mol. Immunol. 2015, 1, 282–285. [Google Scholar] [CrossRef] [Green Version]
- Moussion, C.; Ortega, N.; Girard, J.P. The IL-1-like cytokine IL-33 is constutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: A novel “alarmin”? PLoS ONE 2008, 3, e3331. [Google Scholar] [CrossRef] [Green Version]
- Nishida, A.; Andoh, A.; Imaeda, H.; Inatomi, O.; Shiomi, H.; Fujiyama, Y. Expression of interleukin 1-like cytokine interleukine-33 and its receptor complex (ST2L and IL1RAcP) in human pancreatic myofibroblasts. Gut 2010, 59, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Demyanets, S.; Konya, V.; Kastl, S.P.; Kaun, C.; Rauscher, S.; Niessner, A.; Pentz, R.; Pfaffenberger, S.; Rychli, K.; Lemberger, C.E.; et al. Interleukin-33 induces e xpression of adhesion molecules and inflammatory activation in human endothelial cells and in human atherosclerotic plaques. Arter. Thromb. Vasc. Biol. 2011, 31, 2080–2089. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Martinez, E.; Miana, M.; Jurado-Lopez, R.; Rousseau, E.; Rossignol, P.; Zannad, F.; Cachofeiro, V.; Lopez-Andres, N. A role for soluble ST2 in vascular remodeling associated with obesity in rats. PLoS ONE 2013, 8, e79176. [Google Scholar] [CrossRef] [Green Version]
- Matilla, L.; Arrieta, V.; Jover, E.; Garcia-Pena, A.; Martinez-Martinez, E.; Sadaba, R.; Alvarez, V.; Navarro, A.; Fernandez-Celis, A.; Gainza, A.; et al. Soluble ST2 induces cardiac fibroblast activation and collagen synthesis via neurophilin-1. Cells 2020, 9, 1667. [Google Scholar] [CrossRef]
- Gonciarz, W.; Krupa, A.; Hinc, K.; Obuchowski, M.; Moran, A.P.; Gajewski, A.; Chmiela, M. The effect of Helicobacter pylori infection and different H. pylori components on the proliferation and apoptosis of gastric epithelial cells and fibroblasts. PLoS ONE 2019, 14, e0220636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlessinger, J. Direct binding and activation of receptor tyrosine kinases by collagen. Cell 1997, 91, 869–872. [Google Scholar] [CrossRef] [Green Version]
- Sanders, M.A.; Basson, M.D. Collagen-IV-dependent ERK activation in human Caco-2 intestinal epithelial cells requires focal adhesion kinase. J. Biol. Chem. 2000, 275, 38040–38047. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, A.; Radiziejewski, C.; Campbell, E.; Lubomir, K.; McGlynn, M.; Ryan, T.E.; Davis, S.; Goldfarb, M.P.; Glass, D.J.; Lemke, G.; et al. An orphan receptor tyrosine kinase family whose members serve as nonintegrin collagen receptors. Cell 1997, 1, 25–34. [Google Scholar] [CrossRef]
- Westphal, O.; Lüderitz, O.; Bister, F.; Über, S. Die extraktion von bakterien mit phenol-wasser. Z. Nat. B 1952, 78, 148–155. [Google Scholar] [CrossRef]
- Hitchcock, P.J.; Brown, T.M. Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J. Bacteriol. 1983, 154, 269–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, A.P.; Helander, I.M.; Kosunen, T.U. Compositional analysis of Helicobacter pylori rough-form lipopolysaccharides. J. Bacteriol. 1992, 174, 1370–1377. [Google Scholar] [CrossRef] [Green Version]
- Matter, K.; Aijaz, S.; Tsapara, A.; Balda, M.S. Mammalian tight junctions in the regulation of epithelial differentiation and proliferation. Curr. Opin. Cell Biol. 2005, 17, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Kusters, J.G.; van Vliet, A.H.; Kuipers, E.J. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar] [CrossRef] [Green Version]
- Krzysiek-Maczka, G.; Targosz, A.; Ptak-Belowska, A.; Korbut, E.; Szczyrk, U.; Strzalka, M.; Brzozowski, T. Molecular alterations in fibroblasts exposed to Helicobacter pylori: A missing link in bacterial inflammation progressing into gastric carcinogenesis? J. Physiol. Pharmacol. 2013, 64, 77–87. [Google Scholar]
- Varbanova, M.; Frauenschläger, K.; Malfertheiner, P. Chronic gastritis—An update. Best Pr. Res. Clin. Gastroenterol. 2014, 2, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Ali, S.; Nguyen, D.Q.; Falk, W.; Martin, M.U. Caspase 3 inactivates biologically active full length interleukin-33 as a classical cytokine but does not prohibit nuclear translocation. Biochem. Biophys. Res. Commun. 2010, 91, 1512–1516. [Google Scholar] [CrossRef] [PubMed]
- Slomiany, B.L.; Piotrowski, J.; Slomiany, A. Gastric mucosal inflammatory responses to Helicobacter pylori lipopolysaccharide: Down-regulation of nitric oxide synthase-2 and caspase-3 by sulglycotide. Biochem. Biophys. Res. Commun. 1999, 61, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Gong, C.; Li, G.; Wei, J.; Wang, T.; Meng, W.; Shi, M.; Wang, Y. Ebselen suppresses inflammation induced by Helicobacter pylori lipopolysaccharide via the p38 mitogen-activated protein kinase signaling pathway. Mol. Med. Rep. 2018, 17, 6847–6851. [Google Scholar]
- Florence, J.M.; Krupa, A.; Boosheri, L.M.; Allen, T.C.; Kurdowska, A.K. Metalloproteinase-9 contributes to endothelial dysfunction in atherosclerosis via protease activated receptor-1. PLoS ONE 2017, 12, e0171427. [Google Scholar] [CrossRef]
- Hoyt, D.G.; Mannix, R.J.; Rusnak, J.M.; Pitt, B.R.; Lazo, J.S. Collagen is a survival factor against LPS-induced apoptosis in cultured sheep pulmonary artery endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 1995, 269, L171–L177. [Google Scholar] [CrossRef]
- Krupa, A.; Gonciarz, W.; Rusek-Wala, P.; Rechciński, T.; Gajewski, A.; Samsel, Z.; Dziuba, A.; Śmiech, A.; Chmiela, M. Helicobacter pylori infection acts synergistically with a high-fat diet in the development of a proinflammatory and potentially proatherogenic endothelial cell environment in an experimental model. Int. J. Mol. Sci. 2021, 22, 3394. [Google Scholar] [CrossRef]
- Lopetuso, H.; Scaldaferri, F.; Pizarro, T.T. Emerging role of the interleukin (IL)-23/ST2 axis in gut mucosal wound healing and fibrosis. Fibrogenesis Tissue Repair 2012, 5, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Seppanen, E.; Patel, J.; Rodero, M.P.; Khosrotehrani, K. ST2 receptor invalidation maintains wound inflammation, delays healing and increases fibrosis. Exp. Dermatol. 2016, 25, 71–74. [Google Scholar] [CrossRef] [Green Version]
- Millar, N.L.; O’Donnell, C.; McInnes, I.B.; Brint, E. The role of the IL-33/ST2 pathway in tissue repair and tumorigenesis. Semin. Cell Dev. Biol. 2017, 61, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Sweet, M.J.; Leung, B.P.; Kang, D.; Sogaard, M.; Schulz, K.; Trajkovic, V.; Campbell, C.C.; Xu, D.; Liew, F.Y. A novel pathway regulating lipopolysaccharide- induced shock by ST2/T1 via inhibition of Toll-like receptor 4 expression. J. Immunol. 2011, 166, 6633–6639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takezako, N.; Hayakawa, M.; Hayakawa, H.; Aoki, S.; Yanagisawa, K.; Endo, H.; Tominaga, S. ST2 suppresses IL-6 production via the inhibition of IkappaB degradation induced by the LPS signal in THP-1 cells. Biochem. Biophys. Res. Commun. 2006, 341, 425–432. [Google Scholar] [CrossRef]
- Fagundes, C.T.; Amaral, F.A.; Souza, A.L.; Vieira, A.T.; Xu, D.; Liew, F.Y.; Souza, D.G.; Teixeira, M.M. ST2, an IL-1R family member, attenuates inflammation and lethality after intestinal ischemia and reperfusion. J. Leukoc. Biol. 2007, 81, 492–499. [Google Scholar] [CrossRef]
- Matilla, L.; Ibarololla, J.; Arrieta, V.; Garcia-Pena, A.; Martonez-Martinez, E.; Sadaba, R.; Alvarez, V.; Navarro, A.; Fernandez-Celis, A.; Gainza, A.; et al. Soluble ST2 promotes oxidative stress and inflammation in cardiac fibroblasts: An in vitro and in vivo study in aortic stenosis. Clin. Sci. 2019, 133, 1537–1548. [Google Scholar] [CrossRef]
- Takenaga, K.; Akimoto, M.; Koshikawa, N.; Nagase, H. Cancer cell-derived interleukin 33 decoy receptor sST2 enhances orthotopic tumor growth in a murine pancreatic cancer model. PLoS ONE 2020, 15, e0232230. [Google Scholar] [CrossRef]
- Alimoto, M.; Maruyama, R.; Takamaru, H.; Ochiya, T.; Takenaga, K. Soluble IL-33 receptor sST2 inhibits colorectal cancer malignant growth by modifying the tumor microenvironment. Nat. Commun. 2016, 7, 13589. [Google Scholar] [CrossRef] [PubMed]
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2016, 31 (Suppl. 1), 177–183. [Google Scholar] [CrossRef] [Green Version]
- Endorf, F.W.; Ahrenholz, D. Burn management. Curr. Opin. Crit. Care 2011, 17, 1386–1393. [Google Scholar] [CrossRef]
- Kawaguchi, H.; Hizut, A.; Tanaka, N.; Orita, K. Role of endotoxin in wound healing impairment. Res. Commun. Mol. Pathol. Pharmacol. 1996, 89, 317–327. [Google Scholar]
- Sarrazy, V.; Billet, F.; Micallef, L.; Couloumb, B.; Desmouliere, A. Mechanisms of pathological cell scarring: Role of myofibroblasts and current developments. Wound Repair Regen. 2011, 19, 10–15. [Google Scholar] [CrossRef]
- Yang, H.; Hu, C.; Li, F.; Liang, L.; Liu, L. Effect of lipopolysaccharide on the biological characteristics of human skin fibroblasts and hypertrophic scar tissue formation. IUBMB Life 2013, 65, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.R.; Benman, B. Interferon is the lymphokine and β-interferon the monokine responsible for inhibition of fibroblast collagen production and late but not early fibroblast proliferation. J. Exp. Med. 1985, 162, 516–527. [Google Scholar] [CrossRef]
- Chakravortty, D.; Nanda Kumar, K.S. Induction of cell proliferation and collagen synthesis in human small intestinal lamina propria fibroblasts by lipopolysaccharide: Possible involvement of Nitric Oxide. Biochem. Biophys. Res. Commun. 1997, 240, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Ball, D.H.; Al-Riyami, L.; Harnett, W.; Harnett, M.M. IL-33/ST2 signaling and crosstalk with Fc E RI and TLR-4 is targeted by the parasitic worm product, ES-62. Sci. Rep. 2018, 8, 4497. [Google Scholar] [CrossRef]
- Tarnawski, A.S.; Ahluwalia, A.; Jones, M.K. The mechanisms of gastric mucosal injury: Focus on microvascular endothelium as a key target. Curr. Med. Chem. 2012, 19, 4–15. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonciarz, W.; Krupa, A.; Moran, A.P.; Tomaszewska, A.; Chmiela, M. Interference of LPS H. pylori with IL-33-Driven Regeneration of Caviae porcellus Primary Gastric Epithelial Cells and Fibroblasts. Cells 2021, 10, 1385. https://doi.org/10.3390/cells10061385
Gonciarz W, Krupa A, Moran AP, Tomaszewska A, Chmiela M. Interference of LPS H. pylori with IL-33-Driven Regeneration of Caviae porcellus Primary Gastric Epithelial Cells and Fibroblasts. Cells. 2021; 10(6):1385. https://doi.org/10.3390/cells10061385
Chicago/Turabian StyleGonciarz, Weronika, Agnieszka Krupa, Anthony P. Moran, Agata Tomaszewska, and Magdalena Chmiela. 2021. "Interference of LPS H. pylori with IL-33-Driven Regeneration of Caviae porcellus Primary Gastric Epithelial Cells and Fibroblasts" Cells 10, no. 6: 1385. https://doi.org/10.3390/cells10061385