
Identification of Putative Virulence Genes by DNA Methylation Studies in the Cereal Pathogen Fusarium graminearum

 , , , , and
, , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Fungal Strain and Subculturing
2.2. Virulence Assays
2.2.1. Crown Rot Assay
2.2.2. Fusarium Head Blight Assay
2.3. In Vitro Growth Rate Assay
2.4. In Vitro Conidial Production
2.5. Determination of Secondary Metabolites Biosynthesized In Vitro by F. graminearum Subcultures
2.5.1. F. graminearum Subcultures Preparation
2.5.2. Extraction and Analysis of Secondary Metabolites
2.6. DNA Extraction
2.7. DNA Methylation Analysis
Synteny Block and Statistical Analysis
3. Results
3.1. Subculturing Reduces Fungal Virulence, but Passaging Can Rescue These Defects
3.2. Subculturing Affects Conidial Production but Not an In Vitro Growth Rate
3.3. Subculturing Does Not Affect In Vitro Secondary Metabolite Production
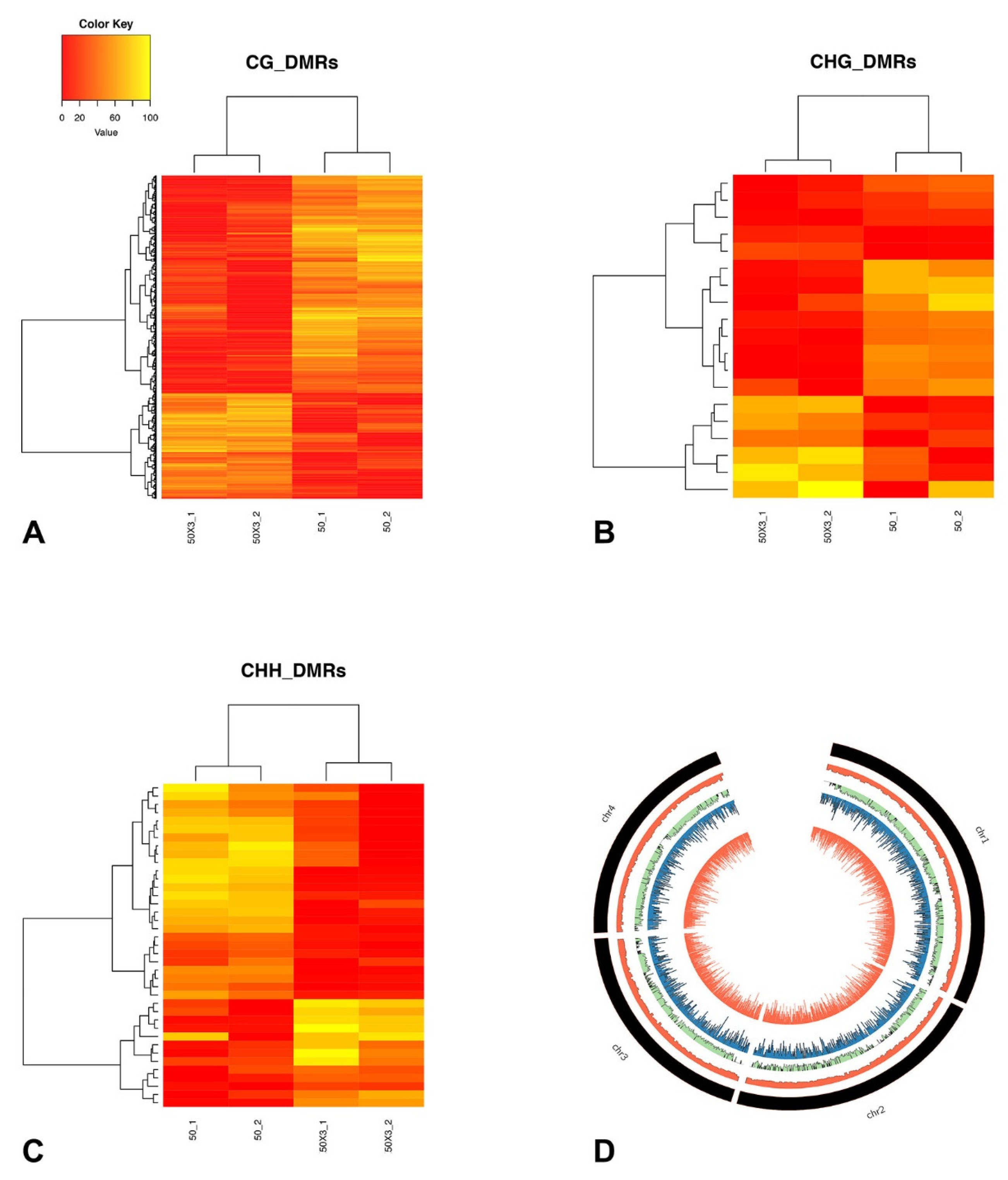
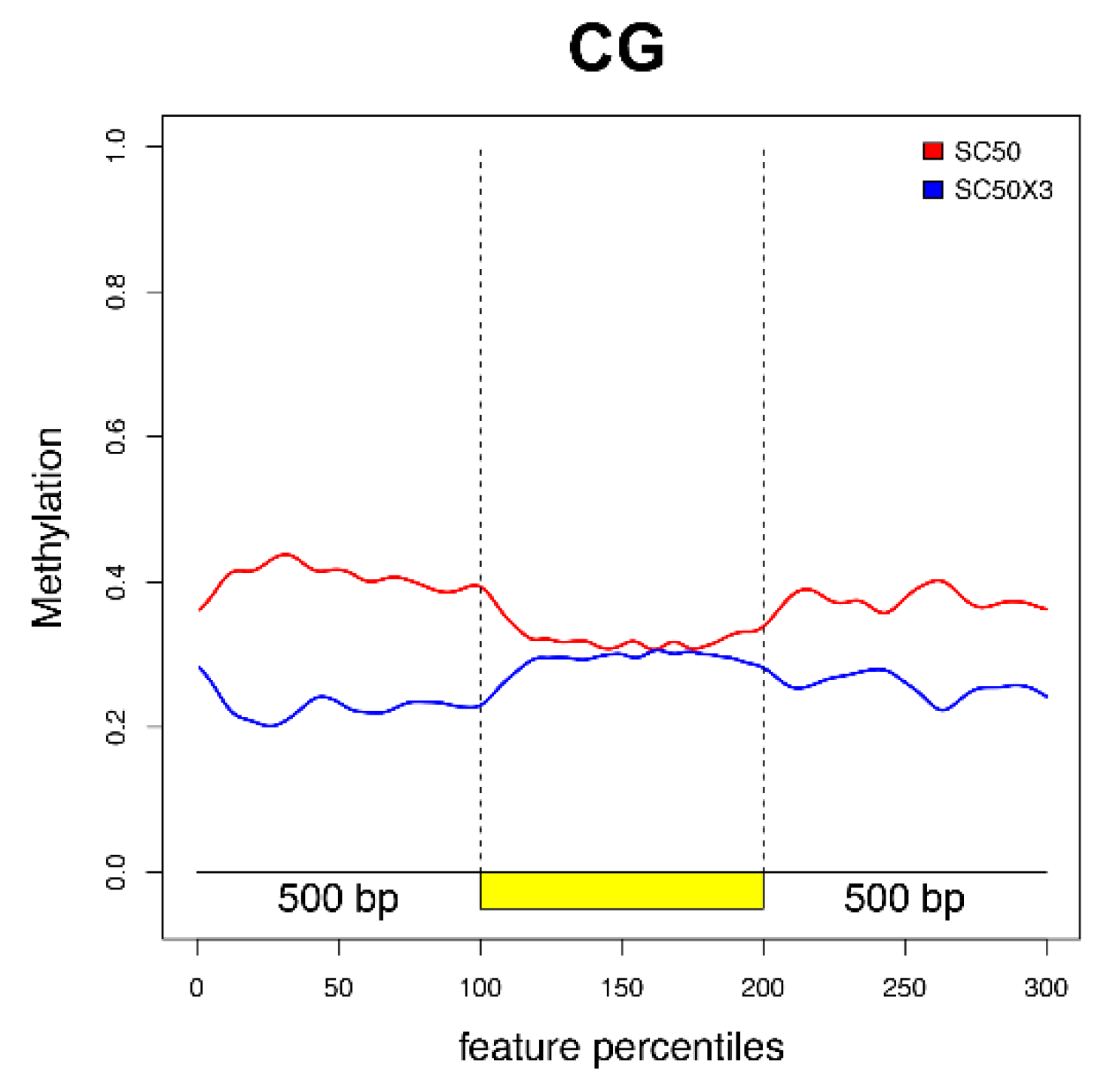
3.4. DNA Methylation Analysis
3.4.1. Identification of Differentially Methylated Positions and Differentially Methylated Regions
3.4.2. Differentially Methylated Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- O’Donnell, K.; Rooney, A.P.; Proctor, R.H.; Brown, D.W.; McComick, S.P.; Ward, T.J.; Frandsen, R.J.N.; Lysoe, E.; Rehner, S.A.; Aoki, T.; et al. Phylogenetic analyses of RPB1 and RPB2 support a middle Cretaceous origin for a clade comprising all agriculturally and medically important fusaria. Fungal Genet. Biol. 2013, 52, 20–31. [Google Scholar] [CrossRef]
- Goswami, R.S.; Kistler, H.C. Heading for disaster: Fusarium graminearum on cereal crops. Mol. Plant Pathol. 2004, 5, 515–525. [Google Scholar] [CrossRef]
- Kazan, K.; Gardiner, D.M.; Manners, J.M. On the trace of a cereal killer: Recent advances in Fusarium graminearum pathogenomics and host resistance. Mol. Plant Pathol. 2012, 13, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Mielniczuk, E.; Skwarylo-Bednarz, B. Fusarium head blight, mycotoxins and strategies for their reduction. Agronomy 2020, 10, 509. [Google Scholar] [CrossRef] [Green Version]
- Geraldo, M.R.F.; Tessmann, D.J.; Kemmelmeier, C. Production of mycotoxins by Fusarium graminearum isolated from small cereal (wheat, triticale and barley) affected with scab disease on Southern Brazil. Braz. J. Microbiol. 2006, 37, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Ward, T.J.; Bielawski, J.P.; Kistler, H.C.; Sullivan, E.; O’Donnel, K. Ancestral polymorphism and adaptive evolution in the trichothecene mycotoxin gene cluster of phytopathogenic Fusarium. Proc. Natl. Acad. Sci. USA 2002, 99, 9278–9283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feil, R.; Fraga, M.F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Causevic, A.; Delaunay, A.; Ounnar, S.; Righezza, M.; Delmotte, F.; Brignolas, F.; Hagege, D.; Maury, S. DNA methylating and demethylating treatments modify phenotype and cell wall differentiation state in sugar beet cell lines. Plant Physiol. Biochem. 2005, 43, 681–691. [Google Scholar] [CrossRef]
- Cao, D.; Gao, X.; Liu, J.; Wang, X.; Geng, S.; Yang, C.; Liu, B.; Shi, D. Root-specific DNA methylation in Chloris virgata, a natural alkaline-resistant halophyte, in response to salt and alkaline stresses. Plant Mol. Biol. Rep. 2012, 30, 1102–1109. [Google Scholar] [CrossRef]
- Angers, B.; Castonguay, E.; Massicotte, R. Environmentally induced phenotypes and DNA methylation: How to deal with unpredictable conditions until the next generation and after. Mol. Ecol. 2010, 19, 1283–1295. [Google Scholar] [CrossRef]
- Lu, Y.; Rong, T.; Cao, M. Analysis of DNA methylation in different maize tissues. J. Genet. Genom. 2008, 35, 41–48. [Google Scholar] [CrossRef]
- Lizal, P.; Relichova, J. The effect of day length, vernalization and DNA demethylation on the flowering time in Arabidopsis thaliana. Physiol. Plant. 2001, 113, 121–127. [Google Scholar] [CrossRef]
- Seidl, M.F. Adenine N6-methylation in diverse fungi. Nat. Genet. 2017, 49, 823–824. [Google Scholar] [CrossRef] [PubMed]
- Mondo, S.J.; Dannebaum, R.O.; Kuo, R.C.; Louie, K.B.; Bewick, A.J.; Labutti, K.; Haridas, S.; Keu, A.; Salamov, A.; Ahrendt, S.R.; et al. Widespread adenine N6- methylation of active genes in fungi. Nat. Genet. 2017, 49, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Lang, Z.; Wang, Y.; Tang, K.; Tang, D.; Datsenka, T.; Cheng, J.; Zhang, J.; Handa, A.K.; Zhu, J.K. Critical roles of DNA demethylation in the activation of ripening-induced genes and inhibition of ripening-repressed genes in tomato fruit. Proc. Natl. Acad. Sci. USA 2017, 114, E4511–E4519. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.L.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cells 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [Green Version]
- Paszkowski, J.; Whitham, S.A. Gene silencing and DNA methylation processes. Curr. Opin. Plant Biol. 2001, 4, 123–129. [Google Scholar] [CrossRef]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- He, C.; Zhang, Z.; Li, B.; Tian, S. The pattern and function of DNA methylation in fungal plant pathogens. Microorganisms 2020, 8, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitag, M.; Williams, R.L.; Kothe, G.O.; Selker, E.U. A cytosine methyltransferase homologue is essential for repeat-induced point mutation in Neurospora crassa. Proc. Natl. Acad. Sci. USA 2002, 99, 8802–8807. [Google Scholar] [CrossRef] [Green Version]
- Malagnac, F.; Wendel, B.; Goyon, C.; Faugeron, G.; Zickler, D.; Rossignol, J.L.; Noyer-Weidner, M.; Vollmayr, P.; Trautner, T.A.; Walter, J. A gene essential for de novo methylation and development in Acobolus reveals a novel type of eukaryotic DNA methyltransferase structure. Cell 1997, 91, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Bewick, A.J.; Hofmeister, B.T.; Powers, R.A.; Mondo, S.J.; Grigoriev, I.V.; James, T.Y.; Stajich, J.E.; Schmitz, R.J. Diversity of cytosine methylation across the fungal tree life. Nat. Ecol. Evol. 2019, 3, 479–490. [Google Scholar] [CrossRef]
- Freitag, M.; Hickey, P.C.; Khlafallah, T.K.; Read, N.D.; Selker, E.U. HP1 is essential for DNA methylation in Neurospora. Mol. Cell 2004, 13, 427–434. [Google Scholar] [CrossRef]
- Tamaru, H.; Selker, E.U. Synthesis of signals for de novo DNA methylation I Neurospora crassa. Mol. Cell Biol. 2003, 23, 2379–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouzminova, E.; Selker, E.U. dim-2 encode a DNA methyltransferase responsible for all known cytosine methylation in Neurospora. EMBO J. 2001, 20, 4309–4323. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.J.; van der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.J.; Di Pietro, A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B.; et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 464, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Pomraning, K.R.; Connolly, L.R.; Whalen, J.P.; Smith, K.M.; Freitag, M. Repeat-induced point mutation, DNA methylation and heterochromatin in Gibberella zeae (Anamorph: Fusarium graminearum. In Fusarium Genomics and Molecular Cellular Biology; Brown, D., Proctor, R.H., Eds.; Horizon Scientific Press: Norwich, UK, 2013; p. 93. [Google Scholar]
- Gijezen, M.; Ishmael, C.; Shrestha, D. Epigenetic control of effectors in plant pathogens. Front. Plant Sci. 2014, 5, 638. [Google Scholar]
- Kim, D.H. Induced change in DNA methylation of Fusarium oxysporum f. sp. niveum due to successive transfer. J. Biochem. Mol. Biol. 1997, 30, 216–221. [Google Scholar]
- Covarelli, L.; Beccari, G.; Prodi, A.; Generotti, S.; Etruschi, F.; Juan, C.; Ferrer, E.; Mañes, J. Fusarium species, chemotype characterization and trichothecene contamination of durum and soft wheat in an area of central Italy. J. Sci. Food Agric. 2015, 95, 540–551. [Google Scholar] [CrossRef]
- Simpson, D.R.; Rezanoor, H.N.; Parry, D.W.; Nicholson, P. Evidence for differential host preference in Microdochium nivale var. majus, M. nivale var. nivale. Plant Pathol. 2000, 49, 261–268. [Google Scholar]
- Beccari, G.; Covarelli, L.; Nicholson, P. Infection processes and soft wheat response to root rot and crown rot caused by Fusarium culmorum. Plant Pathol. 2011, 60, 671–684. [Google Scholar] [CrossRef]
- Covarelli, L.; Beccari, G.; Steed, A.; Nicholson, P. Colonization of soft wheat following infection of the stem base by Fusarium culmorum and translocation of deoxynivalenol to the head. Plant Pathol. 2012, 61, 1121–1129. [Google Scholar] [CrossRef]
- Brunner, K.; Lichtenauer, A.M.; Kratochwill, K.; Delic, M.; Mach, R.L. Xyr1 regulates xylanase but not cellulase formation in the head blight fungus Fusarium graminearum. Curr. Genet. 2007, 52, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Sulyok, M.; Stadler, D.; Steiner, D.; Krska, R. Validation of an LC-MS/MS-based diluted-and-shoot approach for the quantification of >500 mycotoxins and other secondary metabolites in food crops: Challenges and solutions. Anal. Bioanal. Chem. 2020, 412, 2607–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marconi, G.; Capomaccio, S.; Comino, C.; Acquadro, A.; Portis, E.; Porceddu, A.; Albertini, E. Methylation content sensitive enzyme ddRAD (MCSeEd): A reference-free, whole genome profiling system to address cytosine/adenine methylation changes. Sci. Rep. 2019, 9, 14864. [Google Scholar] [CrossRef] [Green Version]
- Di Marsico, M.; Cerruti, E.; Comino, C.; Porceddu, A.; Acquadro, A.; Capomaccio, S.; Marconi, G.; Albertini, E. MCSeEd (Methylation Context Sensitive Enzyme ddRAD): A new method to analyze DNA methylation. In Plant Epigenetics and Epigenomics. Methods in Molecular Biology; Spillane, C., McKeown, P., Eds.; Humana: New York, NY, USA, 2020; Volume 2093, pp. 47–64. [Google Scholar]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Melnick, A.; Mason, C.E. MethylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [Green Version]
- Onofri, A.; Pannacci, E. Spreadsheet tools for biometry classes in crop science programmes. CBCS 2014, 9, 43–53. [Google Scholar]
- Cuomo, C.A.; Guldener, U.; Xu, J.R.; Trail, F.; Turgeon, B.G.; Di Pietro, A.; Walton, J.D.; Baker, S.E.; Rep, M.; Adam, G.; et al. The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 2007, 317, 1400–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Waalwijk, C.; de Wit, P.J.G.M.; Tang, D.; van der Lee, T. RNA-Seq analysis reveals new gene models and alternative splicing in the fungal pathogen Fusarium graminearum. BMC Genom. 2014, 14, 21. [Google Scholar] [CrossRef] [Green Version]
- Li, B.Q.; Zong, Y.Y.; Du, Z.L.; Shang, Y.J.; Chen, Y.; Zhang, Z.Q.; Qin, G.Z.; Zhao, W.M.; Tian, S.P. Genomic characterization reveals insights into patulin biosynthesis and pathogenicity in Penicillium species. Mol. Plant Microbe Interact. 2015, 28, 635–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malcom, G.M.; Kuldau, G.A.; Gugino, B.K.; Jimenez-Gasco, M.D.M. Hidden host plant associations of soilborne fungal pathogens: An ecological perspective. Phytopathology 2013, 103, 538–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, R.; Kan, J.A.V.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Pietro, A.D.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [Green Version]
- Presti, L.L.; Lanver, D.; Schweizer, G.; Tanaka, S.; Liang, L.; Tollot, M.; Zuccaro, A.; Reissmann, S.; Kahmann, R. Fungal effectors and plant susceptibility. Annu. Rev. Plant Biol. 2015, 66, 513–545. [Google Scholar] [CrossRef]
- Presti, L.L.; Kahmann, R. How filamentous plant pathogen effectors are translocated to host cells. Curr. Opin. Plant Biol. 2017, 38, 19–24. [Google Scholar] [CrossRef]
- Oliveira-Garcia, E.; Valent, B. How eukaryotic filamentous pathogens evade plant recognition. Curr. Opin. Microbiol. 2015, 26, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Shalaby, S.; Horwitz, B.A. Plant phenolic compounds and oxidative stress: Integrated signals in fungal-plant interactions. Curr. Genet. 2015, 61, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Reverberi, M.; Punelli, M.; Scala, V.; Scarpari, M.; Uva, P.; Mentzen, W.I.; Dolezal, A.L.; Woloshuk, C.; Pinzari, F.; Fabbri, A.A.; et al. Genotypic and phenotypic versatility of Aspergillus flavus during maize exploitation. PLoS ONE 2013, 8, e68735. [Google Scholar] [CrossRef] [Green Version]
- Slepecky, R.A.; Starmer, W.T. Phenotypic plasticity in fungi: A review with observations on Aureobasidium pullulans. Mycologia 2009, 101, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Bartels, A.; Han, Q.; Nair, P.; Stacey, L.; Gaynier, H.; Mosley, M.; Huang, Q.Q.; Pearson, J.K.; Hsieh, T.F.; An, Y.C.; et al. Dynamic DNA methylation in plant growth and development. Int. J. Mol. Sci. 2018, 19, 2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, S.; Fei, Z.; Chen, Y.R.; Zheng, Y.; Huang, M.; Vrebalov, J.; McQuinn, R.; Gapper, N.; Liu, B.; Xiang, J.; et al. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 2013, 31, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G.P. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.X.; Han, L.; Zhao, Z.M. Conservation and divergence of DNA methylation in eukaryotes: New insights from single base resolution DNA methylomes. Epigenetics 2011, 6, 134–140. [Google Scholar] [CrossRef] [Green Version]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howlett, B.J.; Lowe, R.G.T.; Marcroft, S.J.; van de Wouw, A.P. Evolution of virulence in fungal plant pathogens: Exploiting fungal genomics to control plant disease. Mycologia 2015, 107, 441–451. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.; Choi, J.; Lee, G.W.; Park, S.Y.; Huh, A.; Dean, R.; Lee, Y.H. Genome-wide profiling of DNA methylation provides insights into epigenetic regulation of fungal development in a plant pathogenic fungus, Magnaporthe oryzae. Sci. Rep. 2015, 5, 8567. [Google Scholar] [CrossRef] [Green Version]
- An, Y.C.; Goettel, W.; Han, Q.; Bartels, A.; Liu, Z.; Xiao, W. Dynamic changes of genome-wide DNA methylation during soybean seed development. Sci. Rep. 2017, 7, 12263. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.H.; Zhang, X.Y.; Chen, Z.G.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Nahar, P.B.; Kulkarani, S.A.; Kulye, M.S.; Chavan, S.B.; Kulkarani, G.; Rajendran, A.; Yadav, P.D.; Shouche, Y.; Deshpande, M.V. Effect of repeated in vitro subculturing on the virulence of Metarhizium anisopliae against Helicoverpa armigera (Lepidoptera: Noctuidae). Biocontrol Sci. Technol. 2008, 18, 337–355. [Google Scholar] [CrossRef]
- Shah, F.A.; Allen, N.; Wright, C.J.; Butt, T.M. Repeated in vitro subculturing alters spore surface properties and virulence of Metarhizium anisopliae. FEMS Microbiol. Lett. 2007, 276, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Morrow, B.J.; Boucias, D.G.; Heath, M.A. Loss of virulence in an isolate of an entomopathogenic fungus, Nomuraea rileyi, after serial in vitro passages. J. Econ. Entomol. 1989, 82, 404–407. [Google Scholar] [CrossRef]
- Ignoffo, C.M.; McIntosh, A.H.; Garcia, C.; Kroha, M.; Johnson, J.M. Effects of successive in vitro and in vivo passages on the virulence of the entomopathogenic fungus, Nomuraea rileyi. Entomophaga 1982, 27, 371–378. [Google Scholar] [CrossRef]
- Vandenberg, J.D.; Cantone, F.A. Effect of serial transfer of three strains of Paecilomyces fumosoroseus on growth in vitro, virulence and host specificity. J. Invertebr. Pathol. 2004, 85, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Brownbridge, M.; Costa, S.; Jaronski, S.T. Effects of in vitro passage of Beauveria bassiana on virulence to Bemisia argentifolii. J. Invertebr. Pathol. 2001, 77, 280–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, T.P.; Bidochka, M.J.; Khachatourians, G.C. Entomopathogenicity of several fungi toward the English grain aphid (Homoptera: Aphididae) and enhancement of virulence with host passage of Paecilomyces farinosus. J. Econ. Entomol. 1992, 85, 58–64. [Google Scholar] [CrossRef]
- Hussain, A.; Tian, M.Y.; He, Y.R.; Lei, Y.Y. Differential fluctuation in virulence and VOC profiles among different cultures of entomopathogenic fungi. J. Invertebr. Pathol. 2010, 104, 166–171. [Google Scholar] [CrossRef]
- Butt, T.M.; Wang, C.S.; Shah, F.A.; Hall, R. Degeneration of entomogenous fungi. In An Ecological and Societal Approach to Biological Control; Eilenberg, J., Hokkanen, H.M.T., Eds.; Springer: Cham, The Netherlands, 2006; pp. 213–226. [Google Scholar]
- Shah, F.A.; Wang, C.S.; Butt, T.M. Nutrition influences growth and virulence of the insect-pathogenic fungus Metarhizium anisopliae. FEMS Microbiol. Lett. 2005, 251, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Butt, T.M.; Goettel, M. Bioassays of entomopathogenic fungi. In Bioassays of Entomopathogenic Microbes and Nematodes; Navon, A., Ascher, K.R.S., Eds.; CAB International: Wallingford, UK, 2000; pp. 141–195. [Google Scholar]
- Hussain, A.; Tian, M.Y.; He, Y.R.; Ruan, L.; Ahmed, S. In vitro and in vivo culturing impacts on the virulence of characteristics of serially passed entomopathogenic fungi. J. Food Agric. Environ. 2010, 8, 481–487. [Google Scholar]
- Hutwimmer, S.; Wagner, S.; Affenzeller, M.; Burgstaller, W.; Strasser, H. Algorithmbased design of synthetic growth media stimulating virulence properties of Metarhizium anisopliae conidia. J. Appl. Microbiol. 2008, 105, 2026–2034. [Google Scholar] [CrossRef]
- Rangel, D.; Braga, G.; Flint, S.D. Variations in UV-B tolerance and germination speed of Metarhizium anisopliae conidia produced on insects and artificial substrates. J. Invertebr. Pathol. 2004, 87, 77–83. [Google Scholar] [CrossRef]
- Scala, V.; Grottoli, A.; Cigliano, R.A.; Anzar, I.; Beccacioli, M.; Fanelli, C.; Dall’Asta, C.; Battilani, P.; Reverberi, M.; Sanseverino, W. Carefully with that axe, gene, genome perturbation after a PEG-mediated protoplast transformation in Fusarium verticillioides. Toxins 2017, 9, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safavi, S.A. Attenuation of the entomopathogenic fungus Beauveria bassiana following serial in vitro transfers. Biologia 2012, 67, 1062–1068. [Google Scholar] [CrossRef]
- Ansari, M.A.; Butt, T.M. Effects of successive subculturing on stability, virulence, conidial yield, germination and shelf-life of entomopathogenic fungi. J. Appl. Microbiol. 2011, 110, 1460–1469. [Google Scholar] [CrossRef]
- Kusari, S.; Zuelke, S.; Spiteller, M. Effect of artificial reconstitution of the interaction between the plant Camptotheca acuminata and the fungal endophyte Fusarium solani on camptothecin biosynthesis. J. Nat. Prod. 2011, 74, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Gurudatt, P.S.; Priti, V.; Sweta, S.; Ramesha, B.T.; Ravikanth, G.; Vasudeva, R.; Amna, T.; Deepika, S.; Ganeshaiah, K.N.; Shaanker, U.R.; et al. Attenuation of camptothecin production and negative relation between hyphal biomass and camptothecin content in endophytic fungal strains isolated from Nothapodytes nimmoniana Grahm (Icacinaceae). Curr. Sci. 2010, 98, 1006–1009. [Google Scholar]
- Li, J.; Sidhu, R.S.; Ford, E.J.; Long, D.M.; Hess, W.M.; Strobel, G.A. The induction of taxol production in the endophytic fungus-Periconia sp. from Torreya grandifolia. J. Ind. Microbiol. Biotechnol. 1998, 20, 259–264. [Google Scholar] [CrossRef]
- Priti, V.; Ramesha, B.T.; Shweta, S.; Ravikanth, G.; Ganeshaiah, K.N.; Suryanarayanan, T.S.; Shaanker, U.R. How promising are endophytic fungi as alternative sources of plant secondary metabolites. Curr. Sci. 2009, 97, 4–6. [Google Scholar]
- Sachin, N.; Manjunatha, B.L.; Kumara, M.P.; Ravikanth, G.; Shweta, S.; Suryanarayanan, T.S.; Ganeshaiah, K.N.; Shaanker, U.R. Do endophytic fungi possess pathway genes for plant secondary metabolites? Curr. Sci. 2013, 104, 178–182. [Google Scholar]
- Subramaniam, R.; Narayanan, S.; Walkowiak, S.; Wang, L.; Joshi, M.; Rocheleau, H.; Ouellet, T.; Harris, L.J. Leucine metabolism regulates TRI6 expression and affects deoxynivalenol production and virulence in Fusarium graminearum. Mol. Microbiol. 2015, 98, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Han, Q.; Wang, J.; Wang, X.; Xu, J.; Shi, J. Two FgLEU2 genes with different roles in leucine biosynthesis and infection-related morphogenesis in Fusarium graminearum. PLoS ONE 2016, 11, e0165927. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Zhang, X.; Zhao, L.; Apaliya, M.T.; Yang, Q.; Sun, W.; Zhang, X.; Zhang, H. Screening of deoxynivalenol producing strains and elucidation of possible toxigenic molecular mechanism. Toxins 2017, 9, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, G.H.; Desjardins, A.E.; Plattner, R.D. Deoxynivalenol-nonproducing Fusarium graminearum causes initial infection, but does not cause disease spread in wheat spikes. Mycopathologia 2002, 153, 97–98. [Google Scholar] [CrossRef]
- Eudes, F.; Comeau, A.; Rioux, S.; Collin, J. Impact of trichothecenes on Fusarium head blight (Fusarium graminearum) development in jspring wheat (Triticum aestivum). Can. J. Plant Pathol. 2001, 23, 318–322. [Google Scholar] [CrossRef]
- Kimura, M.; Kaneko, I.; Komiyama, M.; Takatsuki, A.; Koshino, H.; Yoneyama, K.; Yamaguchi, I. Trichothecene 3-O-acetyltransferase protects both the producing organism and transformed yeast from related mycotoxins. Cloning and characterization of Tri101. J. Biol. Chem. 1998, 273, 1654–1661. [Google Scholar] [CrossRef] [Green Version]
- McCormick, S.P.; Alexander, N.J.; Trapp, S.E.; Hohn, T.M. Disruption of TRI101, the gene encoding trichothecene 3-O-acetyltransferase, from Fusarium sporotrichioides. Appl. Environ. Microbiol. 1999, 65, 5252–5256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.; Gu, Q.; Yun, Y.; Yin, Y.; Xu, J.R.; Shim, W.B.; Ma, Z. The TOR signaling pathway regulates vegetative development and virulence in Fusarium graminearum. New Phytol. 2014, 203, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Son, H.; Seo, Y.S.; Min, K.; Park, A.R.; Lee, J.; Jin, J.M.; Lin, Y.; Cao, P.; Hong, S.Y.; Kim, E.K.; et al. A phenome-based functional analysis of transcription factors in the cereal head blight fungus, Fusarium graminearum. PLoS Pathog. 2011, 7, e1002310. [Google Scholar] [CrossRef] [PubMed]
- Puri, K.D.; Yan, C.; Leng, Y.; Zhong, S. RNA-Seq revealed differences in transcriptomes between 3ADON and 15ADON populations of Fusarium graminearum in vitro and in planta. PLoS ONE 2016, 11, e0163803. [Google Scholar]
- Merhej, J.; Urban, M.; Dufresne, M.; Hammond-Kosack, K.E.; Richard-Forget, F.; Barreau, C. The velvet gene, FgVe1, affects fungal development and positively regulates trichothecene biosynthesis and pathogenicity in Fusarium graminearum. Mol. Plant Pathol. 2011, 13, 363–374. [Google Scholar] [CrossRef]
- Jiang, J.; Liu, X.; Yin, Y.; Ma, Z. Involvement of a velvet protein FgVeA in the regulation of asexual development, lipid and secondary metabolisms and virulence in Fusarium graminearum. PLoS ONE 2011, 6, e28291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, M.; Zhu, Q.; Liang, Y.; Li, J.; Fan, X.; Yu, X.; He, F.; Xu, H.; Ling, Y.; Yu, J. Differential roles of three FgPLD genes in regulating development and pathogenicity in Fusarium graminearum. Fungal Genet. Biol. 2017, 109, 46–52. [Google Scholar] [CrossRef]
- Niu, X.W.; Zheng, Z.Y.; Feng, Y.G.; Guo, W.Z.; Wang, X.Y. The Fusarium graminearum virulence factor FGL targets an FKBP12 immunophilin of wheat. Gene 2013, 525, 77–83. [Google Scholar] [CrossRef]
- Gardiner, D.M.; Stephens, A.E.; Munn, A.L.; Manners, J.M. An ABC pleiotropic drug resistance transporter of Fusarium graminearum with a role in crown and root diseases of wheat. FEMS Microbiol. Lett. 2013, 348, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Mara, S.P.; Broz, K.; Boenisch, M.; Zhong, Z.; Dong, Y.; Kistler, H.C. The Fusarium graminearum t-SNARE Sso2 is involved in growth, defense, and DON accumulation and virulence. Mol. Plant-Microbe Int. 2020, 33, 888–901. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Wang, Z.; Cheng, D.; Chen, X.; Chen, Y.; Ma, Z. The ATP-binding protein FgArb1 is essential for penetration, infectious and normal growth of Fusarium graminearum. New Phytol. 2018, 219, 1447–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammar, G.A.; Tryono, R.; Doll, K.; Karlovsky, P.; Deising, H.B.; Wirsel, S.G.R. Identification of ABC transporter genes of Fusarium graminearum with roles in azole tolerance and/or virulence. PLoS ONE 2013, 8, e79042. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Lee, H.J.; Lee, J.; Kim, K.W.; Yun, S.H.; Shim, W.B.; Lee, Y.W. Gibberella zeae chitin synthase genes, GzCHS5 and GzCHS7, are required for hyphal growth, perithecia formation, and pathogenicity. Curr. Genet. 2009, 55, 449–459. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Chen, Q.; Liu, C.H.; Liu, Y.B.; Yi, P.; Niu, K.X.; Wang, Y.Q.; Wang, A.Q.; Yu, H.Y.; Pu, Z.E.; et al. Chitin synthase gene FgCHS8 affects virulence and fungal cell wall sensitivity to environmental stress in Fusarium graminearum. Fungal Biol. 2016, 120, 764–774. [Google Scholar] [CrossRef]
- Seong, K.Y.; Pasquali, M.; Zhou, X.; Song, J.; Hilburn, K.; McCormick, S.; Dong, Y.; Xu, J.R.; Kistler, H.C. Global gene regulation by Fusarium transcription factors Tri6 and Tri10 reveals adaptations for toxin biosynthesis. Mol. Microbiol. 2009, 72, 354–367. [Google Scholar] [CrossRef]
- Guo, L.; Ji, M.; Ye, K. Dynamic network inference and association computation discover gene modules regulating virulence, mycotoxin and sexual reproduction in Fusarium graminearum. BMC Genom. 2020, 21, 176. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Nam, H.; Park, J.; Choi, G.J.; Lee, Y.W.; Son, H. Characterization of the CCAAT-binding transcription factor complex in the plant pathogenic fungus Fusarium graminearum. Sci. Rep. 2020, 10, 4898. [Google Scholar] [CrossRef]
- Talas, F.; Kalih, R.; Miedaner, T.; McDonald, B.A. Genome-wide association study identifies novel candidate genes for aggressiveness, deoxynivalenol production, and azole sensitivity in natural field populations of Fusarium graminearum. Mol. Plant-Microbe Int. 2016, 29, 417–430. [Google Scholar] [CrossRef] [Green Version]
- Lawler, K.; Hammond-Kosack, K.; Brazma, A.; Coulson, R.M.R. Genomic clustering and co-regulation of transcriptional networks in the pathogenic fungus Fusarium graminearum. BMC Syst. Biol. 2013, 7, 52. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Mehrabi, R.; Koten, C.; Kang, Z.; Wei, Y.; Seong, K.; Kistler, H.C.; Xu, J.R. Transduction beta-like gene FTL1 is essential for pathogenesis in Fusarium graminearum. Eukaryot. Cell 2009, 8, 867–876. [Google Scholar] [CrossRef] [Green Version]
- Shin, W.B.; Sagaram, U.S.; Choi, Y.E.; So, J.; Wilkinson, H.H.; Lee, Y.W. FSR1 is essential for fungal virulence and female fertility in Fusarium verticillioides and in Fusarium graminearum. Mol. Plant Microbe Int. 2006, 19, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.Y.; Yang, N.; Xu, Z.; Dai, H.; Tang, S.; Wang, Z.Y. FgHAT2 is involved in regulating vegetative growth, conidiation, DNA damage repair, DON production and virulence in Fusarium graminearum. J. Integr. Agric. 2020, 19, 1813–1824. [Google Scholar] [CrossRef]
- Dufresne, M.; van der Lee, T.; M’Barek, S.B.; Xu, X.; Zhang, X.; Liu, T.; Waalwijk, C.; Zhang, W.; Kema, G.H.J.; Daboussi, M.J. Transposon-tagging identifies novel pathogenicity genes in Fusarium graminearum. Fungal Genet. Biol. 2008, 45, 1552–1561. [Google Scholar] [CrossRef]
- Ren, W.; Zhao, H.; Shao, W.; Ma, W.; Wang, J.; Zhou, M.; Chen, C. Identification of a novel phenamacril-resistance-related gene by the cDNA-RAPD method in Fusarium asiaticum. Pest Manag. Sci. 2016, 72, 1558–1565. [Google Scholar] [CrossRef]
- Menke, J.; Dong, Y.; Kistler, H.C. Fusarium graminearum Tri12p influences virulence to wheat and trichothecene accumulation. Mol. Plant Microbe Int. 2012, 25, 1408–1418. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Woloshuk, C.P. Functional characterization of fst1 in Fusarium verticillioides during colonization of maize kernels. Mol. Plant Microbe Int. 2010, 24, 18–24. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tini, F.; Beccari, G.; Marconi, G.; Porceddu, A.; Sulyok, M.; Gardiner, D.M.; Albertini, E.; Covarelli, L. Identification of Putative Virulence Genes by DNA Methylation Studies in the Cereal Pathogen Fusarium graminearum. Cells 2021, 10, 1192. https://doi.org/10.3390/cells10051192
Tini F, Beccari G, Marconi G, Porceddu A, Sulyok M, Gardiner DM, Albertini E, Covarelli L. Identification of Putative Virulence Genes by DNA Methylation Studies in the Cereal Pathogen Fusarium graminearum. Cells. 2021; 10(5):1192. https://doi.org/10.3390/cells10051192
Chicago/Turabian StyleTini, Francesco, Giovanni Beccari, Gianpiero Marconi, Andrea Porceddu, Micheal Sulyok, Donald M. Gardiner, Emidio Albertini, and Lorenzo Covarelli. 2021. "Identification of Putative Virulence Genes by DNA Methylation Studies in the Cereal Pathogen Fusarium graminearum" Cells 10, no. 5: 1192. https://doi.org/10.3390/cells10051192