
Reduced mGluR5 Activity Modulates Mitochondrial Function

 , ,
, ,
Abstract
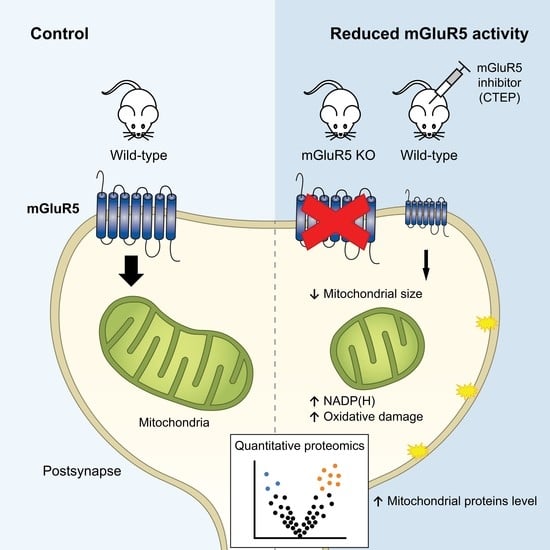
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals and Drug Administration
2.2. Synaptosome Preparation
2.3. Sample Preparation for Mass Spectrometry
2.4. LC-MS Analysis
2.5. SDS-PAGE Immunoblotting
2.6. Electron Microscopy (EM)
2.7. NADP/H and 2-Thiobarbituric Acid Reactive Substances (TBARS) Assay
2.8. Gene Ontology (GO) Enrichment Analysis and Databases
2.9. Cell-Type Enrichment Analysis
2.10. Gene Expression Correlation Analysis
2.11. Experimental Design and Statistical Analyses
3. Results
3.1. Mitochondrial Protein Expression Is Regulated in mGluR5 KO
3.2. Postsynaptic Mitochondrial Morphology Is Altered in mGluR5 KO
3.3. Acute Pharmacological Inhibition of mGluR5 Regulates Mitochondrial Protein Expression
3.4. Reduced mGluR5 Activity Alters Specific Mitochondrial Pathways
3.5. Neuronal Mitochondrial Proteins Are Mostly Regulated by the mGluR5 Inhibition
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shigemoto, R.; Nomura, S.; Ohishi, H.; Sugihara, H.; Nakanishi, S.; Mizuno, N. Immunohistochemical localization of a metabotropic glutamate receptor, mGluR5, in the rat brain. Neurosci. Lett. 1993, 163, 53–57. [Google Scholar] [CrossRef]
- Lu, Y.-M.; Jia, Z.; Janus, C.; Henderson, J.T.; Gerlai, R.; Wojtowicz, J.M.; Roder, J.C. Mice Lacking Metabotropic Glutamate Receptor 5 Show Impaired Learning and Reduced CA1 Long-Term Potentiation (LTP) But Normal CA3 LTP. J. Neurosci. 1997, 17, 5196–5205. [Google Scholar] [CrossRef] [Green Version]
- Schulz, B.; Fendt, M.; Gasparini, F.; Lingenhöhl, K.; Kuhn, R.; Koch, M. The metabotropic glutamate receptor antagonist 2-methyl-6-(phenylethynyl)-pyridine (MPEP) blocks fear conditioning in rats. Neuropharmacology 2001, 41, 1–7. [Google Scholar] [CrossRef]
- Sethna, F.; Wang, H. Acute inhibition of mGluR5 disrupts behavioral flexibility. Neurobiol. Learn. Mem. 2016, 130, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhu, Y.; Contractor, A.; Heinemann, S.F. mGluR5 Has a Critical Role in Inhibitory Learning. J. Neurosci. 2009, 29, 3676–3684. [Google Scholar] [CrossRef]
- She, W.-C.; Quairiaux, C.; Albright, M.J.; Wang, Y.-C.; Sanchez, D.E.; Chang, P.-S.; Welker, E.; Lu, H.-C. Roles of mGluR5 in synaptic function and plasticity of the mouse thalamocortical pathway. Eur. J. Neurosci. 2009, 29, 1379–1396. [Google Scholar] [CrossRef] [Green Version]
- Chokshi, V.; Gao, M.; Grier, B.D.; Owens, A.; Wang, H.; Worley, P.F.; Lee, H.-K. Input-Specific Metaplasticity in the Visual Cortex Requires Homer1a-Mediated mGluR5 Signaling. Neuron 2019, 104, 736–748.e6. [Google Scholar] [CrossRef]
- Abraham, W.C. Metaplasticity: Tuning synapses and networks for plasticity. Nat. Rev. Neurosci. 2008, 9, 387. [Google Scholar] [CrossRef] [PubMed]
- Sidorov, M.S.; Kaplan, E.S.; Osterweil, E.; Lindemann, L.; Bear, M.F. Metabotropic glutamate receptor signaling is required for NMDA receptor-dependent ocular dominance plasticity and LTD in visual cortex. Proc. Natl. Acad. Sci. USA 2015, 112, 12852–12857. [Google Scholar] [CrossRef] [Green Version]
- Waung, M.W.; Huber, K.M. Protein translation in synaptic plasticity: mGluR-LTD, Fragile X. Curr. Opin. Neurobiol. 2009, 19, 319–326. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-C.; Lu, H.-C.; Brumberg, J.C. mGluR5 knockout mice display increased dendritic spine densities. Neurosci. Lett. 2012, 524, 65–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacrosse, A.L.; Taylor, S.B.; Nemirovsky, N.E.; Gass, J.T.; Olive, M.F. mGluR5 Positive and Negative Allosteric Modulators Differentially Affect Dendritic Spine Density and Morphology in the Prefrontal Cortex. CNS Neurol. Disord. Drug Targets 2015, 14, 476–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengmany, K.; Gregory, K.J. Metabotropic glutamate receptor subtype 5: Molecular pharmacology, allosteric modulation and stimulus bias. Br. J. Pharmacol. 2015, 173, 3001–3017. [Google Scholar] [CrossRef] [Green Version]
- Ménard, C.; Quirion, R. Group 1 Metabotropic Glutamate Receptor Function and Its Regulation of Learning and Memory in the Aging Brain. Front. Pharmacol. 2012, 3, 182. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Ji, G.; Neugebauer, V. Mitochondrial Reactive Oxygen Species Are Activated by mGluR5 through IP3 and Activate ERK and PKA to Increase Excitability of Amygdala Neurons and Pain Behavior. J. Neurosci. 2011, 31, 1114–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoppel, L.J.; Auerbach, B.D.; Senter, R.K.; Preza, A.R.; Lefkowitz, R.J.; Bear, M.F. β-Arrestin2 Couples Metabotropic Glutamate Receptor 5 to Neuronal Protein Synthesis and Is a Potential Target to Treat Fragile X. Cell Rep. 2017, 18, 2807–2814. [Google Scholar] [CrossRef]
- Brambilla, C.R.; Veselinović, T.; Rajkumar, R.; Mauler, J.; Orth, L.; Ruch, A.; Ramkiran, S.; Heekeren, K.; Kawohl, W.; Wyss, C.; et al. mGluR5 receptor availability is associated with lower levels of negative symptoms and better cognition in male patients with chronic schizophrenia. Hum. Brain Mapp. 2020, 41, 2762–2781. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Wei, M.; Wu, Y.; Qin, H.; Li, W.; Ma, X.; Cheng, J.; Ren, J.; Shen, Y.; Chen, Z.; et al. Amyloid β oligomers suppress excitatory transmitter release via presynaptic depletion of phosphatidylinositol-4,5-bisphosphate. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Bellozi, P.M.Q.; Gomes, G.F.; Da Silva, M.C.M.; Lima, I.V.D.A.; Batista, C.R.Á.; Junior, W.D.O.C.; Dória, J.G.; Vieira, É.L.M.; Vieira, R.P.; De Freitas, R.P.; et al. A positive allosteric modulator of mGluR5 promotes neuroprotective effects in mouse models of Alzheimer’s disease. Neuropharmacology 2019, 160, 107785. [Google Scholar] [CrossRef]
- Ferreira, D.G.; Temido-Ferreira, M.; Miranda, H.V.; Batalha, V.L.; Coelho, J.; Szegö, É.M.; Marques-Morgado, I.; Vaz, S.H.; Rhee, J.S.; Schmitz, M.; et al. α-synuclein interacts with PrPC to induce cognitive impairment through mGluR5 and NMDAR2B. Nat. Neurosci. 2017, 20, 1569–1579. [Google Scholar] [CrossRef]
- Esterlis, I.; Holmes, S.E.; Sharma, P.; Krystal, J.H.; DeLorenzo, C. Metabotropic Glutamatergic Receptor 5 and Stress Disorders: Knowledge Gained From Receptor Imaging Studies. Biol. Psychiatry 2018, 84, 95–105. [Google Scholar] [CrossRef]
- Dölen, G.; Osterweil, E.; Rao, B.S.; Smith, G.B.; Auerbach, B.D.; Chattarji, S.; Bear, M.F. Correction of Fragile X Syndrome in Mice. Neuron 2007, 56, 955–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemmer, P.; Meredith, R.; Holmgren, C.D.; Klychnikov, O.; Stahl-Zeng, J.; Loos, M.; van der Schors, R.C.; Wortel, J.; de Wit, H.; Spijker, S.; et al. Proteomics, Ultrastructure, and Physiology of Hippocampal Synapses in a Fragile X Syndrome Mouse Model Reveal Presynaptic Phenotype. J. Biol. Chem. 2011, 286, 25495–25504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aloisi, E.; Le Corf, K.; Dupuis, J.; Zhang, P.; Ginger, M.; Labrousse, V.; Spatuzza, M.; Haberl, M.G.; Costa, L.; Shigemoto, R.; et al. Altered surface mGluR5 dynamics provoke synaptic NMDAR dysfunction and cognitive defects in Fmr1 knockout mice. Nat. Commun. 2017, 8, 1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihov, Y.; Hasler, G. Negative Allosteric Modulators of Metabotropic Glutamate Receptors Subtype 5 in Addiction: A Therapeutic Window. Int. J. Neuropsychopharmacol. 2016, 19, pyw002. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Peng, X.-Q.; Jordan, C.; Li, J.; Bi, G.-H.; He, Y.; Yang, H.-J.; Zhang, H.-Y.; Gardner, E.L.; Xi, Z.-X. mGluR5 antagonism inhibits cocaine reinforcement and relapse by elevation of extracellular glutamate in the nucleus accumbens via a CB1 receptor mechanism. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Abd-Elrahman, K.S.; Hamilton, A.; Hutchinson, S.R.; Liu, F.; Russell, R.C.; Ferguson, S.S.G. mGluR5 antagonism increases autophagy and prevents disease progression in thezQ175mouse model of Huntington’s disease. Sci. Signal. 2017, 10, eaan6387. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.V.; Hartmann, J.; Labermaier, C.; Häusl, A.S.; Zhao, G.; Harbich, D.; Schmid, B.; Wang, X.-D.; Santarelli, S.; Kohl, C.; et al. Homer1/mGluR5 Activity Moderates Vulnerability to Chronic Social Stress. Neuropsychopharmacology 2014, 40, 1222–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindemann, L.; Porter, R.H.; Scharf, S.H.; Kuennecke, B.; Bruns, A.; Von Kienlin, M.; Harrison, A.C.; Paehler, A.; Funk, C.; Gloge, A.; et al. Pharmacology of Basimglurant (RO4917523, RG7090), a Unique Metabotropic Glutamate Receptor 5 Negative Allosteric Modulator in Clinical Development for Depression. J. Pharmacol. Exp. Ther. 2015, 353, 213–233. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, A.; Vasefi, M.; Tuin, C.V.; McQuaid, R.J.; Anisman, H.; Ferguson, S.S. Chronic Pharmacological mGluR5 Inhibition Prevents Cognitive Impairment and Reduces Pathogenesis in an Alzheimer Disease Mouse Model. Cell Rep. 2016, 15, 1859–1865. [Google Scholar] [CrossRef] [Green Version]
- Michalon, A.; Sidorov, M.; Ballard, T.M.; Ozmen, L.; Spooren, W.; Wettstein, J.G.; Jaeschke, G.; Bear, M.F.; Lindemann, L. Chronic Pharmacological mGlu5 Inhibition Corrects Fragile X in Adult Mice. Neuron 2012, 74, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Ossowska, K.; Konieczny, J.; Wolfarth, S.; Pilc, A. MTEP, a new selective antagonist of the metabotropic glutamate receptor subtype 5 (mGluR5), produces antiparkinsonian-like effects in rats. Neuropharmacology 2005, 49, 447–455. [Google Scholar] [CrossRef]
- Barnes, S.A.; Sheffler, D.J.; Semenova, S.; Cosford, N.D.; Bespalov, A. Metabotropic Glutamate Receptor 5 as a Target for the Treatment of Depression and Smoking: Robust Preclinical Data but Inconclusive Clinical Efficacy. Biol. Psychiatry 2018, 83, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-W.; Zhang, X.-R.; Zhang, Z.-R.; Wang, X.-S.; Chen, J.; Chen, S.-Y.; Xie, C.-L. Effects of mGluR5 Antagonists on Parkinson’s Patients With L-Dopa-Induced Dyskinesia: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Front. Aging Neurosci. 2018, 10, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youssef, E.A.; FragXis Study Group; Berry-Kravis, E.; Czech, C.; Hagerman, R.J.; Hessl, D.; Wong, C.Y.; Rabbia, M.; Deptula, D.; John, A.; et al. Effect of the mGluR5-NAM Basimglurant on Behavior in Adolescents and Adults with Fragile X Syndrome in a Randomized, Double-Blind, Placebo-Controlled Trial: FragXis Phase 2 Results. Neuropsychopharmacology 2018, 43, 503–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindemann, L.; Jaeschke, G.; Michalon, A.; Vieira, E.; Honer, M.; Spooren, W.; Porter, R.; Hartung, T.; Kolczewski, S.; Büttelmann, B.; et al. CTEP: A Novel, Potent, Long-Acting, and Orally Bioavailable Metabotropic Glutamate Receptor 5 Inhibitor. J. Pharmacol. Exp. Ther. 2011, 339, 474–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Lozano, M.A.; Koopmans, F.; Sullivan, P.F.; Protze, J.; Krause, G.; Verhage, M.; Li, K.W.; Liu, F.; Smit, A.B. Stitching the synapse: Cross-linking mass spectrometry into resolving synaptic protein interactions. Sci. Adv. 2020, 6, eaax5783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koopmans, F.; Pandya, N.J.; Franke, S.K.; Phillippens, I.H.; Paliukhovich, I.; Li, K.W.; Smit, A.B. Comparative Hippocampal Synaptic Proteomes of Rodents and Primates: Differences in Neuroplasticity-Related Proteins. Front. Mol. Neurosci. 2018, 11, 364. [Google Scholar] [CrossRef]
- He, E.; Lozano, M.A.G.; Stringer, S.; Watanabe, K.; Sakamoto, K.; Oudsten, F.D.; Koopmans, F.; Giamberardino, S.N.; Hammerschlag, A.; Cornelisse, L.N.; et al. MIR137 schizophrenia-associated locus controls synaptic function by regulating synaptogenesis, synapse maturation and synaptic transmission. Hum. Mol. Genet. 2018, 27, 1879–1891. [Google Scholar] [CrossRef]
- Bruderer, R.; Bernhardt, O.M.; Gandhi, T.; Miladinović, S.M.; Cheng, L.-Y.; Messner, S.; Ehrenberger, T.; Zanotelli, V.R.T.; Butscheid, Y.; Escher, C.; et al. Extending the Limits of Quantitative Proteome Profiling with Data-Independent Acquisition and Application to Acetaminophen-Treated Three-Dimensional Liver Microtissues. Mol. Cell. Proteom. 2015, 14, 1400–1410. [Google Scholar] [CrossRef] [Green Version]
- Freche, D.; Pannasch, U.; Rouach, N.; Holcman, D. Synapse Geometry and Receptor Dynamics Modulate Synaptic Strength. PLoS ONE 2011, 6, e25122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiemerslage, L.; Lee, D. Quantification of mitochondrial morphology in neurites of dopaminergic neurons using multiple parameters. J. Neurosci. Methods 2016, 262, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.C.; Robinson, A.J. MitoMiner v3.1, an update on the mitochondrial proteomics database. Nucleic Acids Res. 2016, 44, D1258–D1261. [Google Scholar] [CrossRef] [Green Version]
- Giurgiu, M.; Reinhard, J.; Brauner, B.; Dunger-Kaltenbach, I.; Fobo, G.; Frishman, G.; Montrone, C.; Ruepp, A. CORUM: The comprehensive resource of mammalian protein complexes—2019. Nucleic Acids Res. 2019, 47, D559–D563. [Google Scholar] [CrossRef] [Green Version]
- Yim, A.; Koti, P.; Bonnard, A.; Marchiano, F.; Dürrbaum, M.; Garcia-Perez, C.; Villaveces, J.; Gamal, S.; Cardone, G.; Perocchi, F.; et al. mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations. Nucleic Acids Res. 2020, 48, 605–632. [Google Scholar] [CrossRef] [Green Version]
- Slenter, D.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Mélius, J.; Cirillo, E.; Coort, S.L.; Digles, D.; et al. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018, 46, D661–D667. [Google Scholar] [CrossRef] [PubMed]
- Kutmon, M.; Van Iersel, M.P.; Bohler, A.; Kelder, T.; Nunes, N.; Pico, A.; Evelo, C.T. PathVisio 3: An Extendable Pathway Analysis Toolbox. PLoS Comput. Biol. 2015, 11, e1004085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skene, N.; Grant, S.G.N. Identification of Vulnerable Cell Types in Major Brain Disorders Using Single Cell Transcriptomes and Expression Weighted Cell Type Enrichment. Front. Neurosci. 2016, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Tasic, B.; Yao, Z.; Graybuck, L.T.; Smith, K.A.; Nguyen, T.N.; Bertagnolli, D.; Goldy, J.; Garren, E.; Economo, M.N.; Viswanathan, S.; et al. Shared and distinct transcriptomic cell types across neocortical areas. Nat. Cell Biol. 2018, 563, 72–78. [Google Scholar] [CrossRef]
- Watanabe, K.; Mirkov, M.U.; De Leeuw, C.A.; Heuvel, M.P.V.D.; Posthuma, D. Genetic mapping of cell type specificity for complex traits. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Festing, M.F.W. The “completely randomised” and the “randomised block” are the only experimental designs suitable for widespread use in pre-clinical research. Sci. Rep. 2020, 10, 1–5. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.; Maechler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Usinglme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Koopmans, F.; van Nierop, P.; Andres-Alonso, M.; Byrnes, A.; Cijsouw, T.; Coba, M.P.; Cornelisse, L.N.; Farrell, R.J.; Goldschmidt, H.L.; Howrigan, D.P.; et al. SynGO: An Evidence-Based, Expert-Curated Knowledge Base for the Synapse. Neuron 2019, 103, 217–234.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; McConnell, E.; Pare, J.-F.; Xu, Q.; Chen, M.; Peng, W.; Lovatt, D.; Han, X.; Smith, Y.; Nedergaard, M. Glutamate-Dependent Neuroglial Calcium Signaling Differs Between Young and Adult Brain. Science 2013, 339, 197–200. [Google Scholar] [CrossRef] [Green Version]
- Bazargani, N.; Attwell, D. Astrocyte calcium signaling: The third wave. Nat. Neurosci. 2016, 19, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.-E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A Mitochondrial Protein Compendium Elucidates Complex I Disease Biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Fecher, C.; Trovò, L.; Müller, S.A.; Snaidero, N.; Wettmarshausen, J.; Heink, S.; Ortiz, O.; Wagner, I.; Kühn, R.; Hartmann, J.; et al. Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat. Neurosci. 2019, 22, 1731–1742. [Google Scholar] [CrossRef]
- Pandya, N.J.; Klaassen, R.V.; Van Der Schors, R.C.; Slotman, J.A.; Houtsmuller, A.; Smit, A.B.; Li, K.W. Group 1 metabotropic glutamate receptors 1 and 5 form a protein complex in mouse hippocampus and cortex. Proteomics 2016, 16, 2698–2705. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Otaño, I.; Luján, R.; Tavalin, S.J.; Plomann, M.; Modregger, J.; Liu, X.-B.; Jones, E.G.; Heinemann, S.F.; Lo, D.C.; Ehlers, M.D. Endocytosis and synaptic removal of NR3A-containing NMDA receptors by PACSIN1/syndapin1. Nat. Neurosci. 2006, 9, 611–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widagdo, J.; Fang, H.; Jang, S.E.; Anggono, V. PACSIN1 regulates the dynamics of AMPA receptor trafficking. Sci. Rep. 2016, 6, 31070. [Google Scholar] [CrossRef] [PubMed]
- Hanley, J.G. The Regulation of AMPA Receptor Endocytosis by Dynamic Protein-Protein Interactions. Front. Cell. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Raynaud, F.; Janossy, A.; Dahl, J.; Bertaso, F.; Perroy, J.; Varrault, A.; Vidal, M.; Worley, P.F.; Boeckers, T.M.; Bockaert, J.; et al. Shank3-Rich2 Interaction Regulates AMPA Receptor Recycling and Synaptic Long-Term Potentiation. J. Neurosci. 2013, 33, 9699–9715. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Qin, Y.; Zhao, M.; Van Aelst, L.; Malinow, R. Ras and Rap Control AMPA Receptor Trafficking during Synaptic Plasticity. Cell 2002, 110, 443–455. [Google Scholar] [CrossRef] [Green Version]
- Albin, S.D.; Davis, G.W. Coordinating Structural and Functional Synapse Development: Postsynaptic p21-Activated Kinase Independently Specifies Glutamate Receptor Abundance and Postsynaptic Morphology. J. Neurosci. 2004, 24, 6871–6879. [Google Scholar] [CrossRef]
- Jin, D.-Z.; Xue, B.; Mao, L.-M.; Wang, J.Q. Metabotropic glutamate receptor 5 upregulates surface NMDA receptor expression in striatal neurons via CaMKII. Brain Res. 2015, 1624, 414–423. [Google Scholar] [CrossRef] [Green Version]
- Jeanneteau, F.; Arango-Lievano, M. Linking Mitochondria to Synapses: New Insights for Stress-Related Neuropsychiatric Disorders. Neural Plast. 2016, 2016, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Divakaruni, S.S.; Van Dyke, A.M.; Chandra, R.; LeGates, T.A.; Contreras, M.; Dharmasri, P.A.; Higgs, H.N.; Lobo, M.K.; Thompson, S.M.; Blanpied, T.A. Long-Term Potentiation Requires a Rapid Burst of Dendritic Mitochondrial Fission during Induction. Neuron 2018, 100, 860–875.e7. [Google Scholar] [CrossRef] [Green Version]
- Rangaraju, V.; Lauterbach, M.; Schuman, E.M. Spatially Stable Mitochondrial Compartments Fuel Local Translation during Plasticity. Cell 2019, 176, 73–84.e15. [Google Scholar] [CrossRef] [Green Version]
- Cioni, J.-M.; Lin, J.Q.; Holtermann, A.V.; Koppers, M.; Jakobs, M.A.; Azizi, A.; Turner-Bridger, B.; Shigeoka, T.; Franze, K.; Harris, W.A.; et al. Late Endosomes Act as mRNA Translation Platforms and Sustain Mitochondria in Axons. Cell 2019, 176, 56–72.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, G.; Neugebauer, V. Reactive Oxygen Species Are Involved in Group I mGluR-Mediated Facilitation of Nociceptive Processing in Amygdala Neurons. J. Neurophysiol. 2010, 104, 218–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massaad, C.A.; Klann, E. Reactive Oxygen Species in the Regulation of Synaptic Plasticity and Memory. Antioxid. Redox Signal. 2011, 14, 2013–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrigno, A.; Berardo, C.; Di Pasqua, L.G.; Siciliano, V.; Richelmi, P.; Nicoletti, F.; Vairetti, M. Selective Blockade of the Metabotropic Glutamate Receptor mGluR5 Protects Mouse Livers in In Vitro and Ex Vivo Models of Ischemia Reperfusion Injury. Int. J. Mol. Sci. 2018, 19, 314. [Google Scholar] [CrossRef] [Green Version]
- Chantong, B.; Kratschmar, D.V.; Lister, A.; Odermatt, A. Inhibition of metabotropic glutamate receptor 5 induces cellular stress through pertussis toxin-sensitive Gi-proteins in murine BV-2 microglia cells. J. Neuroinflamm. 2014, 11, 190. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Song, D.; Bai, Q.; Zhou, L.; Cai, L.; Hertz, L.; Peng, L. Role of Glycogenolysis in Stimulation of ATP Release from Cultured Mouse Astrocytes by Transmitters and High K+ Concentrations. ASN Neuro 2013, 6, AN20130040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, N.; Zhang, Q.; Wang, S.T.; Gu, L.; Yang, H.M.; Liu, L.; Bakshi, R.; Zhang, H. Blockade of metabotropic glutamate receptor 5 protects against DNA damage in a rotenone-induced Parkinson’s disease model. Free. Radic. Biol. Med. 2015, 89, 567–580. [Google Scholar] [CrossRef]
- Zehnder, T.; Petrelli, F.; Romanos, J.; Figueiredo, E.C.D.O.; Lewis, T.L.; Déglon, N.; Polleux, F.; Santello, M.; Bezzi, P. Mitochondrial biogenesis in developing astrocytes regulates astrocyte maturation and synapse formation. Cell Rep. 2021, 35, 108952. [Google Scholar] [CrossRef]
- Kuzniewska, B.; Cysewski, D.; Wasilewski, M.; Sakowska, P.; Milek, J.; Kulinski, T.M.; Winiarski, M.; Kozielewicz, P.; Knapska, E.; Dadlez, M.; et al. Mitochondrial protein biogenesis in the synapse is supported by local translation. EMBO Rep. 2020, 21, e48882. [Google Scholar] [CrossRef]
- Osterweil, E.; Krueger-Burg, D.; Reinhold, K.; Bear, M.F. Hypersensitivity to mGluR5 and ERK1/2 Leads to Excessive Protein Synthesis in the Hippocampus of a Mouse Model of Fragile X Syndrome. J. Neurosci. 2010, 30, 15616–15627. [Google Scholar] [CrossRef] [Green Version]
- Strosznajder, J.; Wikiel, H.; Sun, G.Y. Effects of Cerebral Ischemia on [3H]Inositol Lipids and [3H]Inositol Phosphates of Gerbil Brain and Subcellular Fractions. J. Neurochem. 1987, 48, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Dave, K.R.; DeFazio, R.A.; Raval, A.P.; Torraco, A.; Saul, I.; Barrientos, A.; Perez-Pinzon, M.A. Ischemic Preconditioning Targets the Respiration of Synaptic Mitochondria via Protein Kinase C. J. Neurosci. 2008, 28, 4172–4182. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Maekawa, T.; Abu Shama, K.M.; Yamashima, T.; Yoshida, K.-I. Translocation and Down-Regulation of Protein Kinase C-α, -β, and -γ Isoforms During Ischemia-Reperfusion in Rat Brain. J. Neurochem. 2002, 72, 2556–2564. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Hirabayashi, Y.; Kwon, S.-K.; Lewis, T.L.; Polleux, F. Emerging roles of mitochondria in synaptic transmission and neurodegeneration. Curr. Opin. Physiol. 2018, 3, 82–93. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez-Lozano, M.A.; Wortel, J.; van der Loo, R.J.; van Weering, J.R.T.; Smit, A.B.; Li, K.W. Reduced mGluR5 Activity Modulates Mitochondrial Function. Cells 2021, 10, 1375. https://doi.org/10.3390/cells10061375
Gonzalez-Lozano MA, Wortel J, van der Loo RJ, van Weering JRT, Smit AB, Li KW. Reduced mGluR5 Activity Modulates Mitochondrial Function. Cells. 2021; 10(6):1375. https://doi.org/10.3390/cells10061375
Chicago/Turabian StyleGonzalez-Lozano, Miguel A., Joke Wortel, Rolinka J. van der Loo, Jan R. T. van Weering, August B. Smit, and Ka Wan Li. 2021. "Reduced mGluR5 Activity Modulates Mitochondrial Function" Cells 10, no. 6: 1375. https://doi.org/10.3390/cells10061375