
Rap1a Regulates Cardiac Fibroblast Contraction of 3D Diabetic Collagen Matrices by Increased Activation of the AGE/RAGE Cascade


Abstract
:1. Introduction
2. Methods and Materials
2.1. Animal Models
2.2. Animal Care
2.3. Cardiac Fibroblast Isolation and Culture
2.4. Collagen Extraction
2.5. 3-Dimensional Collagen Matrix
2.6. Matrix Contraction Analysis
2.7. Western Blot Analysis
2.8. Immunofluorescence
2.9. Hydrogen Peroxide Assay
2.10. Matrix Transmission Assay
2.11. Matrix Compaction Assay
2.12. Statistical Analysis
3. Results
3.1. The Presence of 3D Non-Diabetic Collagen Matrix-Induced Changes in Cell Morphology and Rearrangment of Cardiac Fibroblasts Independent of RAGE
3.2. The Presence of RAGE, Rap1a, and Isolated ECM Altered the Area of the Contractile Ring in Cardiac Fibroblasts
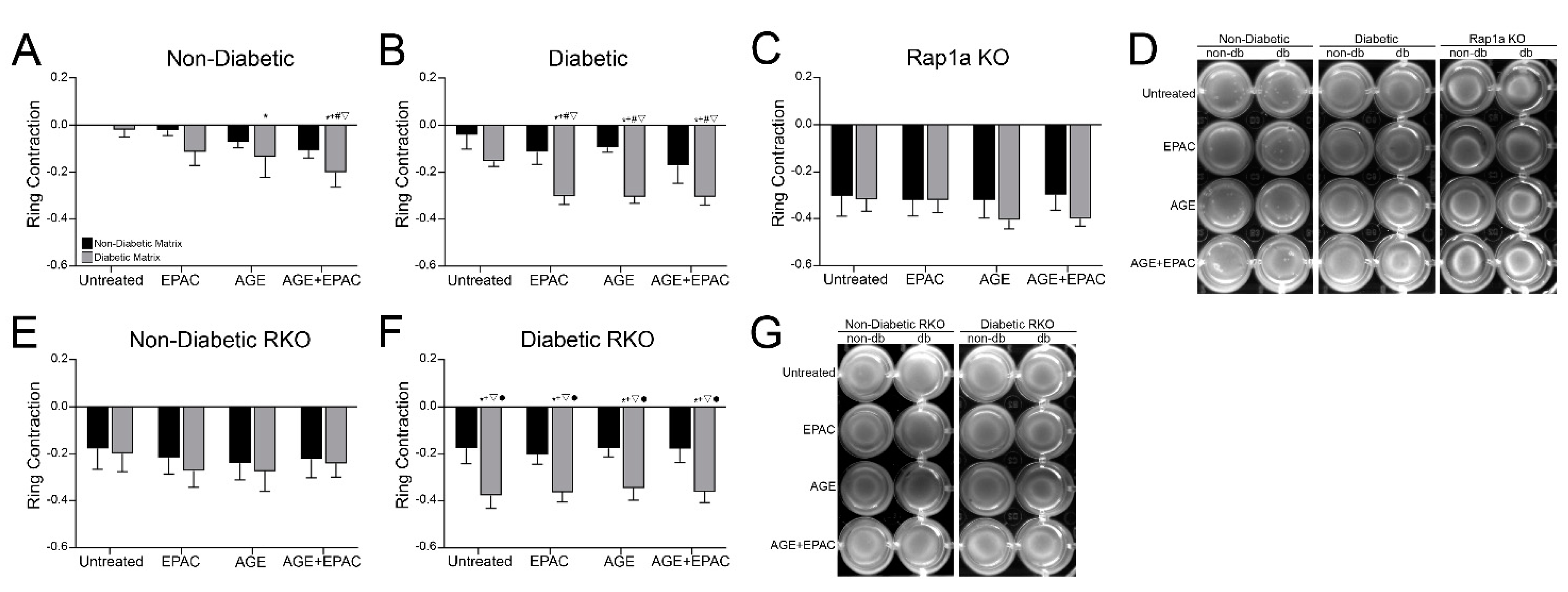
3.3. Cardiac Fibroblast Matrix Contraction Was Impacted by AGE/RAGE Signaling and Diabetic Matrix Components
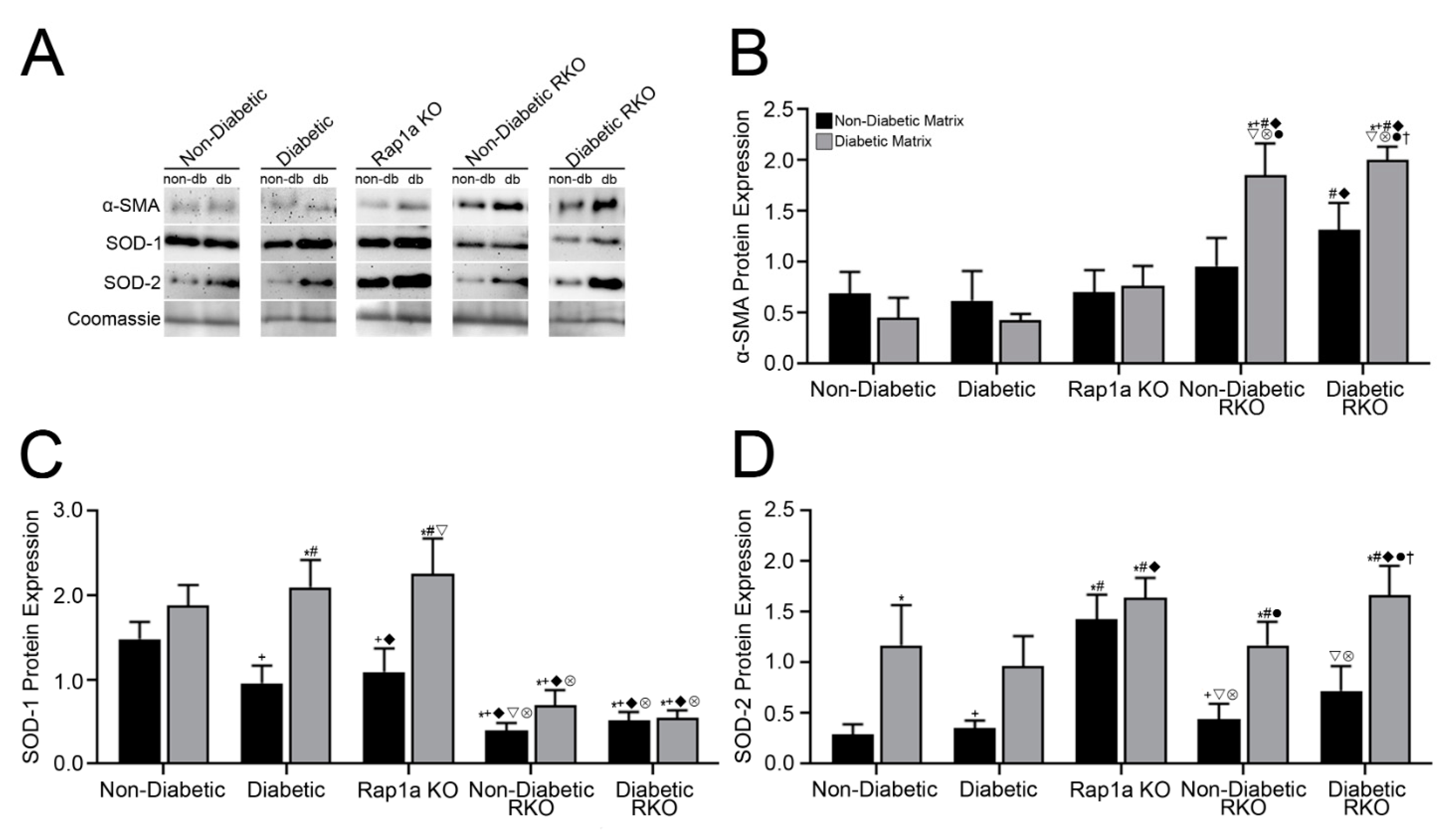
3.4. Isolated ECM and Genotype Variations Induced Significant Changes in Expression of Proteins Associated with RAGE Signaling
3.5. Isolated Collagen and Rap1a-Mediated AGE/RAGE-Induced Increased α-SMA Expression in Cardiac Fibroblasts Wildtype for RAGE
3.6. AGEs in Diabetic Collagen Induced Changes in SOD-1 Expression Utilizing Both the AGE/RAGE Signaling Cascade and Rap1a
3.7. Diabetic Matrix and AGE/RAGE Signaling Impacted Expression of SOD-2 in Cardiac Fibroblasts
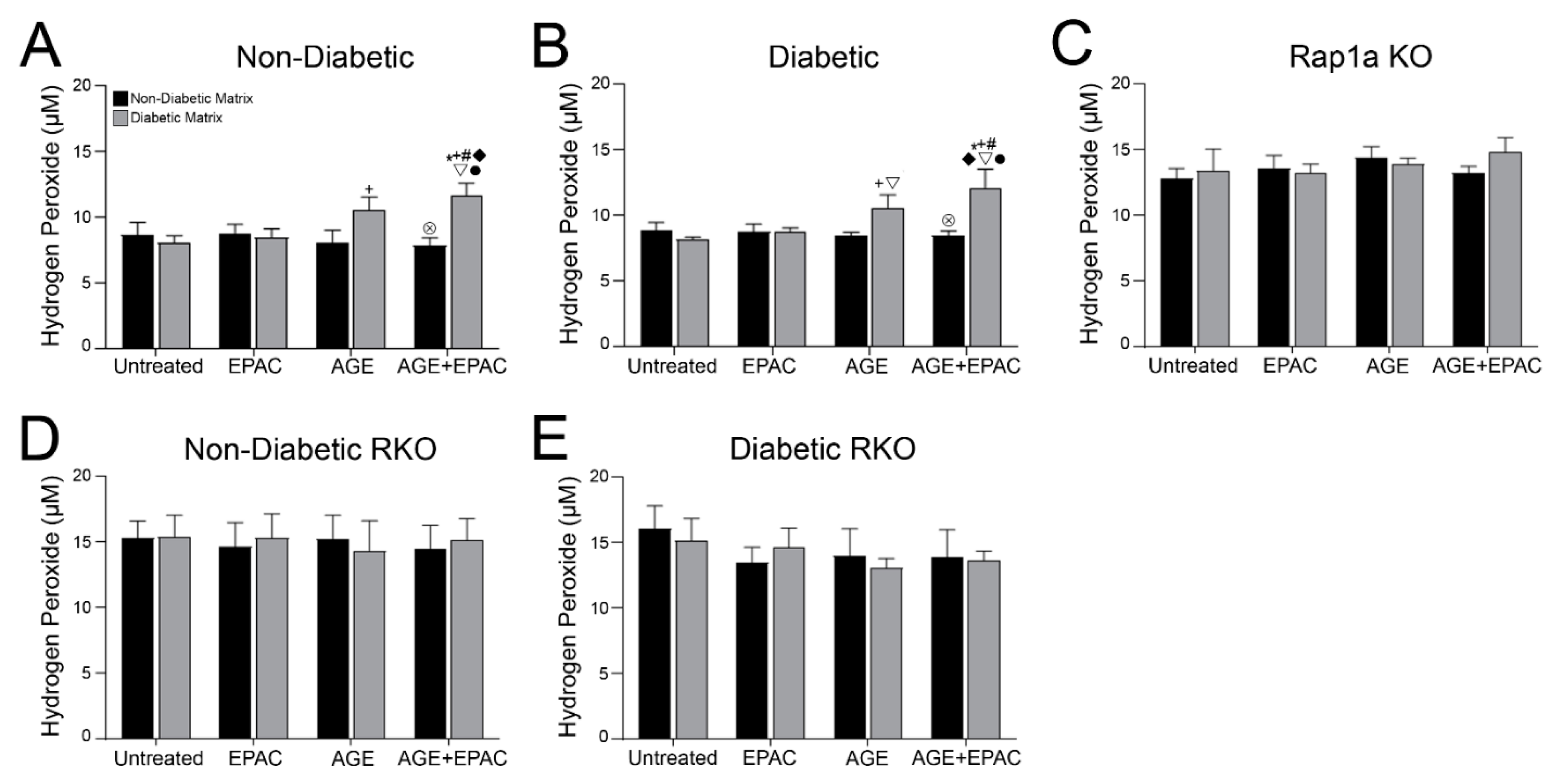
3.8. Cardiac Fibroblasts with Knocked out RAGE Exhibited Significantly Higher Concentration of Hydrogen Peroxide
3.9. AGE/RAGE Signaling and Rap1a Expression Altered Hydrogen Peroxide Levels in Cardiac Fibroblasts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fowlkes, V.; Clark, J.; Fix, C.; Law, B.A.; Morales, M.O.; Qiao, X.; Ako-Asare, K.; Goldsmith, J.G.; Carver, W.; Murray, D.B.; et al. Type II diabetes promotes a myofibroblast phenotype in cardiac fibroblasts. Life Sci. 2013, 92, 669–676. [Google Scholar] [CrossRef] [Green Version]
- Shamhart, P.E.; Luther, D.J.; Adapala, R.K.; Bryant, J.E.; Petersen, K.A.; Meszaros, J.G.; Thodeti, C.K. Hyperglycemia enhances function and differentiation of adult rat cardiac fibroblasts. Can. J. Physiol. Pharmacol. 2014, 92, 598–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchinson, K.R.; Lord, C.K.; West, T.A.; Stewart, J.A.S., Jr. Cardiac Fibroblast-Dependent Extracellular Matrix Accumulation Is Associated with Diastolic Stiffness in Type 2 Diabetes. PLoS ONE 2013, 8, e72080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, C.K.; Won, D.; Pruthi, S.; Lin, S.S.; Barnard, R.J. Effect of a diet and exercise intervention on oxidative stress, inflammation and monocyte adhesion in diabetic men. Diabetes Res. Clin. Pract. 2006, 73, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-L.; Wang, F.-S.; Kuo, Y.-R.; Huang, Y.-T.; Huang, H.-C.; Sun, Y.-C.; Kuo, Y.-H. Ras modulation of superoxide activates ERK-dependent fibronectin expression in diabetes-induced renal injuries. Kidney Int. 2006, 69, 1593–1600. [Google Scholar] [CrossRef] [Green Version]
- Shinde, A.V.; Humeres, C.; Frangogiannis, N.G. The role of α-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 298–309. [Google Scholar] [CrossRef]
- Burr, S.D.; Stewart, J.A. Extracellular matrix components isolated from diabetic mice alter cardiac fibroblast function through the AGE/RAGE signaling cascade. Life Sci. 2020, 250, 117569. [Google Scholar] [CrossRef]
- Burr, S.D.; Harmon, M.B.; Stewart, J.A.S., Jr. The Impact of Diabetic Conditions and AGE / RAGE Signaling on Cardiac Fibroblast Migration. Front. Cell Dev. Biol. 2020, 8, 112. [Google Scholar] [CrossRef]
- Kato, T.; Yamashita, T.; Sekiguchi, A.; Tsuneda, T.; Sagara, K.; Takamura, M.; Kaneko, S.; Aizawa, T.; Fu, L.-T. AGEs-RAGE System Mediates Atrial Structural Remodeling in the Diabetic Rat. J. Cardiovasc. Electrophysiol. 2008, 19, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Umadevi, S.; Gopi, V.; Elangovan, V. Regulatory mechanism of gallic acid against advanced glycation end products induced cardiac remodeling in experimental rats. Chem. Interact. 2014, 208, 28–36. [Google Scholar] [CrossRef]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Torreggiani, M.; Zhu, L.; Chen, X.; He, J.C.; Striker, G.E.; Vlassara, H. AGER1 regulates endothelial cell NADPH oxidase-dependent oxidant stress via PKC-δ: Implications for vascular disease. Am. J. Physiol. Physiol. 2010, 298, C624–C634. [Google Scholar] [CrossRef] [Green Version]
- Nitti, M.; Furfaro, A.L.; Traverso, N.; Odetti, P.; Storace, D.; Cottalasso, D.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. PKC delta and NADPH oxidase in AGE-induced neuronal death. Neurosci. Lett. 2007, 416, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.J.; Niu, H.X.; Hou, F.F.; Zhang, L.; Fu, N.; Nagai, R.; Lu, X.; Chen, B.H.; Shan, Y.X.; Tian, J.W.; et al. Advanced Oxidation Protein Products Activate Vascular Endothelial Cells via a RAGE-Mediated Signaling Pathway. Antioxid. Redox Signal. 2008, 10, 1699–1712. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, D.B.; Siwik, D.A.; Xiao, L.; Pimentel, D.R.; Singh, K.; Colucci, W.S. Role of Oxidative Stress in Myocardial Hypertrophy and Failure. J. Mol. Cell. Cardiol. 2002, 34, 379–388. [Google Scholar] [CrossRef]
- Wei, X.F.; Zhou, Q.G.; Hou, F.F.; Liu, B.Y.; Liang, M. Advanced oxidation protein products induce mesangial cell perturbation through PKC-dependent activation of NADPH oxidase. Am. J. Physiol. Physiol. 2009, 296, F427–F437. [Google Scholar] [CrossRef] [Green Version]
- Adamopoulos, C.; Piperi, C.; Gargalionis, A.N.; Dalagiorgou, G.; Spilioti, E.; Korkolopoulou, P.; Papavassiliou, A.G. Advanced glycation end products upregulate lysyl oxidase and endothelin-1 in human aortic endothelial cells via parallel activation of ERK1/2-NF-??B and JNK-AP-1 signaling pathways. Cell. Mol. Life Sci. 2016, 73, 1685–1698. [Google Scholar] [CrossRef] [PubMed]
- Kameda, K.; Matsunaga, T.; Abe, N.; Hanada, H.; Ishizaka, H.; Ono, H.; Saitoh, M.; Fukui, K.; Fukuda, I.; Osanai, T.; et al. Correlation of oxidative stress with activity of matrix metalloproteinase in patients with coronary artery disease Possible role for left ventricular remodelling. Eur. Heart J. 2003, 24, 2180–2185. [Google Scholar] [CrossRef]
- Piperi, C.; Goumenos, A.; Adamopoulos, C.; Papavassiliou, A.G. AGE/RAGE signalling regulation by miRNAs: Associations with diabetic complications and therapeutic potential. Int. J. Biochem. Cell Biol. 2015, 60, 197–201. [Google Scholar] [CrossRef]
- Leuner, B.; Ruhs, S.; Brömme, H.-J.; Bierhaus, A.; Sel, S.; Silber, R.-E.; Somoza, V.; Simm, A.; Nass, N. RAGE-dependent activation of gene expression of superoxide dismutase and vanins by AGE-rich extracts in mice cardiac tissue and murine cardiac fibroblasts. Food Funct. 2012, 3, 1091. [Google Scholar] [CrossRef]
- Fujiwara, T.; Dohi, T.; Maan, Z.N.; Rustad, K.C.; Kwon, S.H.; Padmanabhan, J.; Whittam, A.J.; Suga, H.; Duscher, D.; Rodrigues, M.; et al. Age-associated intracellular superoxide dismutase deficiency potentiates dermal fibroblast dysfunction during wound healing. Exp. Dermatol. 2017, 28, 485–492. [Google Scholar] [CrossRef]
- Arora, P.D.; McCulloch, C.A.G. Dependence of collagen remodelling on -smooth muscle actin expression by fibroblasts. J. Cell. Physiol. 1994, 159, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Celetta, G.; Tomasek, J.J.; Gabbiani, G.; Chaponnier, C. Alpha-Smooth Muscle Actin Expression Upregulates Fibroblast Contractile Activity. Mol. Biol. Cell 2001, 12, 2730–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avery, D.; Govindaraju, P.; Jacob, M.; Todd, L.; Monslow, J.; Puré, E. Extracellular matrix directs phenotypic heterogeneity of activated fibroblasts. Matrix Biol. 2018, 67, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Randive, R.; Stewart, J.A. Molecular mechanisms of AGE/RAGE-mediated fibrosis in the diabetic heart. World J. Diabetes 2014, 5, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Machahua, C.; Montes-Worboys, A.; Llatjos, R.; Escobar, I.; Dorca, J.; Molina-Molina, M.; Vicens-Zygmunt, V. Increased AGE-RAGE ratio in idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, I.; Frangogiannis, N.G. Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 2016, 90, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Simard, E.; Söllradl, T.; Maltais, J.-S.; Boucher, J.; D’Orléans-Juste, P.; Grandbois, M. Receptor for Advanced Glycation End-Products Signaling Interferes with the Vascular Smooth Muscle Cell Contractile Phenotype and Function. PLoS ONE 2015, 10, e0128881. [Google Scholar] [CrossRef] [Green Version]
- Oldfield, M.D.; Bach, L.A.; Forbes, J.M.; Nikolic-Paterson, D.; McRobert, A.; Thallas, V.; Atkins, R.C.; Osicka, T.; Jerums, G.; Cooper, M.E. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J. Clin. Investig. 2001, 108, 1853–1863. [Google Scholar] [CrossRef]
- Burr, S.D.; Stewart, J.A. Rap1a Overlaps the AGE/RAGE Signaling Cascade to Alter Expression of α-SMA, p-NF-κ B, and p-PKC-ζ in Cardiac Fibroblasts Isolated from Type 2 Diabetic Mice. Cells 2021, 10, 557. [Google Scholar] [CrossRef]
- Burgstaller, G.; Sengupta, A.; Vierkotten, S.; Preissler, G.; Lindner, M.; Behr, J.; Königshoff, M.; Eickelberg, O. Distinct niches within the extracellular matrix dictate fibroblast function in (cell free) 3D lung tissue cultures. Am. J. Physiol. Cell. Mol. Physiol. 2018, 314, L708–L723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, G.S.; Wysocki, A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen. 2009, 17, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Mott, J.D.; Werb, Z. Regulation of matrix biology by matrix metalloproteinases. Curr. Opin. Cell Biol. 2004, 16, 558–564. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, S.; Pawelek, P.; Grinnell, F. Extracellular matrix organization modulates fibroblast growth and growth factor responsiveness. Exp. Cell Res. 1989, 182, 572–582. [Google Scholar] [CrossRef]
- Berthod, F.; Hayek, D.; Damour, O.; Collombel, C. Collagen synthesis by fibroblasts cultured within a collagen sponge. Biomaterials 1993, 14, 749–754. [Google Scholar] [CrossRef]
- Pizzo, A.M.; Kokini, K.; Vaughn, L.C.; Waisner, B.Z.; Voytik-Harbin, S.L. HIGHLIGHTED TOPIC Biomechanics and Mechanotransduction in Cells and Tissues Extracellular matrix (ECM) microstructural composition regulates local cell-ECM biomechanics and fundamental fibroblast behavior: A multidimensional perspective. J. Appl. Physiol. 1909, 98, 1909–1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loftis, M.J.; Sexton, D.; Carver, W. Effects of collagen density on cardiac fibroblast behavior and gene expression. J. Cell. Physiol. 2003, 196, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Krieg, T.; Bankert, R.B.; Weber, L. Integrin a2ß1 Is Upregulated in Fibroblasts and Highly Aggressive Melanoma Cells in Three-Dimensional Collagen Lattices and Mediates the Reorganization of Collagen I Fibrils. J. Cell Biol. 1991, 115, 1427–1436. [Google Scholar]
- Eckes, B.; Zweers, M.C.; Zhang, Z.G.; Hallinger, R.; Mauch, C.; Aumailley, M.; Krieg, T. Mechanical tension and integrin α2β1 regulate fibroblast functions. J. Investig. Dermatol. Symp. Proc. 2006, 11, 66–72. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Yang, Z.; Yang, F.; Wang, J.; Zhuang, Y.; Xu, C.; Zhang, B.; Tian, X.-L.; Liu, D. Suppression of Rap1 Impairs Cardiac Myofibrils and Conduction System in Zebrafish. PLoS ONE 2012, 7, e50960. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Li, F.; Ingram, D.A.; Quilliam, L.A. Rap1a Is a Key Regulator of Fibroblast Growth Factor 2-Induced Angiogenesis and Together with Rap1b Controls Human. Mol. Cell. Biol. 2008, 28, 5803–5810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, O.L.; Kamishima, T.; Barrett-Jolley, R.; Quayle, J.M.; Dart, C. Exchange protein activated by cAMP (Epac) induces vascular relaxation by activating Ca 2+ -sensitive K + channels in rat mesenteric artery. J. Physiol. 2013, 591, 5107–5123. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Fotheringham, L.; Wittchen, E.S.; Hartnett, M.E. Rap1 GTPase inhibits tumor necrosis factor-α-induced choroidal endothelial migration via NADPH oxidase- and NF-κB-dependent activation of Rac1. Am. J. Pathol. 2015, 185, 3316–3325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panchatcharam, M.; Miriyala, S.; Yang, F.; Leitges, M.; Chrzanowska-Wodnicka, M.; Quilliam, L.A.; Anaya, P.; Morris, A.J.; Smyth, S.S. Enhanced proliferation and migration of vascular smooth muscle cells in response to vascular injury under hyperglycemic conditions is controlled by β3 integrin signaling. Int. J. Biochem. Cell Biol. 2010, 42, 965–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokoch, G.M.; Quilliam, L.A.; Bohl, B.P.; Jesaitis, A.J.; Quinn, M.T. Inhibition of Rap1A binding to cytochrome b558 of NADPH oxidase by phosphorylation of Rap1A. Science 1991, 254, 1794–1796. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Wang, H.; Munk, S.; Kwan, J.; Goldberg, H.J.; Fantus, I.G.; Whiteside, C.I. High glucose activates PKC-ζ and NADPH oxidase through autocrine TGF-β1 signaling in mesangial cells. Am. J. Physiol. Physiol. 2008, 295, F1705–F1714. [Google Scholar] [CrossRef]
- Fontayne, A.; Dang, P.M.-C.; Gougerot-Pocidalo, A.M.-A.; El Benna, J. Phosphorylation of p47phoxSites by PKC α, βΙΙ, δ, and ζ: Effect on Binding to p22phoxand on NADPH Oxidase Activation. Biochemistry 2002, 41, 7743–7750. [Google Scholar] [CrossRef]
- Regier, D.S.; Waite, K.A.; Wallin, R.; Mcphail, L.C. A Phosphatidic Acid-activated Protein Kinase and Conventional Protein Kinase C Isoforms Phosphorylate p22 phox, an NADPH Oxidase Component. J. Biol. Chem. 1999, 274, 36601–36608. [Google Scholar] [CrossRef] [Green Version]
- Thallas-Bonke, V.; Coughlan, M.T.; Tan, A.L.; Harcourt, B.E.; Morgan, P.E.; Davies, M.J.; Bach, L.A.; Cooper, M.E.; Forbes, J.M. Targeting the AGE-RAGE axis improves renal function in the context of a healthy diet low in advanced glycation end-product content. Nephrology 2012, 18, 47–56. [Google Scholar] [CrossRef]
- Constien, R.; Forde, A.; Liliensiek, B.; Gröne, H.-J.; Nawroth, P.; Hämmerling, G.; Arnold, B. Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis 2001, 30, 36–44. [Google Scholar] [CrossRef]
- Liliensiek, B.; Weigand, M.A.; Bierhaus, A.; Nicklas, W.; Kasper, M.; Hofer, S.; Plachky, J.; Gröne, H.-J.; Kurschus, F.C.; Schmidt, A.M.; et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J. Clin. Investig. 2004, 113, 1641–1650. [Google Scholar] [CrossRef]
- Brodeur, M.R.; Bouvet, C.; Bouchard, S.; Moreau, S.; Leblond, J.; Deblois, D.; Moreau, P. Reduction of Advanced-Glycation End Products Levels and Inhibition of RAGE Signaling Decreases Rat Vascular Calcification Induced by Diabetes. PLoS ONE 2014, 9, e85922. [Google Scholar] [CrossRef] [Green Version]
- Schwenk, F.; Baron, U.; Rajewsky, K. A cremtransgenic mouse strain for te ubiquitous deletion of Ioxflanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995, 23, 5080–5081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Yan, J.; De, P.; Chang, H.-C.; Yamauchi, A.; Christopherson, K.W.; Paranavitana, N.C.; Peng, X.; Kim, C.; Munugulavadla, V.; et al. Rap1a Null Mice Have Altered Myeloid Cell Functions Suggesting Distinct Roles for the Closely Related Rap1a and 1b Proteins. J. Immunol. 2007, 179, 8322–8331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NIH. Guide for the Care and Use of Laboratory Animals; The National Academies Press: Washington, DC, USA, 2011; ISBN 9780309154000. [Google Scholar]
- Rajan, N.; Habermehl, J.; Coté, M.-F.; Doillon, C.J.; Mantovani, D. Preparation of ready-to-use, storable and reconstituted type I collagen from rat tail tendon for tissue engineering applications. Nat. Protoc. 2006, 1, 2753–2758. [Google Scholar] [CrossRef]
- Stewart, J.A.; Massey, E.P.; Fix, C.; Zhu, J.; Goldsmith, E.C.; Carver, W. Temporal alterations in cardiac fibroblast function following induction of pressure overload. Cell Tissue Res. 2010, 340, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Treiber, N.; Peters, T.; Sindrilaru, A.; Huber, R.; Kohn, M.; Menke, A.; Briviba, K.; Kreppel, F.; Basu, A.; Maity, P.; et al. Overexpression of manganese superoxide dismutase in human dermal fibroblasts enhances the contraction of free floating collagen lattice: Implications for ageing and hyperplastic scar formation. Arch. Dermatol. Res. 2009, 301, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Howard, E.W.; Benton, R.; Ahern-Moore, J.; Tomasek, J.J. Cellular Contraction of Collagen Lattices Is Inhibited by Nonenzymatic Glycation. Exp. Cell Res. 1996, 228, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Welinder, C.; Ekblad, L. Coomassie Staining as Loading Control in Western Blot Analysis. J. Proteome Res. 2011, 10, 1416–1419. [Google Scholar] [CrossRef]
- Nie, X.; Li, C.; Hu, S.; Xue, F.; Kang, Y.J.; Zhang, W. An appropriate loading control for western blot analysis in animal models of myocardial ischemic infarction. Biochem. Biophys. Rep. 2017, 12, 108–113. [Google Scholar] [CrossRef]
- Jeyaraj, S.C.; Unger, N.T.; Chotani, M.A. Rap1 GTPases: An emerging role in the cardiovasculature. Life Sci. 2011, 88, 645–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmedo, I.; Muñoz, C.; Guzmán, N.; Catalán, M.; Vivar, R.; Ayala, P.; Humeres, C.; Aranguiz, P.; Garcia, L.; Velarde, V.; et al. EPAC expression and function in cardiac fibroblasts and myofibroblasts. Toxicol. Appl. Pharmacol. 2013, 272, 414–422. [Google Scholar] [CrossRef]
- Tillmanns, J.; Hoffmann, D.; Habbaba, Y.; Schmitto, J.D.; Sedding, D.G.; Fraccarollo, D.; Galuppo, P.; Bauersachs, J. Fibroblast activation protein alpha expression identifies activated fibroblasts after myocardial infarction. J. Mol. Cell. Cardiol. 2015, 87, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, E.C.; Bradshaw, A.D.; Zile, M.R.; Spinale, F.G. Myocardial fibroblast–matrix interactions and potential therapeutic targets. J. Mol. Cell. Cardiol. 2014, 70, 92–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiura, H.; Liu, X.; Kobayashi, T.; Togo, S.; Ertl, R.F.; Kawasaki, S.; Kamio, K.; Wang, X.Q.; Mao, L.; Shen, L.; et al. Reactive Nitrogen Species Augment Fibroblast-Mediated Collagen Gel Contraction, Mediator Production, and Chemotaxis. Am. J. Respir. Cell Mol. Biol. 2006, 34, 592–599. [Google Scholar] [CrossRef]
- Sorescu, D.; Griendling, K.K. Reactive Oxygen Species, Mitochondria, and NAD(P)H Oxidases in the Development and Progression of Heart Failure. Congest. Heart Fail. 2002, 8, 132–140. [Google Scholar] [CrossRef]
- Rodiño-Janeiro, B.K.; González-Peteiro, M.; Ucieda-Somoza, R.; González-Juanatey, J.R.; Álvarez, E. Glycated albumin, a precursor of advanced glycation end-products, up-regulates NADPH oxidase and enhances oxidative stress in human endothelial cells: Molecular correlate of diabetic vasculopathy. Diabetes/Metab. Res. Rev. 2010, 26, 550–558. [Google Scholar] [CrossRef]
- Lakshmikanthan, S.; Zieba, B.J.; Ge, Z.-D.; Momotani, K.; Zheng, X.; Lund, H.; Artamonov, M.V.; Maas, J.E.; Szabo, A.; Zhang, D.X.; et al. Rap1b in Smooth Muscle and Endothelium Is Required for Maintenance of Vascular Tone and Normal Blood Pressure. Arter. Thromb. Vasc. Biol. 2014, 34, 1486–1494. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Wang, L.; Delguste, F.; Durand, A.; Guilbaud, A.; Rousselin, C.; Schmidt, A.M.; Tessier, F.; Boulanger, E.; Neviere, R. Advanced glycation end products receptor RAGE controls myocardial dysfunction and oxidative stress in high-fat fed mice by sustaining mitochondrial dynamics and autophagy-lysosome pathway. Free Radic. Biol. Med. 2017, 112, 397–410. [Google Scholar] [CrossRef]
- Zieba, B.J.; Artamonov, M.V.; Jin, L.; Momotani, K.; Ho, R.; Franke, A.S.; Neppl, R.L.; Stevenson, A.S.; Khromov, A.S.; Chrzanowska-Wodnicka, M.; et al. The cAMP-responsive Rap1 Guanine Nucleotide Exchange Factor, Epac, Induces Smooth Muscle Relaxation by Down-regulation of RhoA Activity. J. Biol. Chem. 2011, 286, 16681–16692. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Zhang, Q.; Zhao, Y.; Schwarz, B.J.; Stallone, J.N.; Heaps, C.L.; Han, G. Activation of G protein-coupled estrogen receptor 1 induces coronary artery relaxation via Epac/Rap1-mediated inhibition of RhoA/Rho kinase pathway in parallel with PKA. PLoS ONE 2017, 12, e0173085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Hirose, A.; Tanikawa, T.; Mori, H.; Okada, Y.; Tanaka, Y. Advanced glycation end products increase endothelial permeability through the RAGE/Rho signaling pathway. FEBS Lett. 2009, 584, 61–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Liu, H.; Chen, B.; Li, Q.; Huang, X.; Wang, L.; Huang, Q. RhoA/ROCK-dependent moesin phosphorylation regulates AGE-induced endothelial cellular response. Cardiovasc. Diabetol. 2012, 11, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Tsapara, A.; Luthert, P.; Greenwood, J.S.; Hill, C.S.; Matter, K.; Balda, M.S. The RhoA Activator GEF-H1/Lfc Is a Transforming Growth Factor-β Target Gene and Effector That Regulates α-Smooth Muscle Actin Expression and Cell Migration. Mol. Biol. Cell 2010, 21, 860–870. [Google Scholar] [CrossRef] [Green Version]
- Serban, A.I.; Stanca, L.; Geicu, O.I.; Munteanu, M.C.; Dinischiotu, A. RAGE and TGF-β1 Cross-Talk Regulate Extracellular Matrix Turnover and Cytokine Synthesis in AGEs Exposed Fibroblast Cells. PLoS ONE 2016, 11, e0152376. [Google Scholar] [CrossRef]
- Song, J.S.; Kang, C.M.; Park, C.K.; Yoon, H.K.; Lee, S.Y.; Ahn, J.H.; Moon, H.-S. Inhibitory effect of receptor for advanced glycation end products (RAGE) on the TGF-β-induced alveolar epithelial to mesenchymal transition. Exp. Mol. Med. 2011, 43, 517–524. [Google Scholar] [CrossRef] [Green Version]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.A.; Rahmani, A.H.; Aldebasi, Y.H.; Aly, S.M. Biochemical and Pathological Studies on Peroxidases—An Updated Review. Glob. J. Health Sci. 2014, 6, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Zeller, K.S.; Riaz, A.; Sarve, H.; Li, J.; Tengholm, A.; Johansson, S. The Role of Mechanical Force and ROS in Integrin-Dependent Signals. PLoS ONE 2013, 8, e64897. [Google Scholar] [CrossRef] [Green Version]
- MI Peixoto, E.B.; Sevá Pessôa, B.; Kumar Biswas, S.; Lopes de Faria, J. Antioxidant SOD Mimetic Prevents NADPH Oxidase-Induced Oxidative Stress and Renal Damage in the Early Stage of Experimental Diabetes and Hypertension. Artic. Am. J. Nephrol. 2008, 29, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Brennan, L.A.; Steinhorn, R.H.; Wedgwood, S.; Mata-Greenwood, E.; Roark, E.A.; Russell, J.A.; Black, S.M. Increased Superoxide Generation Is Associated with Pulmonary Hypertension in Fetal Lambs. Circ. Res. 2003, 92, 683–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenk, J.; Brenneisen, P.; Wlaschek, M.; Poswig, A.; Briviba, K.; Oberley, T.D.; Scharffetter-Kochanek, K. Stable Overexpression of Manganese Superoxide Dismutase in Mitochondria Identifies Hydrogen Peroxide as a Major Oxidant in the AP-1-mediated Induction of Matrix-degrading Metalloprotease-1. J. Biol. Chem. 1999, 274, 25869–25876. [Google Scholar] [CrossRef] [PubMed]
- Sadi, G.; Güray, T. Gene expressions of Mn-SOD and GPx-1 in streptozotocin-induced diabetes: Effect of antioxidants. Mol. Cell. Biochem. 2009, 327, 127–134. [Google Scholar] [CrossRef]
- Muratsu-Ikeda, S.; Nangaku, M.; Ikeda, Y.; Tanaka, T.; Wada, T.; Inagi, R. Downregulation of miR-205 Modulates Cell Susceptibility to Oxidative and Endoplasmic Reticulum Stresses in Renal Tubular Cells. PLoS ONE 2012, 7, e41462. [Google Scholar] [CrossRef] [Green Version]
- Kojima, I.; Tanaka, T.; Inagi, R.; Kato, H.; Yamashita, T.; Sakiyama, A.; Ohneda, O.; Takeda, N.; Sata, M.; Miyata, T.; et al. Protective Role of Hypoxia-Inducible Factor-2α against Ischemic Damage and Oxidative Stress in the Kidney. J. Am. Soc. Nephrol. 2007, 18, 1218–1226. [Google Scholar] [CrossRef]
- Shimizu, M.; Umeda, K.; Sugihara, N.; Yoshio, H.; Ino, H.; Takeda, R.; Nakanishi, I. Collagen remodelling in myocardia of patients with diabetes Second Department of Internal Medicine. Clin. Pathol. 1993, 46, 32–36. [Google Scholar] [CrossRef] [Green Version]
- Monnier, V.M.; Mustata, G.T.; Biemel, K.L.; Reihl, O.; Lederer, M.O.; Zhenyu, D.; Sell, D.R. Cross-Linking of the Extracellular Matrix by the Maillard Reaction in Aging and Diabetes: An Update on “a Puzzle Nearing Resolution”. Ann. N. Y. Acad. Sci. 2005, 1043, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Chandran, P.L.; Paik, D.C.; Holmes, J.W. Structural Mechanism for Alteration of Collagen Gel Mechanics by Glutaraldehyde Crosslinking. Connect. Tissue Res. 2012, 53, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Sajithlal, G.; Chithra, P.; Chandrakasan, G. Advanced glycation end products induce crosslinking of collagen in vitro. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1998, 1407, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Taddei, M.L.; Parri, M.; Mello, T.; Catalano, A.; Levine, A.D.; Raugei, G.; Ramponi, G.; Chiarugi, P. Integrin-Mediated Cell Adhesion and Spreading Engage Different Sources of Reactive Oxygen Species. Antioxid. Redox Signal. 2007, 9, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Sangrar, W.; Gao, Y.; Scott, M.; Truesdell, P.; Greer, P.A. Fer-Mediated Cortactin Phosphorylation Is Associated with Efficient Fibroblast Migration and Is Dependent on Reactive Oxygen Species Generation during Integrin-Mediated Cell Adhesion. Mol. Cell. Biol. 2007, 27, 6140–6152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, E.; Werb, Z. Integrins engage mitochondrial function for signal transduction by a mechanism dependent on Rho GTPases. J. Cell Biol. 2002, 158, 357–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiaschi, T.; Magherini, F.; Gamberi, T.; Lucchese, G.; Faggian, G.; Modesti, A.; Modesti, P.A. Hyperglycemia and angiotensin II cooperate to enhance collagen I deposition by cardiac fibroblasts through a ROS-STAT3-dependent mechanism. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1843, 2603–2610. [Google Scholar] [CrossRef] [Green Version]
- Gratzer, P.F.; Santerre, J.P.; Lee, J.M. The effect of chemical modification of amino acid side-chains on collagen degradation by enzymes. J. Biomed. Mater. Res. Part B Appl. Biomater. 2007, 81, 1–11. [Google Scholar] [CrossRef]
- Kunkemoeller, B.; Kyriakides, T.R. Redox Signaling in Diabetic Wound Healing Regulates Extracellular Matrix Deposition. Antioxid. Redox Signal. 2017, 27, 823–838. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Heart Weight | Blood Glucose (mg/dL) | |
|---|---|---|
| Body Weight | ||
| Non-Diabetic (n = 25) | 0.0038 ± 7.9128 × 10−5 | 190.6 ± 4.323 |
| Diabetic (n = 18) | 0.0017 ± 6.269 × 10−5 **** | 546.1 ± 24.07 **** |
| Rap1a KO (n = 22) | 0.0041 ± 11.18 × 10−5 * | 180.0 ± 8.334 |
| Non-Diabetic RKO (n = 20) | 0.0037 ± 6.256 × 10−5 | 204.2 ± 4.454 |
| Diabetic RKO (n = 13) | 0.0022 ± 3.534 × 10−5 **** | 456.4 ± 29.24 **** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burr, S.D.; Stewart, J.A., Jr. Rap1a Regulates Cardiac Fibroblast Contraction of 3D Diabetic Collagen Matrices by Increased Activation of the AGE/RAGE Cascade. Cells 2021, 10, 1286. https://doi.org/10.3390/cells10061286
Burr SD, Stewart JA Jr. Rap1a Regulates Cardiac Fibroblast Contraction of 3D Diabetic Collagen Matrices by Increased Activation of the AGE/RAGE Cascade. Cells. 2021; 10(6):1286. https://doi.org/10.3390/cells10061286
Chicago/Turabian StyleBurr, Stephanie D., and James A. Stewart, Jr. 2021. "Rap1a Regulates Cardiac Fibroblast Contraction of 3D Diabetic Collagen Matrices by Increased Activation of the AGE/RAGE Cascade" Cells 10, no. 6: 1286. https://doi.org/10.3390/cells10061286