
Sphingosine 1 Phosphate (S1P) Receptor 1 Is Decreased in Human Lung Microvascular Endothelial Cells of Smokers and Mediates S1P Effect on Autophagy

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Isolation and Culture
2.2. Animal Studies
2.3. Protein Isolation and Western Blotting
2.4. RNA Isolation, Reverse Transcription, and Real-Time Quantitative Polymerase Chain Reaction (RTqPCR)
2.5. Immunohistochemistry
2.6. Immunofluorescence
2.7. siRNA Transfection
2.8. Quantitative Sphingolipid Determination
2.9. Statistical Analysis
3. Results
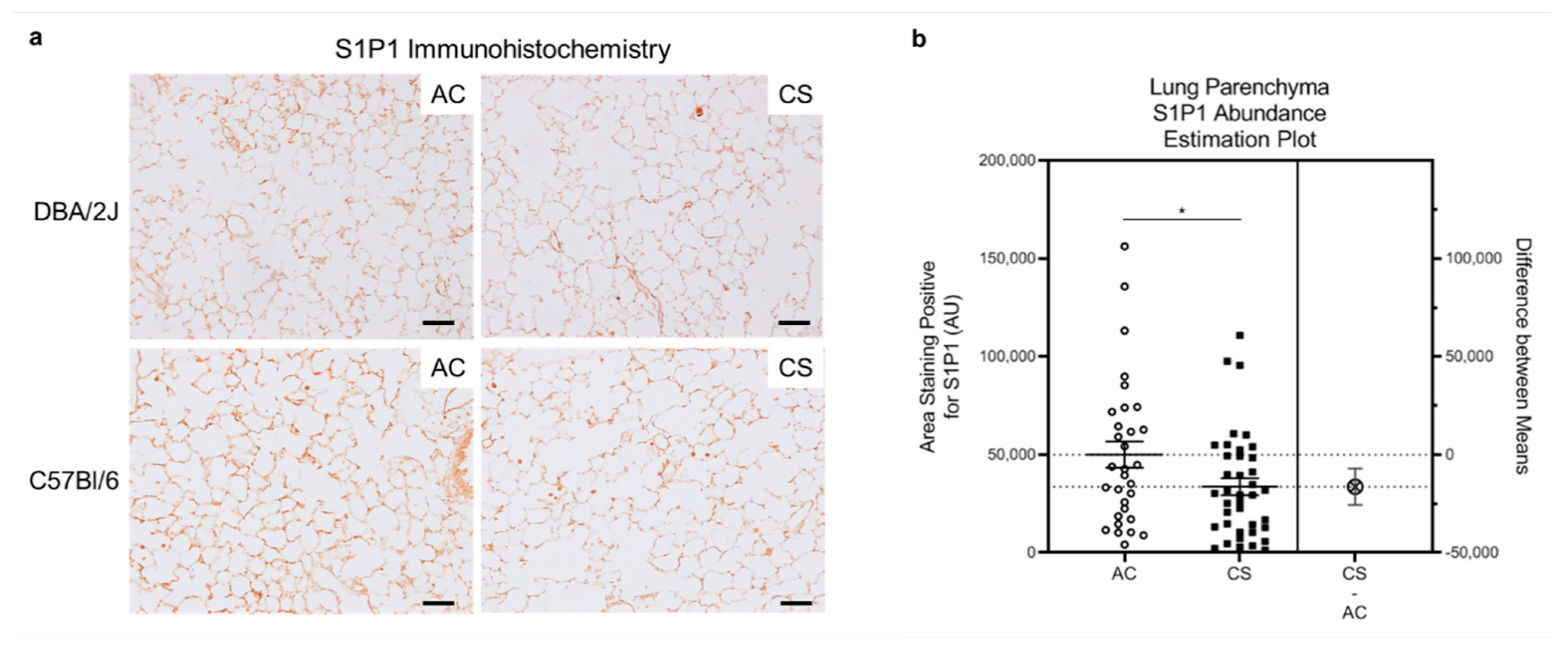
3.1. Effect of Smoking on S1P1 Expression and Abundance in HLMVECs
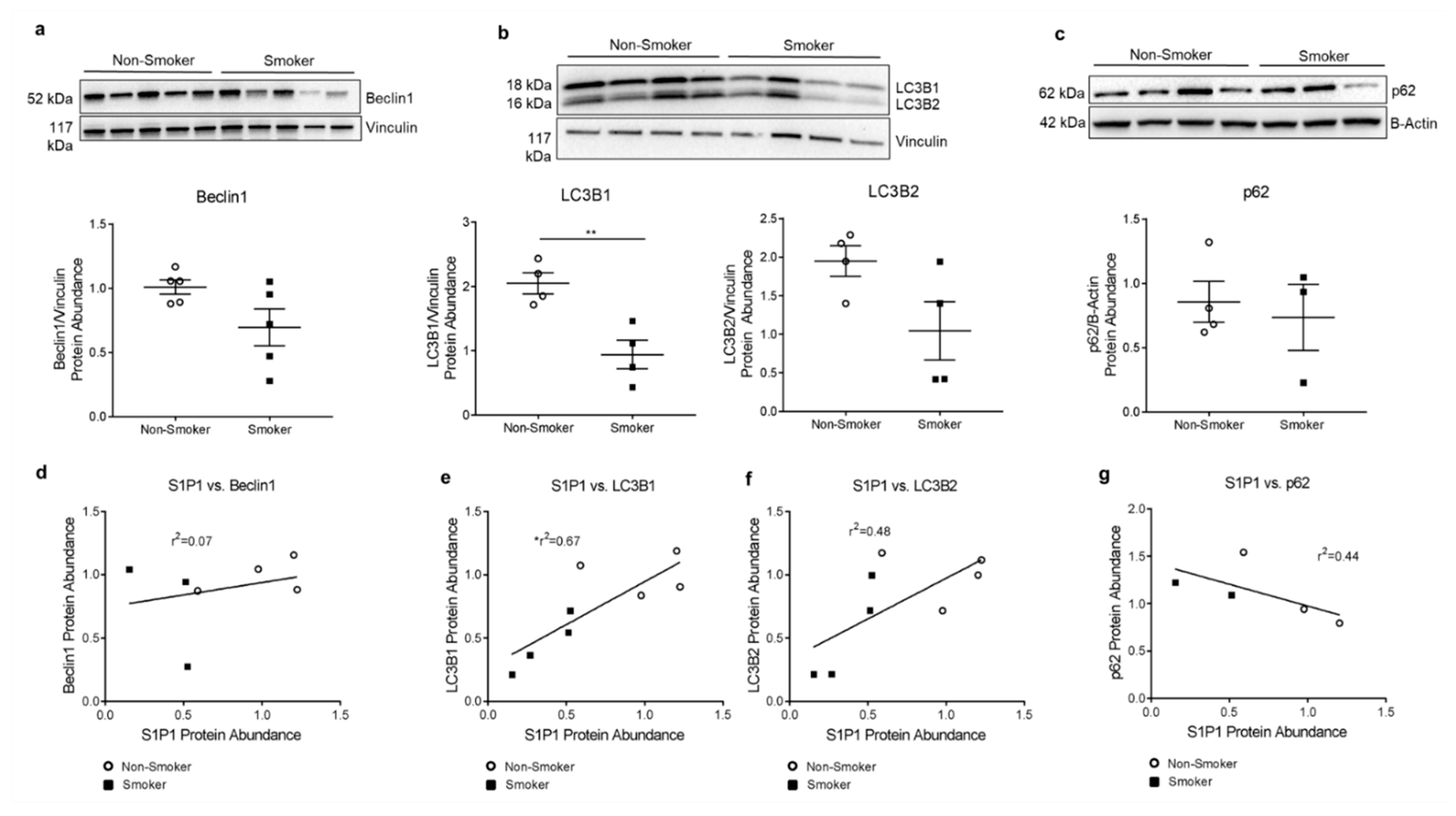
3.2. S1P1 Role in HLMVEC Autophagy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- National Center for Chronic Disease Prevention and Health Promotion (US) Office on Smoking and Health. The Health Consequences of Smoking-50 Years of Progress: A Report of the Surgeon General; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2014.
- Tuder, R.M.; Petrache, I. Pathogenesis of chronic obstructive pulmonary disease. J. Clin. Investig. 2012, 122, 2749–2755. [Google Scholar] [CrossRef]
- Cornelius, M.E.; Wang, T.W.; Jamal, A.; Loretan, C.G.; Neff, L.J. Tobacco Product Use Among Adults—United States, 2019. Morb. Mortal. Wkly. Rep. 2020, 69, 1736–1742. [Google Scholar] [CrossRef]
- Suzuki, Y.; Yoshimura, K.; Uto, T.; Sato, J.; Imokawa, S.; Suda, T. Morphological changes in small pulmonary vessels are associated with severe acute exacerbation in chronic obstructive pulmonary disease. Int. J. Chronic Obstr. Pulm. Dis. 2016, 11, 1435–1445. [Google Scholar] [CrossRef] [Green Version]
- King, J.; Hamil, T.; Creighton, J.; Wu, S.; Bhat, P.; McDonald, F.; Stevens, T. Structural and functional characteristics of lung macro- and microvascular endothelial cell phenotypes. Microvasc. Res. 2004, 67, 139–151. [Google Scholar] [CrossRef]
- Garcia, J.G. Concepts in microvascular endothelial barrier regulation in health and disease. Microvasc. Res. 2009, 77, 1–3. [Google Scholar] [CrossRef]
- Boucher, R.C.; Johnson, J.; Inoue, S.; Hulbert, W.; Hogg, J.C. The effect of cigarette smoke on the permeability of guinea pig airways. Lab. Investig. 1980, 43, 94–100. [Google Scholar]
- Low, B.; Liang, M.; Fu, J. p38 mitogen-activated protein kinase mediates sidestream cigarette smoke-induced endothelial permeability. J. Pharmacol. Sci. 2007, 104, 225–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serikov, V.B.; Leutenegger, C.; Krutilina, R.; Kropotov, A.; Pleskach, N.; Suh, J.H.; Tomilin, N.V. Cigarette smoke extract inhibits expression of peroxiredoxin V and increases airway epithelial permeability. Inhal. Toxicol. 2006, 18, 79–92. [Google Scholar] [CrossRef]
- Petrache, I.; Natarajan, V.; Zhen, L.; Medler, T.R.; Richter, A.; Cho, C.; Hubbard, W.C.; Berdyshev, E.V.; Tuder, R.M. Ceramide Upregulation Causes Pulmonary Cell Apoptosis and Emphysema. Nat. Med. 2005, 11, 491–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diab, K.J.; Adamowicz, J.J.; Kamocki, K.; Rush, N.I.; Garrison, J.; Gu, Y.; Schweitzer, K.S.; Skobeleva, A.; Rajashekhar, G.; Hubbard, W.C.; et al. Stimulation of sphingosine 1-phosphate signaling as an alveolar cell survival strategy in emphysema. Am. J. Respir. Crit. Care Med. 2010, 181, 344–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrusca, D.N.; Van Demark, M.; Gu, Y.; Justice, M.J.; Rogozea, A.; Hubbard, W.C.; Petrache, I. Smoking exposure induces human lung endothelial cell adaptation to apoptotic stress. Am. J. Respir. Cell Mol. Biol. 2014, 50, 513–525. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Zhang, X.-J.; Chen, S.; Huang, K.-X.; Le, W.-D. Why should autophagic flux be assessed? Acta Pharmacol. Sin. 2013, 34, 595–599. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Petrache, I.; Birukova, A.; Ramirez, S.I.; Garcia, J.G.N.; Verin, A.D. The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am. J. Respir. Cell Mol. Biol. 2003, 28, 574–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Cartier, A.; Hla, T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019, 366, eaar5551. [Google Scholar] [CrossRef] [PubMed]
- Takuwa, Y.; Takuwa, N.; Sugimoto, N. The Edg family G protein-coupled receptors for lysophospholipids: Their signaling properties and biological activities. J. Biochem. 2002, 131, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Comhair, S.A.; Xu, W.; Mavrakis, L.; Aldred, M.A.; Asosingh, K.; Erzurum, S.C. Human Primary Lung Endothelial Cells in Culture. Am. J. Respir. Cell Mol. Biol. 2012, 46, 723–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuder, R.M.; Marecki, J.C.; Richter, A.; Fijalkowska, I.; Flores, S. Pathology of Pulmonary Hypertension. Clin. Chest Med. 2007, 28, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, K.S.; Johnstone, B.H.; Garrison, J.; Rush, N.I.; Cooper, S.; Traktuev, D.O.; Feng, D.; Adamowicz, J.J.; Van Demark, M.; Fisher, A.J.; et al. Adipose stem cell treatment in mice attenuates lung and systemic injury induced by cigarette smoking. Am. J. Respir. Crit. Care Med. 2011, 183, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Cruickshank-Quinn, C.I.; Mahaffey, S.; Justice, M.J.; Hughes, G.; Armstrong, M.; Bowler, R.P.; Reisdorph, R.; Petrache, I.; Reisdorph, N. Transient and persistent metabolomic changes in plasma following chronic cigarette smoke exposure in a mouse model. PLoS ONE 2014, 9, e101855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clauss, M.; Voswinckel, R.; Rajashekhar, G.; Sigua, N.L.; Fehrenbach, H.; Rush, N.I.; Schweitzer, K.S.; Yildirim, A.Ö.; Kamocki, K.; Fisher, A.J.; et al. Lung endothelial monocyte-activating protein 2 is a mediator of cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2011, 121, 2470–2479. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Justice, M.J.; Bronova, I.; Schweitzer, K.S.; Poirier, C.; Blum, J.S.; Berdyshev, E.V.; Petrache, I. Inhibition of acid sphingomyelinase disrupts LYNUS signaling and triggers autophagy. J. Lipid Res. 2018, 59, 596–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoura, A.; Hla, T. Regulation of vascular physiology and pathology by the S1P2 receptor subtype. Cardiovasc. Res. 2008, 82, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Reeves, P.M.; Kang, Y.-L.; Kirchhausen, T. Endocytosis of Ligand-Activated Sphingosine 1-Phosphate Receptor 1 Mediated by the Clathrin-Pathway. Traffic 2016, 17, 40–52. [Google Scholar] [CrossRef]
- Martínez-Morales, J.C.; Romero-Ávila, M.T.; Reyes-Cruz, G.; García-Sáinz, J.A. S1P1 receptor phosphorylation, internalization, and interaction with Rab proteins: Effects of sphingosine 1-phosphate, FTY720-P, phorbol esters, and paroxetine. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, K.; Lee, Y.-M.; Michaud, J.; Thangada, S.; Ai, Y.; Bonkovsky, H.L.; Parikh, N.S.; Habrukowich, C.; Hla, T. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ. Res. 2008, 102, 669–676. [Google Scholar] [CrossRef] [Green Version]
- Camp, S.M.; Chiang, E.T.; Sun, C.; Usatyuk, P.V.; Bittman, R.; Natarajan, V.; Garcia, J.G.N.; Dudek, S.M. Pulmonary Endothelial Cell Barrier Enhancement by Novel FTY720 Analogs: Methoxy-FTY720, Fluoro-FTY720, and beta-Glucuronide-FTY720. Chem. Phys. Lipids 2016, 194, 85–93. [Google Scholar] [CrossRef]
- Chavez, A.; Schmidt, T.T.; Yazbeck, P.; Rajput, C.; Desai, B.; Sukriti, S.; Giantsos-Adams, K.; Knezevic, N.; Malik, A.B.; Mehta, D. S1PR1 Tyr143 phosphorylation downregulates endothelial cell surface S1PR1 expression and responsiveness. J. Cell Sci. 2015, 128, 878–887. [Google Scholar] [CrossRef] [Green Version]
- Oo, M.L.; Thangada, S.; Wu, M.-T.; Liu, C.H.; Macdonald, T.L.; Lynch, K.R.; Lin, C.-Y.; Hla, T. Immunosuppressive and anti-angiogenic sphingosine 1-phosphate receptor-1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. J. Biol. Chem. 2007, 282, 9082–9089. [Google Scholar] [CrossRef] [Green Version]
- Watterson, K.R.; Johnston, E.; Chalmers, C.; Pronin, A.; Cook, S.J.; Benovic, J.L.; Palmer, T.M. Dual regulation of EDG1/S1P(1) receptor phosphorylation and internalization by protein kinase C and G-protein-coupled receptor kinase 2. J. Biol. Chem. 2002, 277, 5767–5777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, J.; Erwin, P.A.; Dantas, A.P.V.; Chen, H.; Michel, T. VEGF induces S1P1 receptors in endothelial cells: Implications for cross-talk between sphingolipid and growth factor receptors. Proc. Natl. Acad. Sci. USA 2003, 100, 10664–10669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josipovic, I.; Pflüger, B.; Fork, C.; Vasconez, A.E.; Oo, J.A.; Hitzel, J.; Seredinski, S.; Gamen, E.; Zu Heringdorf, D.M.; Chen, W.; et al. Long noncoding RNA LISPR1 is required for S1P signaling and endothelial cell function. J. Mol. Cell. Cardiol. 2018, 116, 57–68. [Google Scholar] [CrossRef]
- Carlson, C.M.; Endrizzi, B.T.; Wu, J.; Ding, X.; Weinreich, M.A.; Walsh, E.R.; Wani, M.A.; Lingrel, J.B.; Hogquist, K.A.; Jameson, S.C. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature 2006, 442, 299–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, J.S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.G.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate: Dual messenger functions. FEBS Lett. 2002, 531, 54–57. [Google Scholar] [CrossRef]
- Koike, K.; Berdyshev, E.V.; Mikosz, A.M.; Bronova, I.A.; Bronoff, A.S.; Jung, J.P.; Beatman, E.L.; Ni, K.; Cao, D.; Scruggs, A.K.; et al. Role of Glucosylceramide in Lung Endothelial Cell Fate and Emphysema. Am. J. Respir. Crit. Care Med. 2019, 200, 1113–1125. [Google Scholar] [CrossRef]
- Chen, Q.; Rehman, J.; Chan, M.; Fu, P.; Dudek, S.M.; Natarajan, V.; Malik, A.B.; Liu, Y. Angiocrine Sphingosine-1-Phosphate Activation of S1PR2-YAP Signaling Axis in Alveolar Type II Cells Is Essential for Lung Repair. Cell Rep. 2020, 31, 107828. [Google Scholar] [CrossRef]
- Schweitzer, K.S.; Chen, S.X.; Law, S.; Van Demark, M.; Poirier, C.; Justice, M.J.; Hubbard, W.C.; Kim, E.S.; Lai, X.; Wang, M.; et al. Endothelial disruptive proinflammatory effects of nicotine and e-cigarette vapor exposures. Am. J. Physiol. Cell. Mol. Physiol. 2015, 309, L175–L187. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goel, K.; Beatman, E.L.; Egersdorf, N.; Scruggs, A.; Cao, D.; Berdyshev, E.V.; Schweitzer, K.S.; Petrache, I. Sphingosine 1 Phosphate (S1P) Receptor 1 Is Decreased in Human Lung Microvascular Endothelial Cells of Smokers and Mediates S1P Effect on Autophagy. Cells 2021, 10, 1200. https://doi.org/10.3390/cells10051200
Goel K, Beatman EL, Egersdorf N, Scruggs A, Cao D, Berdyshev EV, Schweitzer KS, Petrache I. Sphingosine 1 Phosphate (S1P) Receptor 1 Is Decreased in Human Lung Microvascular Endothelial Cells of Smokers and Mediates S1P Effect on Autophagy. Cells. 2021; 10(5):1200. https://doi.org/10.3390/cells10051200
Chicago/Turabian StyleGoel, Khushboo, Erica L. Beatman, Nicholas Egersdorf, April Scruggs, Danting Cao, Evgeny V. Berdyshev, Kelly S. Schweitzer, and Irina Petrache. 2021. "Sphingosine 1 Phosphate (S1P) Receptor 1 Is Decreased in Human Lung Microvascular Endothelial Cells of Smokers and Mediates S1P Effect on Autophagy" Cells 10, no. 5: 1200. https://doi.org/10.3390/cells10051200