
B Cell Activation and Escape of Tolerance Checkpoints: Recent Insights from Studying Autoreactive B Cells

 , , and
, , and
Abstract
:1. Introduction
2. General B Cell Development and B Cell Tolerance
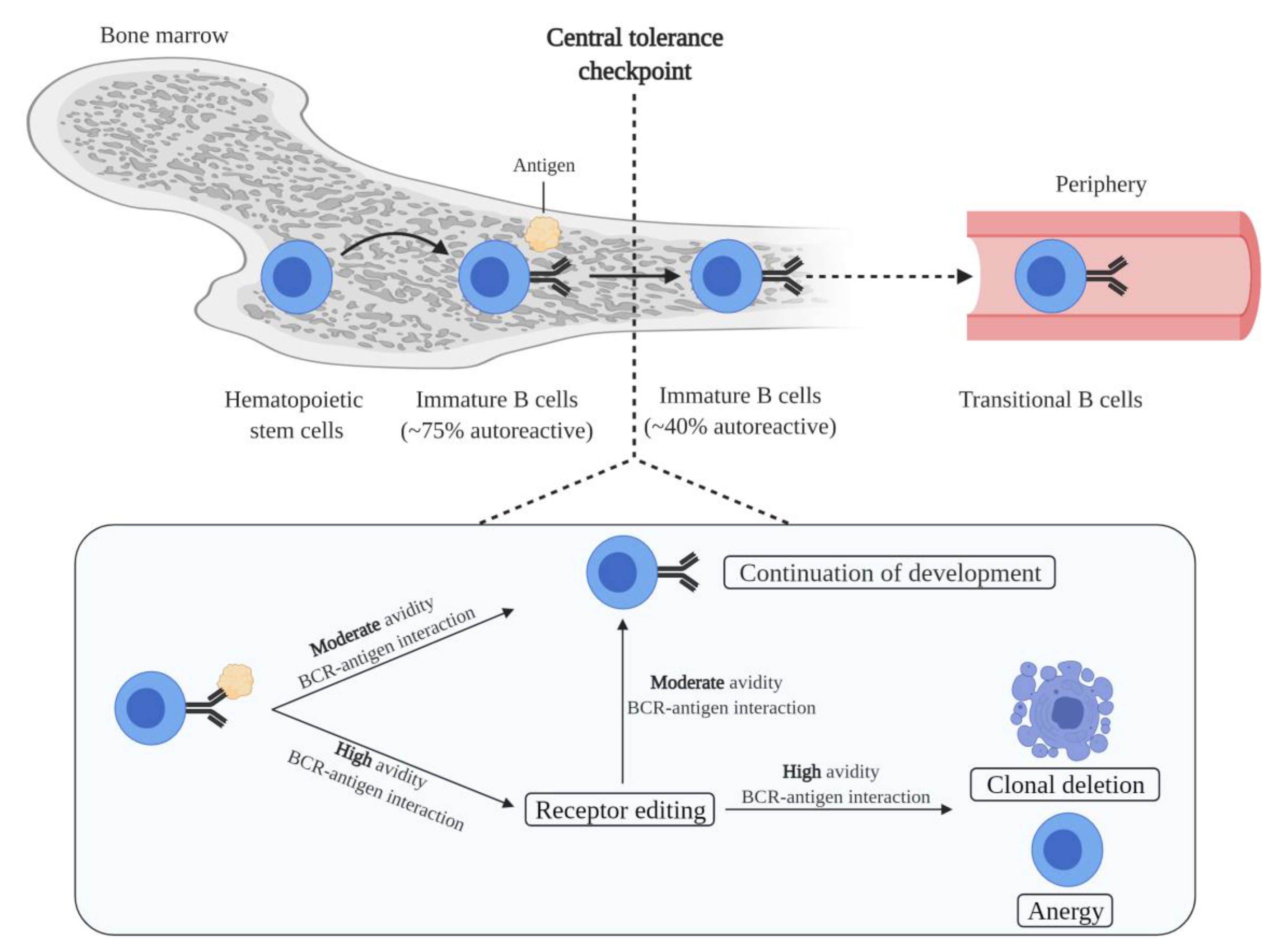
2.1. B Cell Generation and Central Tolerance
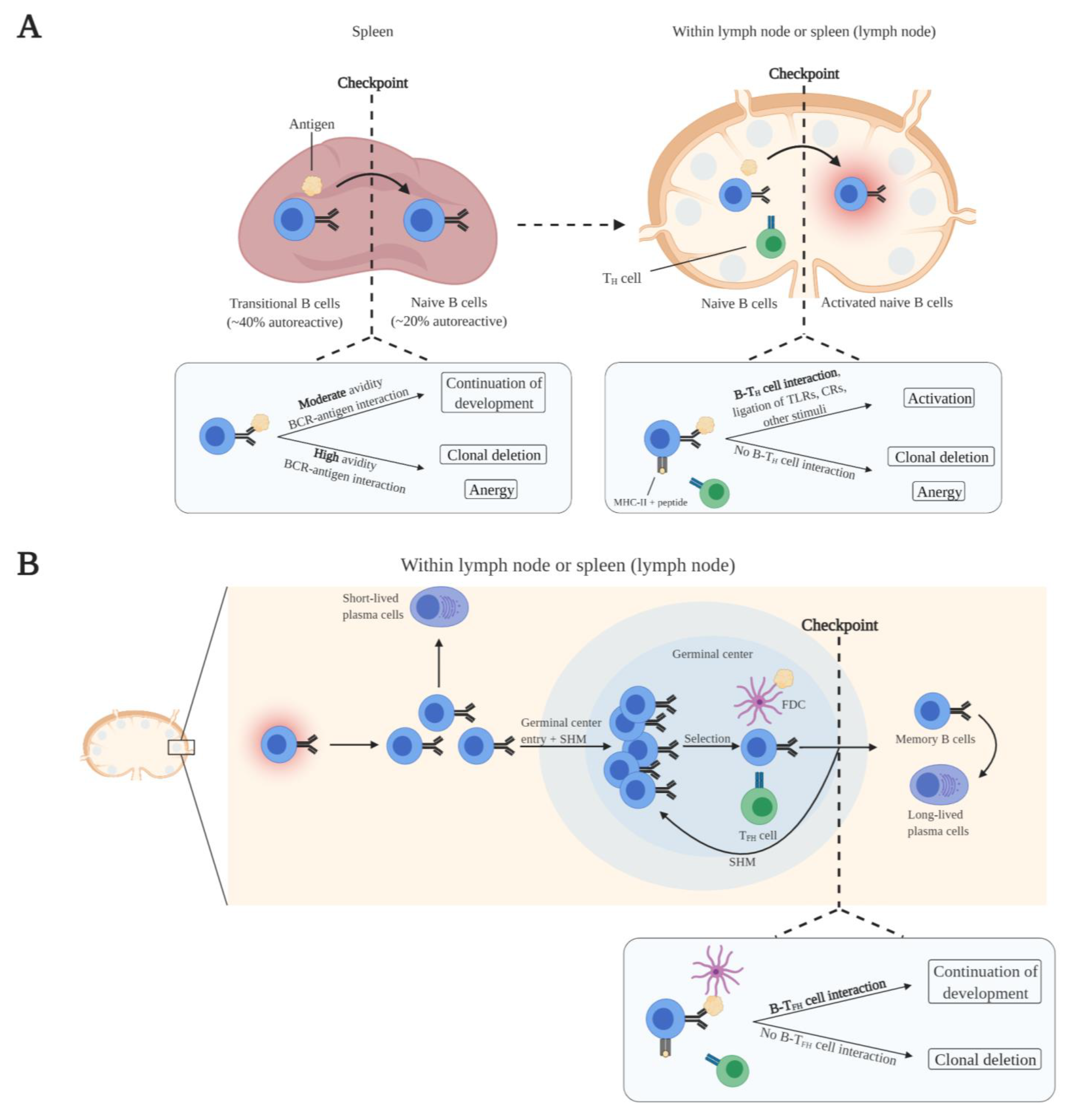
2.2. Peripheral B Cell Maturation and Tolerance
3. Autoreactive B Cell Development in Autoimmune Diseases
3.1. Compromised Self-Tolerance Checkpoints and Precursor Cells of Autoreactive B Cells
3.2. Activation of Autoreactive B Cells
3.3. Escape of Tolerance
3.4. Effector Functions of Autoreactive B Cells
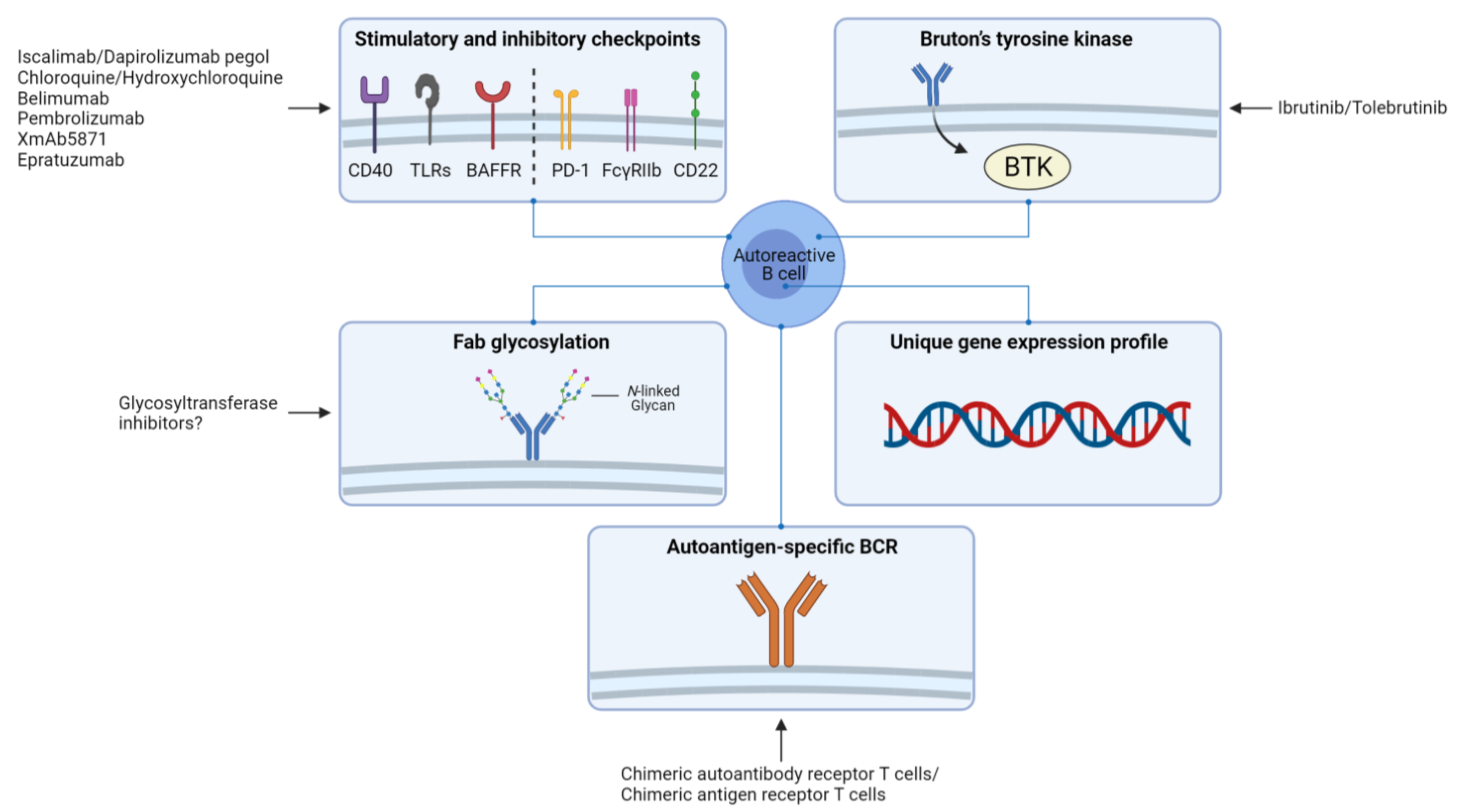
4. Implications for Therapy and Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cooper, G.S.; Bynum, M.L.; Somers, E.C. Recent insights in the epidemiology of autoimmune diseases: Improved prevalence estimates and understanding of clustering of diseases. J. Autoimmun. 2009, 33, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Lerner, A.; Jeremias, P.; Matthias, T. The World Incidence and Prevalence of Autoimmune Diseases is Increasing. Int. J. Celiac Dis. 2016, 3, 151–155. [Google Scholar] [CrossRef] [Green Version]
- Rosenblum, M.D.; Remedios, K.A.; Abbas, A.K. Mechanisms of human autoimmunity. J. Clin. Investig. 2015, 125, 2228–2233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaegi, C.; Wuest, B.; Schreiner, J.; Steiner, U.C.; Vultaggio, A.; Matucci, A.; Crowley, C.; Boyman, O. Systematic Review of Safety and Efficacy of Rituximab in Treating Immune-Mediated Disorders. Front. Immunol. 2019, 10, 1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, P.; Fillatreau, S. Antibody-independent functions of B cells: A focus on cytokines. Nat. Rev. Immunol. 2015, 15, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, W.; Lakkis, F.G.; Chalasani, G. B Cells, Antibodies, and More. Clin. J. Am. Soc. Nephrol. 2016, 11, 137–154. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001. [Google Scholar] [CrossRef]
- Tavakolpour, S.; AleSaeidi, S.; Darvishi, M.; Ghasemiadl, M.; Darabi-Monadi, S.; Akhlaghdoust, M.; Behjati, S.E.; Jafarieh, A. A comprehensive review of rituximab therapy in rheumatoid arthritis patients. Clin. Rheumatol. 2019, 38, 2977–2994. [Google Scholar] [CrossRef]
- Kaul, A.; Gordon, C.; Crow, M.K.; Touma, Z.; Urowitz, M.B.; Van Vollenhoven, R.; Ruiz-Irastorza, G.; Hughes, G. Systemic lupus erythematosus. Nat. Rev. Dis. Primers 2016, 2, 16039. [Google Scholar] [CrossRef]
- Kitching, A.R.; Anders, H.-J.; Basu, N.; Brouwer, E.; Gordon, J.; Jayne, D.R.; Kullman, J.; Lyons, P.A.; Merkel, P.A.; Savage, C.O.S.; et al. ANCA-associated vasculitis. Nat. Rev. Dis. Primers 2020, 6, 1–27. [Google Scholar] [CrossRef]
- Kasperkiewicz, M.; Ellebrecht, C.T.; Takahashi, H.; Yamagami, J.; Zillikens, D.; Payne, A.S.; Amagai, M. Pemphigus. Nat. Rev. Dis. Primers 2017, 3, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, G.; Di Zenzo, G.; Cozzani, E.; Berti, E.; Cugno, M.; Marzano, A.V. New Insights into the Pathogenesis of Bullous Pemphigoid: 2019 Update. Front. Immunol. 2019, 10, 1506. [Google Scholar] [CrossRef] [PubMed]
- Kremer, N.; Snast, I.; Cohen, E.S.; Hodak, E.; Mimouni, D.; Lapidoth, M.; Mazor, S.; Levi, A. Rituximab and Omalizumab for the Treatment of Bullous Pemphigoid: A Systematic Review of the Literature. Am. J. Clin. Dermatol. 2019, 20, 209–216. [Google Scholar] [CrossRef]
- Nocturne, G.; Mariette, X. B cells in the pathogenesis of primary Sjögren syndrome. Nat. Rev. Rheumatol. 2018, 14, 133–145. [Google Scholar] [CrossRef]
- Gilhus, N.E.; Tzartos, S.; Evoli, A.; Palace, J.; Burns, T.M.; Verschuuren, J.J.G.M. Myasthenia gravis. Nat. Rev. Dis. Primers 2019, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Swinkels, M.; Rijkers, M.; Voorberg, J.; Vidarsson, G.; Leebeek, F.W.G.; Jansen, A.J.G. Emerging Concepts in Immune Thrombocytopenia. Front. Immunol. 2018, 9, 880. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Fraser, W.; Lebowitz, D. Treatments for Primary Immune Thrombocytopenia: A Review. Cureus 2019, 11, e5849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, T.F.; Andersen, S.; Latif, R.; Nagayama, Y.; Barbesino, G.; Brito, M.; Eckstein, A.K.; Stagnaro-Green, A.; Kahaly, G.J. Graves’ disease. Nat. Rev. Dis. Primers 2020, 6, 1–23. [Google Scholar] [CrossRef]
- McAdoo, S.P.; Pusey, C.D. Anti-Glomerular Basement Membrane Disease. Clin. J. Am. Soc. Nephrol. 2017, 12, 1162–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenfield, A.L.; Hauser, S.L. B-cell Therapy for Multiple Sclerosis: Entering an era. Ann. Neurol. 2018, 83, 13–26. [Google Scholar] [CrossRef]
- Lehmann, H.C.; Burke, D.; Kuwabara, S. Chronic inflammatory demyelinating polyneuropathy: Update on diagnosis, immunopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2019, 90, 981–987. [Google Scholar] [CrossRef] [Green Version]
- Querol, L.; Devaux, J.; Rojas-Garcia, R.; Illa, I. Autoantibodies in chronic inflammatory neuropathies: Diagnostic and therapeutic implications. Nat. Rev. Neurol. 2017, 13, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Pai, A.R.; Kamath, U.; Rao, P. Pathophysiology and diagnosis of Guillain–Barré syndrome–challenges and needs. Int. J. Neurosci. 2014, 125, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Motamed-Gorji, N.; Matin, N.; Tabatabaie, O.; Pavone, P.; Romano, C.; Falsaperla, R.; Vitaliti, G. Biological Drugs in Guillain-Barré Syndrome: An Update. Curr. Neuropharmacol. 2017, 15, 938–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boonyaratanakornkit, J.; Taylor, J.J. Techniques to Study Antigen-Specific B Cell Responses. Front. Immunol. 2019, 10, 1694. [Google Scholar] [CrossRef]
- LeBien, T.W. Bien, T.W. B Cell Development. In Fetal and Neonatal Physiology; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1202–1207. [Google Scholar]
- Wardemann, H.; Yurasov, S.; Schaefer, A.; Young, J.W.; Meffre, E.; Nussenzweig, M.C. Predominant Autoantibody Production by Early Human B Cell Precursors. Science 2003, 301, 1374–1377. [Google Scholar] [CrossRef] [Green Version]
- Nemazee, D. Mechanisms of central tolerance for B cells. Nat. Rev. Immunol. 2017, 17, 281–294. [Google Scholar] [CrossRef]
- Prak, E.T.L.; Monestier, M.; Eisenberg, R.A. B cell receptor editing in tolerance and autoimmunity. Ann. N. Y. Acad. Sci. 2011, 1217, 96–121. [Google Scholar] [CrossRef]
- Gauld, S.B.; Merrell, K.T.; Cambier, J.C. Silencing of autoreactive B cells by anergy: A fresh perspective. Curr. Opin. Immunol. 2006, 18, 292–297. [Google Scholar] [CrossRef]
- Nemazee, D.; Buerki, K. Clonal deletion of autoreactive B lymphocytes in bone marrow chimeras. Proc. Natl. Acad. Sci. USA 1989, 86, 8039–8043. [Google Scholar] [CrossRef] [Green Version]
- Elgueta, R.; Benson, M.J.; De Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef] [Green Version]
- Eertwegh, A.J.V.D.; Noelle, R.J.; Roy, M.; Shepherd, D.M.; Aruffo, A.; A Ledbetter, J.; Boersma, W.J.; Claassen, E. In vivo CD40-gp39 interactions are essential for thymus-dependent humoral immunity. I. In vivo expression of CD40 ligand, cytokines, and antibody production delineates sites of cognate T-B cell interactions. J. Exp. Med. 1993, 178, 1555–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, Z.; Hou, B. TLR signaling in B-cell development and activation. Cell. Mol. Immunol. 2013, 10, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Dunkelberger, J.R.; Song, W.-C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [Green Version]
- Crotty, S. T Follicular Helper Cell Differentiation, Function, and Roles in Disease. Immunity 2014, 41, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesin, L.; Ersching, J.; Victora, G.D. Germinal Center B Cell Dynamics. Immunity 2016, 45, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Gatto, D.; Brink, R. The germinal center reaction. J. Allergy Clin. Immunol. 2010, 126, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Brink, R.; Phan, T.G. Self-Reactive B Cells in the Germinal Center Reaction. Annu. Rev. Immunol. 2018, 36, 339–357. [Google Scholar] [CrossRef] [PubMed]
- Lobo, P.I. Role of Natural Autoantibodies and Natural IgM Anti-Leucocyte Autoantibodies in Health and Disease. Front. Immunol. 2016, 7, 198. [Google Scholar] [CrossRef] [Green Version]
- Maddur, M.S.; Lacroix-Desmazes, S.; Dimitrov, J.D.; Kazatchkine, M.D.; Bayry, J.; Kaveri, S.V. Natural Antibodies: From First-Line Defense Against Pathogens to Perpetual Immune Homeostasis. Clin. Rev. Allergy Immunol. 2019, 58, 213–228. [Google Scholar] [CrossRef]
- Kerkman, P.F.; Fabre, E.; Van Der Voort, E.I.H.; Zaldumbide, A.; Rombouts, Y.; Rispens, T.; Wolbink, G.; Hoeben, R.C.; Spits, H.; Baeten, D.L.P.; et al. Identification and characterisation of citrullinated antigen-specific B cells in peripheral blood of patients with rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Cornec, D.; Berti, A.; Hummel, A.; Peikert, T.; Pers, J.-O.; Specks, U. Identification and phenotyping of circulating autoreactive proteinase 3-specific B cells in patients with PR3-ANCA associated vasculitis and healthy controls. J. Autoimmun. 2017, 84, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Pollmann, R.; Walter, E.; Schmidt, T.; Waschke, J.; Hertl, M.; Möbs, C.; Eming, R. Identification of Autoreactive B Cell Subpopulations in Peripheral Blood of Autoimmune Patients With Pemphigus Vulgaris. Front. Immunol. 2019, 10, 1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, A.; Caldara, A.L.; Ran, N.A.; Menne, Z.; Kauffman, R.C.; Affer, M.; Llovet, A.; Norwood, C.; Scanlan, A.; Mantus, G.; et al. Single-Cell Analysis Suggests that Ongoing Affinity Maturation Drives the Emergence of Pemphigus Vulgaris Autoimmune Disease. Cell Rep. 2019, 28, 909–922.e6. [Google Scholar] [CrossRef] [Green Version]
- Steen, J.; Forsström, B.; Sahlström, P.; Odowd, V.; Israelsson, L.; Krishnamurthy, A.; Badreh, S.; Alm, L.M.; Compson, J.; Ramsköld, D.; et al. Recognition of Amino Acid Motifs, Rather Than Specific Proteins, by Human Plasma Cell–Derived Monoclonal Antibodies to Posttranslationally Modified Proteins in Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 196–209. [Google Scholar] [CrossRef]
- Malkiel, S.; Jeganathan, V.; Wolfson, S.; Orduño, N.M.; Marasco, E.; Aranow, C.; Mackay, M.; Gregersen, P.K.; Diamond, B. Checkpoints for Autoreactive B Cells in the Peripheral Blood of Lupus Patients Assessed by Flow Cytometry. Arthritis Rheumatol. 2016, 68, 2210–2220. [Google Scholar] [CrossRef] [PubMed]
- De La Varga-Martínez, R.; Rodríguez-Bayona, B.; Campos-Caro, A.; A Añez, G.; Medina-Varo, F.; Rodríguez, C.; La Varga-Martínez, R. Autoreactive B-lymphocytes in SLE and RA patients: Isolation and characterisation using extractable nuclear and citrullinated antigens bound to immunobeads. Eur. J. Immunol. 2019, 49, 1107–1116. [Google Scholar] [CrossRef]
- Wellmann, U.; Letz, M.; Herrmann, M.; Angermüller, S.; Kalden, J.R.; Winkler, T.H. The evolution of human anti-double-stranded DNA autoantibodies. Proc. Natl. Acad. Sci. USA 2005, 102, 9258–9263. [Google Scholar] [CrossRef] [Green Version]
- Mietzner, B.; Tsuiji, M.; Scheid, J.; Velinzon, K.; Tiller, T.; Abraham, K.; Gonzalez, J.B.; Pascual, V.; Stichweh, D.; Wardemann, H.; et al. Autoreactive IgG memory antibodies in patients with systemic lupus erythematosus arise from nonreactive and polyreactive precursors. Proc. Natl. Acad. Sci. USA 2008, 105, 9727–9732. [Google Scholar] [CrossRef] [Green Version]
- Yuseff, M.-I.; Pierobon, P.; Reversat, A.; Lennon-Duménil, A.-M. How B cells capture, process and present antigens: A crucial role for cell polarity. Nat. Rev. Immunol. 2013, 13, 475–486. [Google Scholar] [CrossRef]
- Rozario, T.; DeSimone, D.W. The extracellular matrix in development and morphogenesis: A dynamic view. Dev. Biol. 2010, 341, 126–140. [Google Scholar] [CrossRef] [Green Version]
- Suurmond, J.; Diamond, B. Autoantibodies in systemic autoimmune diseases: Specificity and pathogenicity. J. Clin. Investig. 2015, 125, 2194–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darrah, E.; Andrade, F. NETs: The missing link between cell death and systemic autoimmune diseases? Front. Immunol. 2013, 3, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, A.; Herrmann, M.; Muñoz, L.E. Clearance Deficiency and Cell Death Pathways: A Model for the Pathogenesis of SLE. Front. Immunol. 2016, 7, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, N.; Wehner, N.; Marshall, B.; Gray, W.; Gray, B.; Hoidal, J. Characterization of proteinase-3 (PR-3), a neutrophil serine proteinase. Structural and functional properties. J. Biol. Chem. 1991, 266, 9540–9548. [Google Scholar] [CrossRef]
- Lombard, C.; Bouchu, D.; Wallach, J.; Saulnier, J. Proteinase 3 hydrolysis of peptides derived from human elastin exon. Amino Acids 2005, 28, 403–408. [Google Scholar] [CrossRef]
- Pham, C.T.N. Neutrophil serine proteases fine-tune the inflammatory response. Int. J. Biochem. Cell Biol. 2008, 40, 1317–1333. [Google Scholar] [CrossRef] [Green Version]
- Gerstner, C.; Turcinov, S.; Hensvold, A.H.; Chemin, K.; Uchtenhagen, H.; Ramwadhdoebe, T.H.; Dubnovitsky, A.; Kozhukh, G.; Rönnblom, L.; Kwok, W.W.; et al. Multi-HLA class II tetramer analyses of citrulline-reactive T cells and early treatment response in rheumatoid arthritis. BMC Immunol. 2020, 21, 1–14. [Google Scholar] [CrossRef]
- Abdirama, D.; Tesch, S.; Grießbach, A.-S.; von Spee-Mayer, C.; Humrich, J.Y.; Stervbo, U.; Babel, N.; Meisel, C.; Alexander, T.; Biesen, R.; et al. Nuclear antigen–reactive CD4+ T cells expand in active systemic lupus erythematosus, produce effector cytokines, and invade the kidneys. Kidney Int. 2021, 99, 238–246. [Google Scholar] [CrossRef]
- Winek, J.; Mueller, A.; Csernok, E.; Gross, W.L.; Lamprecht, P. Frequency of proteinase 3 (PR3)-specific autoreactive T cells determined by cytokine flow cytometry in Wegener’s granulomatosis. J. Autoimmun. 2004, 22, 79–85. [Google Scholar] [CrossRef]
- Veldman, C.; Eming, R.; Wolff-Franke, S.; Sonderstrup, G.; Kwok, W.W.; Hertl, M. Detection of low avidity desmoglein 3-reactive T cells in pemphigus vulgaris using HLA-DRβ1⁎0402 tetramers. Clin. Immunol. 2007, 122, 330–337. [Google Scholar] [CrossRef]
- Suthers, A.N.; Sarantopoulos, S. TLR7/TLR9- and B Cell Receptor-Signaling Crosstalk: Promotion of Potentially Dangerous B Cells. Front. Immunol. 2017, 8, 775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K. Toll-like receptors in innate immunity. Int. Immunol. 2004, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Leadbetter, E.A.; Rifkin, I.R.; Hohlbaum, A.M.; Beaudette, B.C.; Shlomchik, M.J.; Marshak-Rothstein, A. Chromatin–IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nat. Cell Biol. 2002, 416, 603–607. [Google Scholar] [CrossRef]
- Lau, C.M.; Broughton, C.; Tabor, A.S.; Akira, S.; Flavell, R.A.; Mamula, M.J.; Christensen, S.R.; Shlomchik, M.J.; Viglianti, G.A.; Rifkin, I.R.; et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J. Exp. Med. 2005, 202, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- A Viglianti, G.; Lau, C.M.; Hanley, T.M.; A Miko, B.; Shlomchik, M.J.; Marshak-Rothstein, A. Activation of Autoreactive B Cells by CpG dsDNA. Immunity 2003, 19, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Groom, J.R.; Fletcher, C.A.; Walters, S.N.; Grey, S.T.; Watt, S.V.; Sweet, M.J.; Smyth, M.J.; Mackay, C.R.; Mackay, F. BAFF and MyD88 signals promote a lupuslike disease independent of T cells. J. Exp. Med. 2007, 204, 1959–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herlands, R.A.; Christensen, S.R.; Sweet, R.A.; Hershberg, U.; Shlomchik, M.J. T Cell-Independent and Toll-like Receptor-Dependent Antigen-Driven Activation of Autoreactive B Cells. Immunity 2008, 29, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Gregersen, P.K.; Olsson, L.M. Recent Advances in the Genetics of Autoimmune Disease. Annu. Rev. Immunol. 2009, 27, 363–391. [Google Scholar] [CrossRef] [Green Version]
- Joshua, V.; Schobers, L.; Titcombe, P.J.; Israelsson, L.; Rönnelid, J.; Hansson, M.; Catrina, A.I.; Pruijn, G.J.M.; Malmström, V. Antibody responses to de novo identified citrullinated fibrinogen peptides in rheumatoid arthritis and visualization of the corresponding B cells. Arthritis Res. Ther. 2016, 18, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, D.J.; Metzler, G.; Wray-Dutra, D.J.R.G.M.M.; Jackson, D.J.R.G.M.M.W.-D.S.W. Altered B cell signalling in autoimmunity. Nat. Rev. Immunol. 2017, 17, 421–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corneth, O.B.J.; Wolterink, R.G.J.K.; Hendriks, R.W. BTK Signaling in B Cell Differentiation and Autoimmunity. Curr. Top. Microbiol. Immunol. 2015, 393, 67–105. [Google Scholar] [CrossRef]
- Rip, J.; Van Der Ploeg, E.K.; Hendriks, R.W.; Corneth, O.B.J. The Role of Bruton’s Tyrosine Kinase in Immune Cell Signaling and Systemic Autoimmunity. Crit. Rev. Immunol. 2018, 38, 17–62. [Google Scholar] [CrossRef]
- Kil, L.P.; De Bruijn, M.J.W.; Van Nimwegen, M.; Corneth, O.B.J.; Van Hamburg, J.P.; Dingjan, G.M.; Thaiss, F.; Rimmelzwaan, G.F.; Elewaut, D.; Delsing, D.; et al. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood 2012, 119, 3744–3756. [Google Scholar] [CrossRef]
- Corneth, O.B.J.; Verstappen, G.M.P.; Paulissen, S.M.J.; De Bruijn, M.J.W.; Rip, J.; Lukkes, M.; Van Hamburg, J.P.; Lubberts, E.; Bootsma, H.; Kroese, F.G.M.; et al. Enhanced Bruton’s Tyrosine Kinase Activity in Peripheral Blood B Lymphocytes From Patients With Autoimmune Disease. Arthritis Rheumatol. 2017, 69, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- von Borstel, A.; Abdulahad, W.H.; Sanders, J.S.; Rip, J.; Neys, S.F.H.; Hendriks, R.W.; A Stegeman, C.; Heeringa, P.; Rutgers, A.; Corneth, O.B.J. Evidence for enhanced Bruton’s tyrosine kinase activity in transitional and naïve B cells of patients with granulomatosis with polyangiitis. Rheumatology 2019, 58, 2230–2239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vletter, E.M.; Koning, M.T.; Scherer, H.U.; Veelken, H.; Toes, R.E.M. A Comparison of Immunoglobulin Variable Region N-Linked Glycosylation in Healthy Donors, Autoimmune Disease and Lymphoma. Front. Immunol. 2020, 11, 241. [Google Scholar] [CrossRef] [Green Version]
- Rombouts, Y.; Willemze, A.; Van Beers, J.J.B.C.; Shi, J.; Kerkman, P.F.; Van Toorn, L.; Janssen, G.M.C.; Zaldumbide, A.; Hoeben, R.C.; Pruijn, G.J.M.; et al. Extensive glycosylation of ACPA-IgG variable domains modulates binding to citrullinated antigens in rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Dunn-Walters, D. Effect of somatic hypermutation on potential N-glycosylation sites in human immunoglobulin heavy chain variable regions. Mol. Immunol. 2000, 37, 107–113. [Google Scholar] [CrossRef]
- Zhu, D.; McCarthy, H.; Ottensmeier, C.H.; Johnson, P.; Hamblin, T.J.; Stevenson, F.K. Acquisition of potential N-glycosylation sites in the immunoglobulin variable region by somatic mutation is a distinctive feature of follicular lymphoma. Blood 2002, 99, 2562–2568. [Google Scholar] [CrossRef]
- Hafkenscheid, L.; Bondt, A.; Scherer, H.U.; Huizinga, T.W.J.; Wuhrer, M.; Toes, R.E.M.; Rombouts, Y. Structural Analysis of Variable Domain Glycosylation of Anti-Citrullinated Protein Antibodies in Rheumatoid Arthritis Reveals the Presence of Highly Sialylated Glycans. Mol. Cell. Proteom. 2017, 16, 278–287. [Google Scholar] [CrossRef] [Green Version]
- Van De Bovenkamp, F.S.; Hafkenscheid, L.; Rispens, T.; Rombouts, Y. The Emerging Importance of IgG Fab Glycosylation in Immunity. J. Immunol. 2016, 196, 1435–1441. [Google Scholar] [CrossRef] [Green Version]
- Hamza, N.; Hershberg, U.; Kallenberg, C.G.M.; Vissink, A.; Spijkervet, F.K.L.; Bootsma, H.; Kroese, F.G.M.; Bos, N.A. Ig Gene Analysis Reveals Altered Selective Pressures on Ig-Producing Cells in Parotid Glands of Primary Sjögren’s Syndrome Patients. J. Immunol. 2014, 194, 514–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coelho, V.; Krysov, S.; Ghaemmaghami, A.M.; Emara, M.; Potter, K.N.; Johnson, P.; Packham, G.; Martinez-Pomares, L.; Stevenson, F.K. Glycosylation of surface Ig creates a functional bridge between human follicular lymphoma and microenvironmental lectins. Proc. Natl. Acad. Sci. USA 2010, 107, 18587–18592. [Google Scholar] [CrossRef] [Green Version]
- Germar, K.; Fehres, C.M.; Scherer, H.U.; Van Uden, N.; Pollastro, S.; Yeremenko, N.; Hansson, M.; Kerkman, P.F.; Van Der Voort, E.I.H.; Reed, E.; et al. Generation and Characterization of Anti–Citrullinated Protein Antibody–Producing B Cell Clones From Rheumatoid Arthritis Patients. Arthritis Rheumatol. 2019, 71, 340–350. [Google Scholar] [CrossRef]
- Mahendra, A.; Yang, X.; Abnouf, S.; Ms, J.R.T.A.; Park, D.; Soomro, S.; Roszik, J.; Coarfa, C.; Romain, G.; Wanzeck, K.; et al. Beyond Autoantibodies: Biologic Roles of Human Autoreactive B Cells in Rheumatoid Arthritis Revealed by RNA-Sequencing. Arthritis Rheumatol. 2019, 71, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Leandro, M.J. B-cell subpopulations in humans and their differential susceptibility to depletion with anti-CD20 monoclonal antibodies. Arthritis Res. Ther. 2013, 15, S3–S8. [Google Scholar] [CrossRef] [Green Version]
- Cambridge, G.; Leandro, M.J.; Edwards, J.C.W.; Ehrenstein, M.R.; Salden, M.; Bodman-Smith, M.; Webster, A.D.B. Serologic changes following B lymphocyte depletion therapy for rheumatoid arthritis. Arthritis Rheum. 2003, 48, 2146–2154. [Google Scholar] [CrossRef] [PubMed]
- Cambridge, G.; Perry, H.; Nogueira, L.; Serre, G.; Parsons, H.; De La Torre, I.; Dickson, M.; Leandro, M.; Edwards, J. The effect of B-cell depletion therapy on serological evidence of B-cell and plasmablast activation in patients with rheumatoid arthritis over multiple cycles of rituximab treatment. J. Autoimmun. 2014, 50, 67–76. [Google Scholar] [CrossRef]
- Jones, R.B.; Tervaert, J.W.C.; Hauser, T.; Luqmani, R.; Morgan, M.D.; Peh, C.A.; Savage, C.O.; Segelmark, M.; Tesar, V.; Van Paassen, P.; et al. Rituximab versus Cyclophosphamide in ANCA-Associated Renal Vasculitis. N. Engl. J. Med. 2010, 363, 211–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, J.H.; Merkel, P.A.; Spiera, R.; Seo, P.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.M.; Clair, E.W.S.; Turkiewicz, A.; Tchao, N.K.; et al. Rituximab versus Cyclophosphamide for ANCA-Associated Vasculitis. N. Engl. J. Med. 2010, 363, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Joly, P.; Mouquet, H.; Roujeau, J.-C.; D’Incan, M.; Gilbert, D.; Jacquot, S.; Gougeon, M.-L.; Bedane, C.; Muller, R.; Dreno, B.; et al. A Single Cycle of Rituximab for the Treatment of Severe Pemphigus. N. Engl. J. Med. 2007, 357, 545–552. [Google Scholar] [CrossRef]
- Eming, R.; Nagel, A.; Wolff-Franke, S.; Podstawa, E.; Debus, D.; Hertl, M. Rituximab exerts a Dual Effect in Pemphigus Vulgaris. J. Investig. Dermatol. 2008, 128, 2850–2858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joly, P.; Maho-Vaillant, M.; Prost-Squarcioni, C.; Hebert, V.; Houivet, E.; Calbo, S.; Caillot, F.; Golinski, M.L.; Labeille, B.; Picard-Dahan, C.; et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): A prospective, multicentre, parallel-group, open-label randomised trial. Lancet 2017, 389, 2031–2040. [Google Scholar] [CrossRef]
- Devauchelle-Pensec, V.; Mariette, X.; Jousse-Joulin, S.; Berthelot, J.-M.; Perdriger, A.; Puéchal, X.; Le Guern, V.; Sibilia, J.; Gottenberg, J.-E.; Chiche, L.; et al. Treatment of Primary Sjögren Syndrome With Rituximab. Ann. Intern. Med. 2014, 160, 233–242. [Google Scholar] [CrossRef]
- Bowman, S.J.; Everett, C.C.; O’Dwyer, J.L.; Emery, P.; Pitzalis, C.; Ng, W.-F.; Pease, C.T.; Price, E.J.; Sutcliffe, N.; Gendi, N.S.T.; et al. Randomized Controlled Trial of Rituximab and Cost-Effectiveness Analysis in Treating Fatigue and Oral Dryness in Primary Sjögren’s Syndrome. Arthritis Rheumatol. 2017, 69, 1440–1450. [Google Scholar] [CrossRef] [Green Version]
- Carubbi, F.; Cipriani, P.; Marrelli, A.; Benedetto, P.D.; Ruscitti, P.; Berardicurti, O.; Pantano, I.; Liakouli, V.; Alvaro, S.; Alunno, A.; et al. Efficacy and safety of rituximab treatment in early primary Sjögren’s syndrome: A prospective, multi-center, follow-up study. Arthritis Res. Ther. 2013, 15, R172. [Google Scholar] [CrossRef] [Green Version]
- St. Clair, E.W.; Levesque, M.C.; Prak, E.T.L.; Vivino, F.B.; Alappatt, C.J.; Spychala, M.E.; Wedgwood, J.; McNamara, J.; Sivils, K.L.M.; Fisher, L.; et al. Rituximab Therapy for Primary Sjögren’s Syndrome: An Open-Label Clinical Trial and Mechanistic Analysis. Arthritis Rheum. 2013, 65, 1097–1106. [Google Scholar] [CrossRef] [Green Version]
- Devauchelle-Pensec, V.; Pennec, Y.; Morvan, J.; Pers, J.-O.; Daridon, C.; Jousse-Joulin, S.; Roudaut, A.; Jamin, C.; Renaudineau, Y.; Roué, I.Q.; et al. Improvement of Sjögren’s syndrome after two infusions of rituximab (anti-CD20). Arthritis Rheum. 2007, 57, 310–317. [Google Scholar] [CrossRef]
- Hébert, V.; Petit, M.; Maho-Vaillant, M.; Golinski, M.-L.; Riou, G.; Derambure, C.; Boyer, O.; Joly, P.; Calbo, S. Modifications of the Transcriptomic Profile of Autoreactive B Cells From Pemphigus Patients After Treatment with Rituximab or a Standard Corticosteroid Regimen. Front. Immunol. 2019, 10, 1794. [Google Scholar] [CrossRef] [Green Version]
- Néel, A.; Bucchia, M.; Néel, M.; Tilly, G.; Caristan, A.; Yap, M.; Rimbert, M.; Bruneau, S.; Cadoux, M.; Agard, C.; et al. Dampening of CD8+ T Cell Response by B Cell Depletion Therapy in Antineutrophil Cytoplasmic Antibody–Associated Vasculitis. Arthritis Rheumatol. 2019, 71, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Rubin, S.J.S.; Bloom, M.S.; Robinson, W.H. B cell checkpoints in autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2019, 15, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Nezhad, M.S.; Seifalian, A.; Bagheri, N.; Yaghoubi, S.; Karimi, M.H.; Adbollahpour-Alitappeh, M. Chimeric Antigen Receptor Based Therapy as a Potential Approach in Autoimmune Diseases: How Close Are We to the Treatment? Front. Immunol. 2020, 11, 3062. [Google Scholar] [CrossRef]
- Haddadi, M.-H.; Hajizadeh-Saffar, E.; Khosravi-Maharlooei, M.; Basiri, M.; Negahdari, B.; Baharvand, H. Autoimmunity as a target for chimeric immune receptor therapy: A new vision to therapeutic potential. Blood Rev. 2020, 41, 100645. [Google Scholar] [CrossRef]
- Ellebrecht, C.T.; Bhoj, V.G.; Nace, A.; Choi, E.J.; Mao, X.; Cho, M.J.; Di Zenzo, G.; Lanzavecchia, A.; Seykora, J.T.; Cotsarelis, G.; et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 2016, 353, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Lundgren, D.K.; Mao, X.; Manfredo-Vieira, S.; Nunez-Cruz, S.; Williams, E.F.; Assenmacher, C.-A.; Radaelli, E.; Oh, S.; Wang, B.; et al. Antigen-specific B cell depletion for precision therapy of mucosal pemphigus vulgaris. J. Clin. Investig. 2020, 130, 6317–6324. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, Y.; Yuan, Y.; Sun, J.; Liu, L.; Huang, D.; Hu, J.; Wang, M.; Li, S.; Song, W.; et al. In vitro elimination of autoreactive B cells from rheumatoid arthritis patients by universal chimeric antigen receptor T cells. Ann. Rheum. Dis. 2021, 80, 176–184. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Disease | Major Affected Tissue(s) | Predominant Autoantigen(s) | Location of Autoantigen(s) | Predominant Autoantibodies | B Cell-Depleting Therapy Effective? | References |
|---|---|---|---|---|---|---|
| Rheumatoid arthritis (RA) | Joints | Cyclic citrullinated peptides/proteins (CCP), IgG | Intra- and extracellular | Anti-citrullinated protein antibodies (ACPAs), rheumatoid factor (RF) | Yes | [7,8] |
| Systemic lupus erythematosus (SLE) | Multiple | Double-stranded DNA, Smith, SSA(Ro), SSB(La) | Nucleus | Anti-nuclear (ANA), anti-double-stranded DNA, anti-Smith, anti-SSA(Ro), anti-SSB(La) | Inconclusive | [4,9] |
| Granulomatosis with polyangiitis (GPA) | Airways, kidneys | Proteinase-3 (PR3) | Cytoplasm | PR3-anti-neutrophil cytoplasmic antibody (PR3-ANCA) | Yes | [10] |
| Microscopic polyangiitis | Kidneys, skin | Myeloperoxidase (MPO) | Cytoplasm | MPO-ANCA | Yes | [10] |
| Pemphigus vulgaris | Oral mucosa and/or skin | Desmoglein-3 (Dsg3), Desmoglein-1 (Dsg1) | Cell surface | Anti-Dsg3, Anti-Dsg1 | Yes | [11] |
| Pemphigus foliaceus | Skin | Dsg1 | Cell surface | Anti-Dsg1 | Yes | [11] |
| Bullous pemphigoid | Skin | BP180 BP230 | Cell surface Intracellular | Anti-BP180 Anti-BP230 | Yes | [12,13] |
| Sjögren’s syndrome | Salivary glands, lacrimal glands | SSA(Ro), SSB(La) | Nucleus | Anti-SSA(Ro), anti-SSB(La) | Inconclusive | [14] |
| Myasthenia gravis | Muscles | Acetylcholine receptor (AChR), muscle-specific kinase (MuSK) | Cell surface | Anti-AChR, anti-MuSK | Yes | [15] |
| Immune thrombocytopenia | Platelets | Platelet glycoprotein (GP) IIb/IIIa, GPIb-IX-V | Surface of platelets | Anti-Ib/IIIa, anti-GPIb–IX-V | Yes | [16,17] |
| Graves’ disease | Thyroid gland | Thyroid-stimulating hormone receptor (TSHR) | Cell surface | Anti-TSHR | Inconclusive | [18] |
| Anti–glomerular basement membrane disease | Kidneys, lungs | Type IV collagen | Extracellular | Anti–glomerular basement membrane antibody | Inconclusive | [19] |
| Multiple sclerosis | Central nervous system | Unknown | - | Unknown | Yes | [20] |
| Chronic inflammatory demyelinating polyneuropathy | Peripheral nervous system | Contactin-1, neurofascin-155/140/186 | Cell surface | Anti-contactin-1, anti-neurofascin-155/140/186 | Possibly | [21,22] |
| Guillain–Barré syndrome | Peripheral nervous system | GD1a, GM1b, GM1, GD1b, GT1a GalNac-GD1a, GQ1b | Cell surface | Anti-ganglioside | Unknown | [23,24] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonasia, C.G.; Abdulahad, W.H.; Rutgers, A.; Heeringa, P.; Bos, N.A. B Cell Activation and Escape of Tolerance Checkpoints: Recent Insights from Studying Autoreactive B Cells. Cells 2021, 10, 1190. https://doi.org/10.3390/cells10051190
Bonasia CG, Abdulahad WH, Rutgers A, Heeringa P, Bos NA. B Cell Activation and Escape of Tolerance Checkpoints: Recent Insights from Studying Autoreactive B Cells. Cells. 2021; 10(5):1190. https://doi.org/10.3390/cells10051190
Chicago/Turabian StyleBonasia, Carlo G., Wayel H. Abdulahad, Abraham Rutgers, Peter Heeringa, and Nicolaas A. Bos. 2021. "B Cell Activation and Escape of Tolerance Checkpoints: Recent Insights from Studying Autoreactive B Cells" Cells 10, no. 5: 1190. https://doi.org/10.3390/cells10051190