
Non-Tumorigenic Pluripotent Reparative Muse Cells Provide a New Therapeutic Approach for Neurologic Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
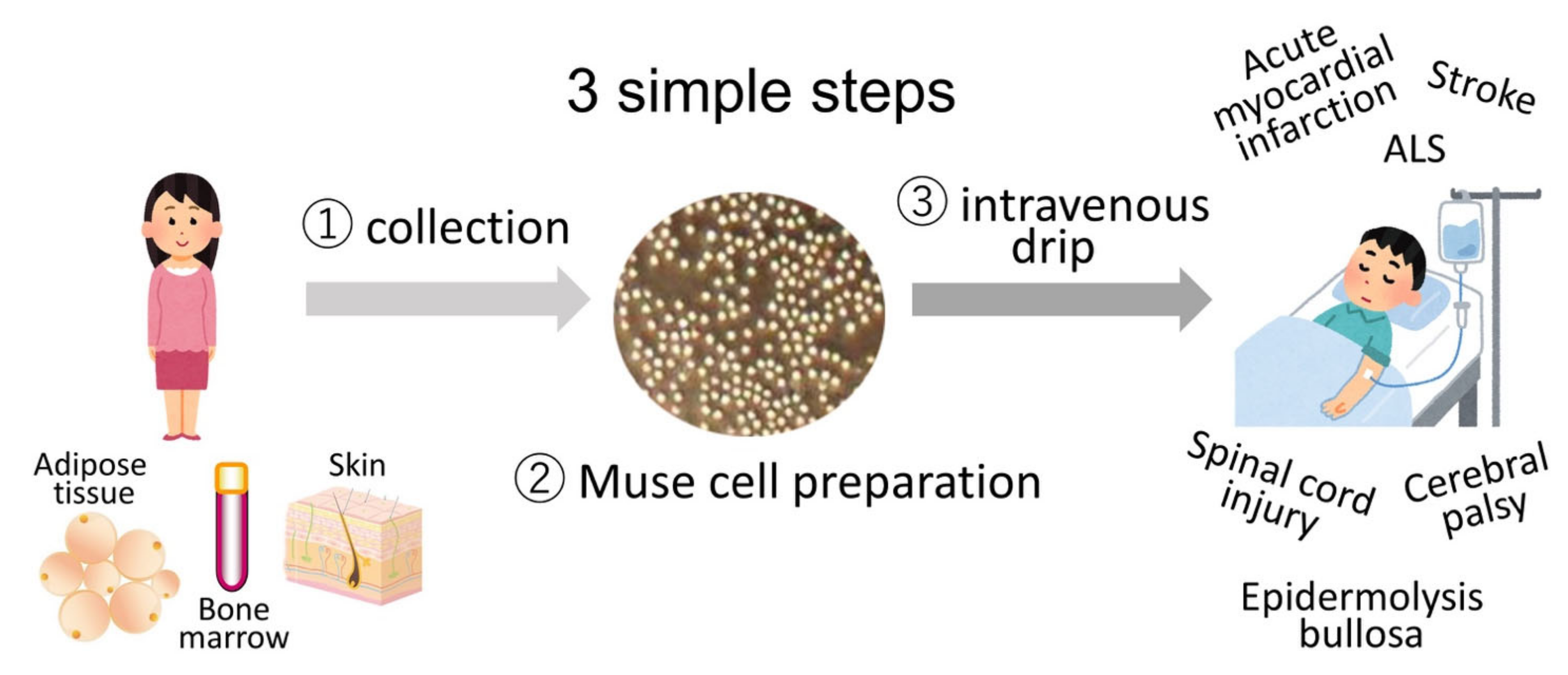{kind=link}
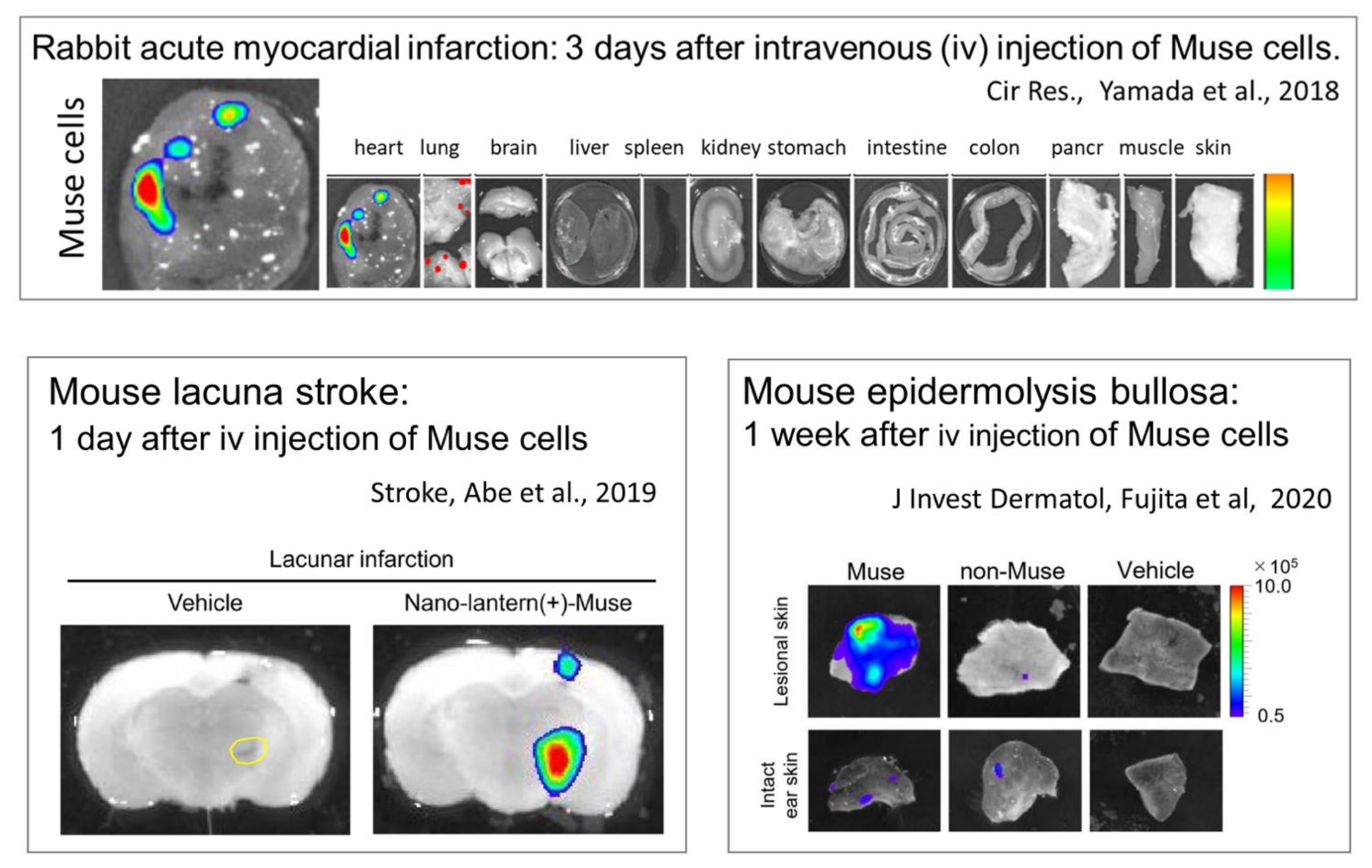{kind=link}
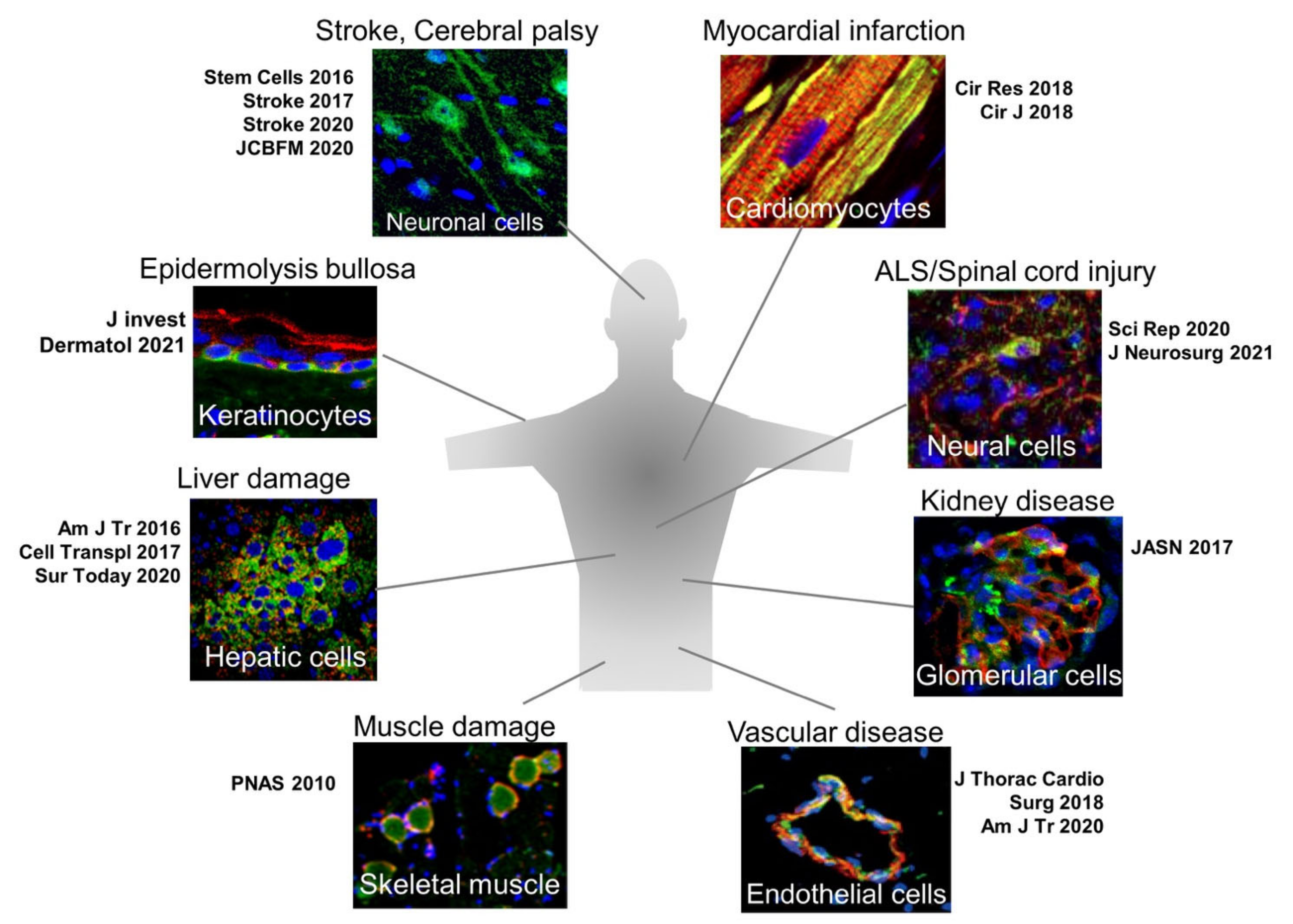{kind=link}
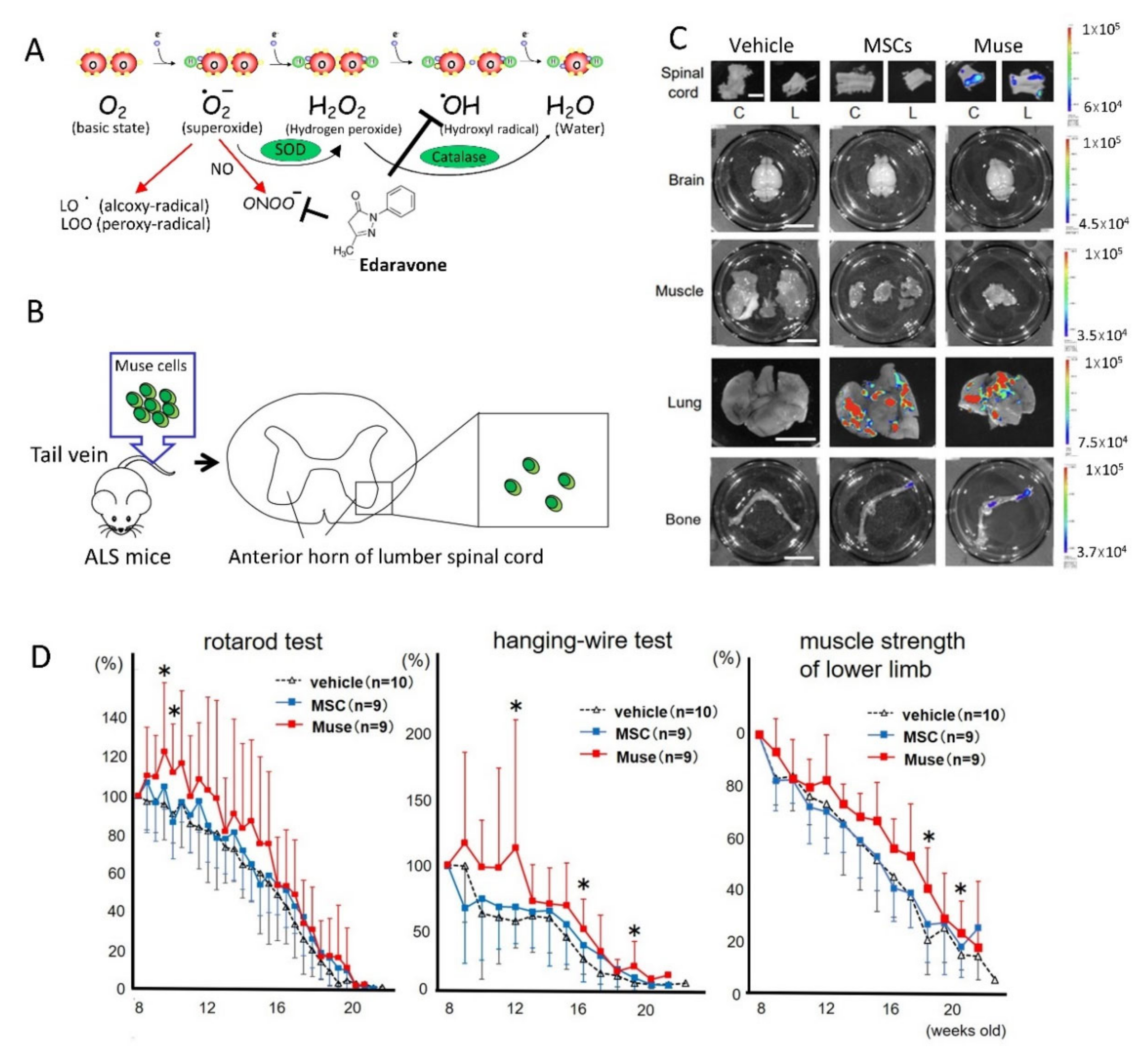{kind=link}
{kind=link}
Abstract
:1. Introduction
- Muse cells are endogenous and therefore elicit few safety concerns.
- Muse cells can be delivered intravenously and do not require surgery for their administration.
- Muse cells do not require gene introduction or cytokine treatment to be rendered pluripotent and induce differentiation.
- Donor Muse cells can be used without HLA-matching or immunosuppressant treatment.
- Muse cells remain incorporated as functional cells in the host tissue for an extended period of time, making their anti-inflammatory, anti-apoptotic, and trophic effects long-lasting.
2. Basic Characteristics of Muse Cells
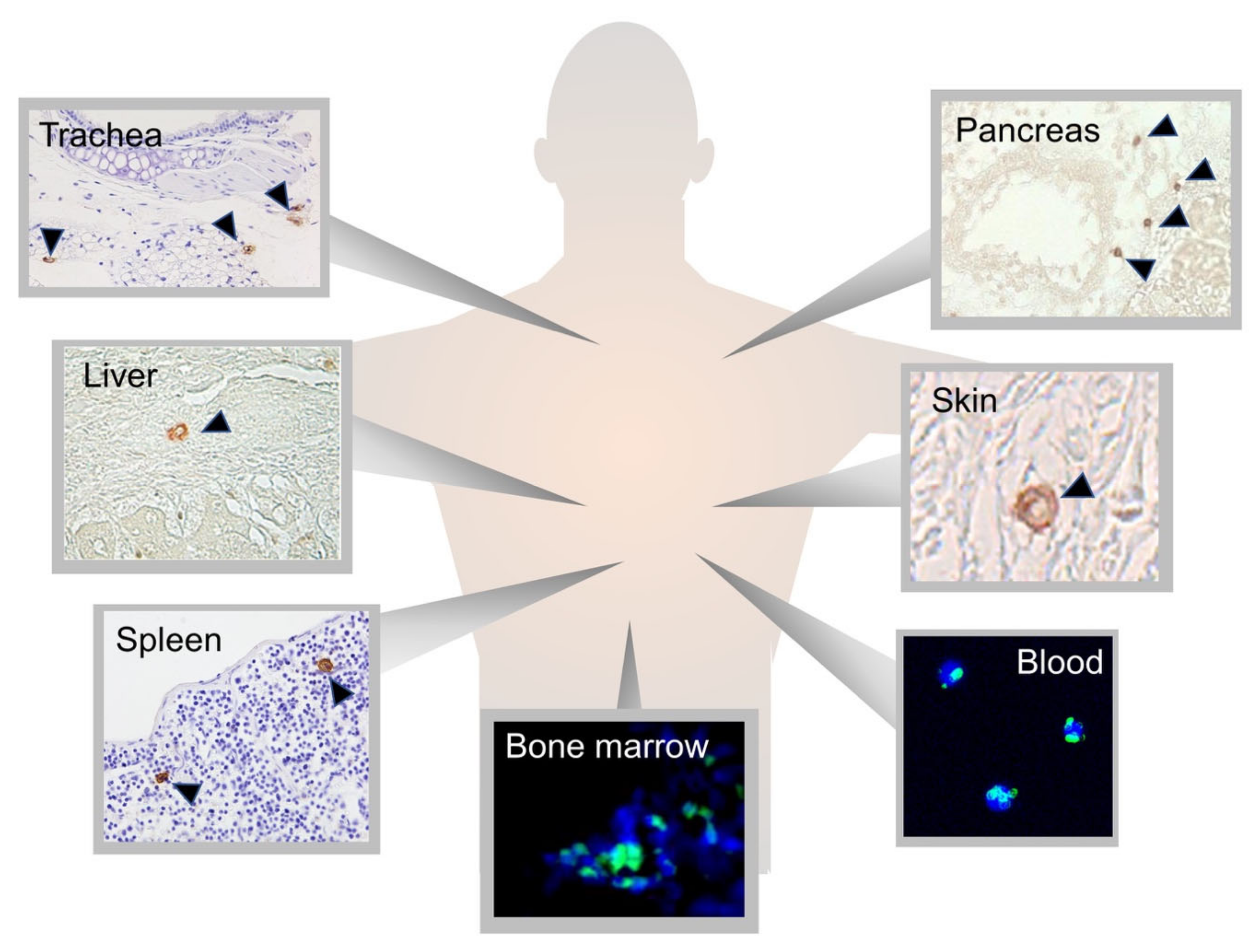
2.1. Muse Cells as Endogenous Reparative Stem Cells Are Widely Distributed in the Body
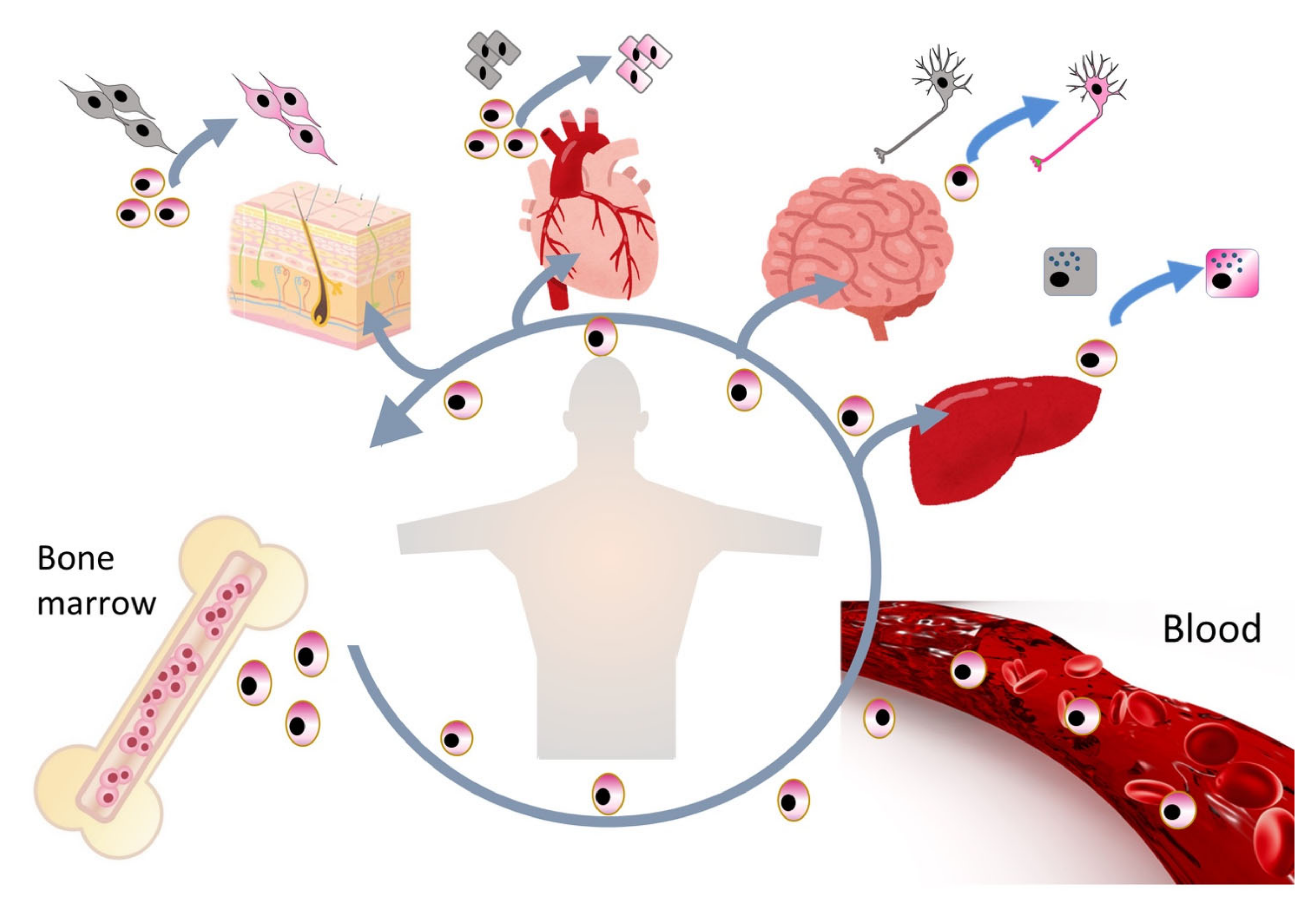
2.2. Sources of Muse Cells
2.3. Stress Tolerance, High DNA Repair Ability, and Non-Tumorigenicity
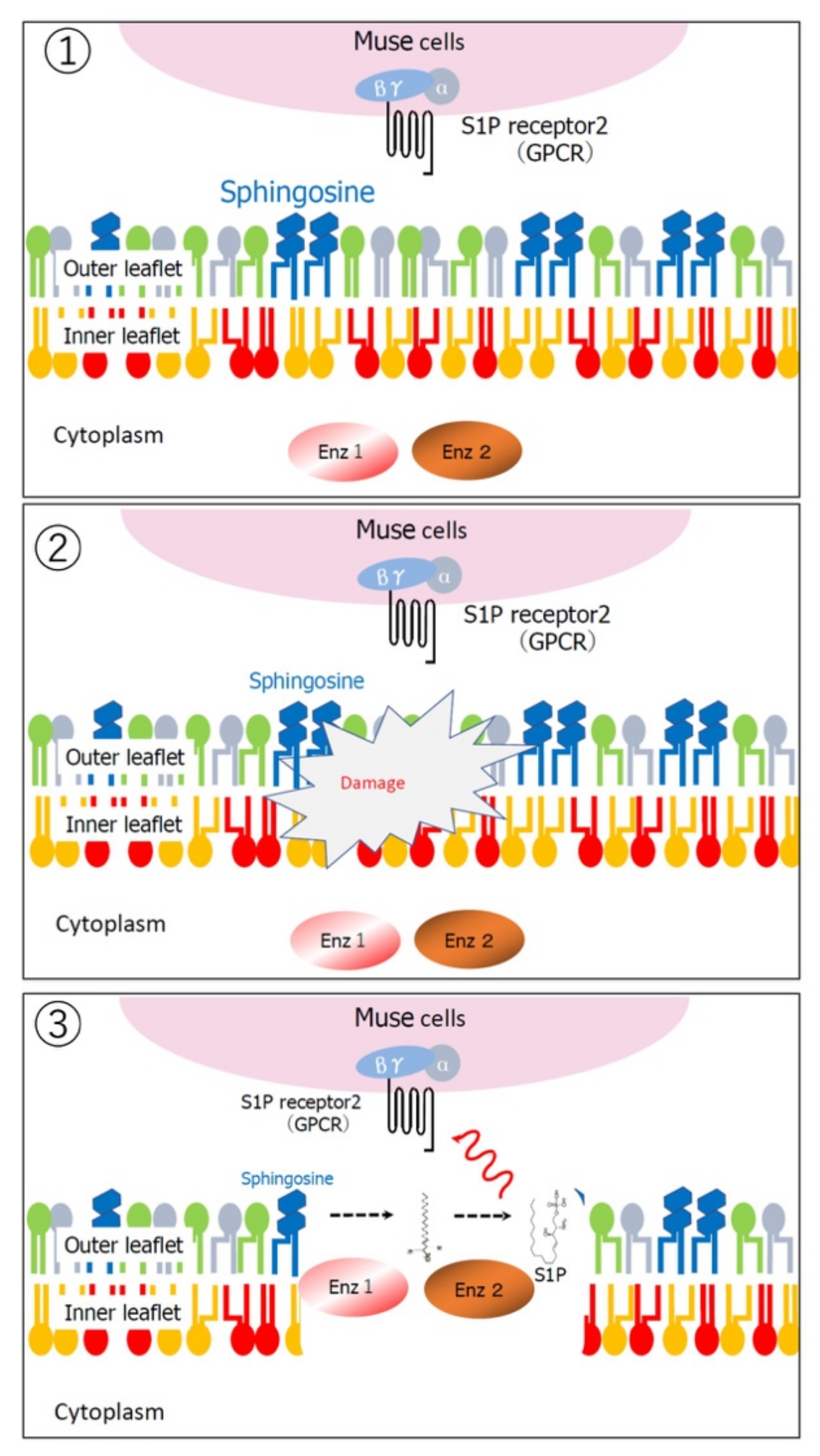
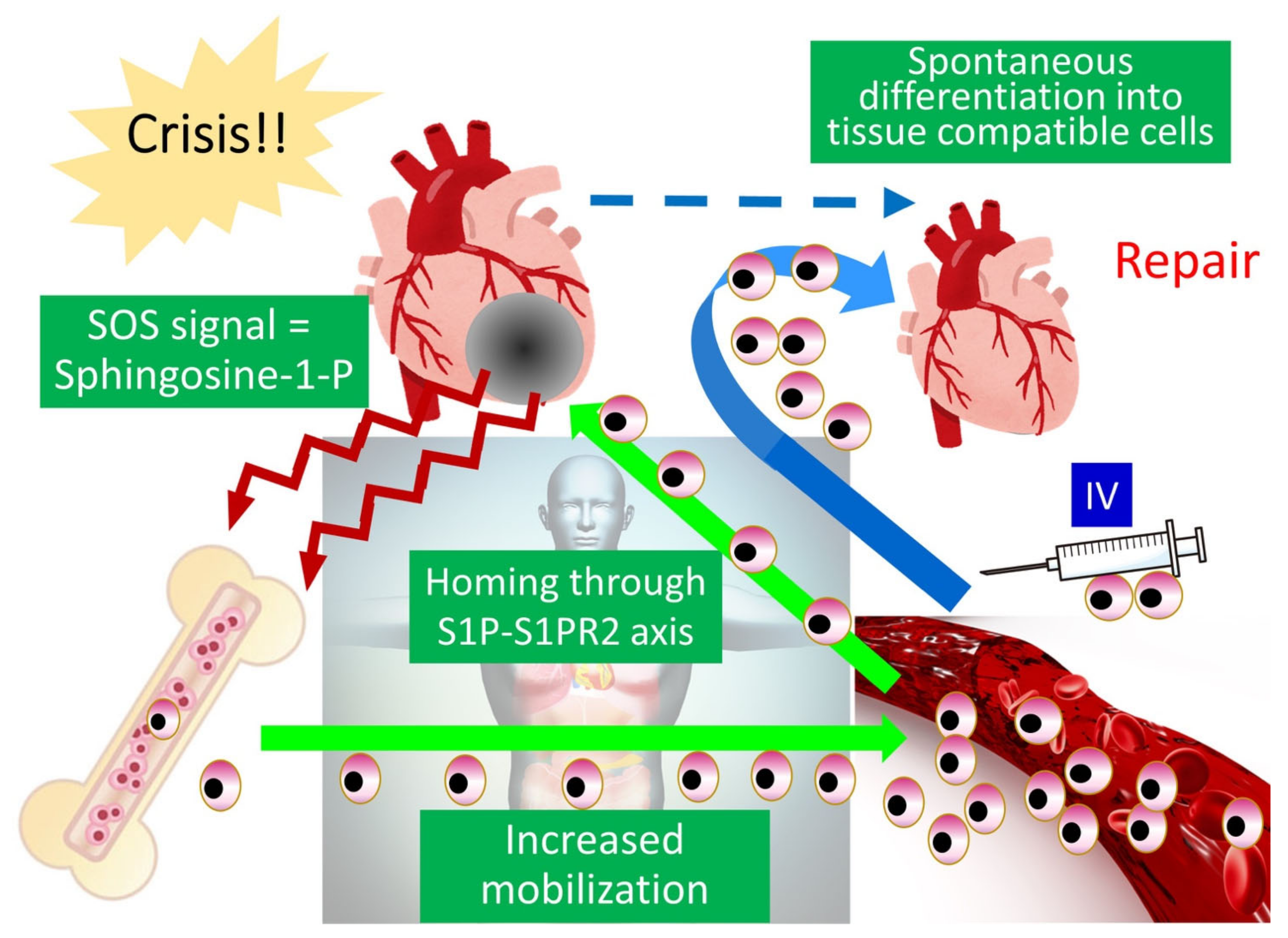
2.4. Ability to Selectively Home to Sites of Damage
2.5. Replacement of Damaged/Apoptotic Cells by Spontaneous Differentiation of Muse Cells into the Damaged/Apoptotic Cell Type
2.6. Immune Privilege of Muse Cells
2.7. Bystander Effects of Muse Cells on Tissue Repair
3. Comparison of the Reparative Effects of Muse Cells and MSCs
Comparison with Other Stem Cells
4. Muse Cell-Based Preclinical Studies in Neural Diseases
4.1. ALS
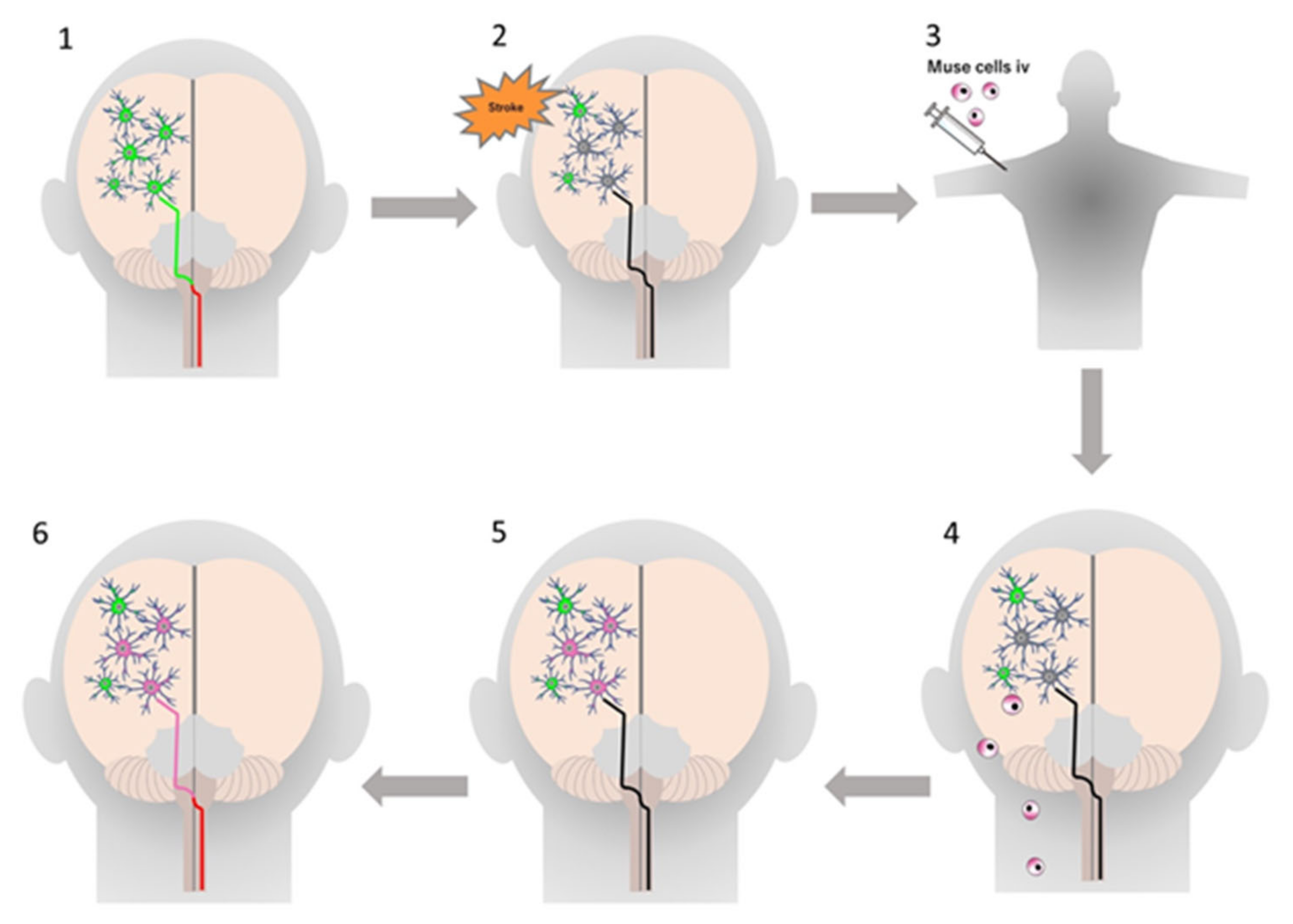
4.2. Stroke
4.3. Perinatal Hypoxic Ischemic Encephalopathy
4.4. Intracerebral Hemorrhage
4.5. Shiga Toxin-Producing E. coli-Associated Acute Encephalopathy
5. Clinical Trials and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kuroda, Y.; Kitada, M.; Wakao, S.; Nishikawa, K.; Tanimura, Y.; Makinoshima, H.; Goda, M.; Akashi, H.; Inutsuka, A.; Niwa, A.; et al. Unique multipotent cells in adult human mesenchymal cell populations. Proc. Natl. Acad. Sci. USA 2010, 107, 8639–8643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushida, Y.; Wakao, S.; Dezawa, M. Muse Cells Are Endogenous Reparative Stem Cells. Adv. Exp. Med. Biol. 2018, 1103, 43–68. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nishigaki, K.; Minatoguchi, S.; Nawa, T.; Yamada, Y.; Kanamori, H.; Mikami, A.; Ushikoshi, H.; Kawasaki, M.; Dezawa, M.; et al. Mobilized Muse Cells After Acute Myocardial Infarction Predict Cardiac Function and Remodeling in the Chronic Phase. Circ. J. 2018, 82, 561–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Wakao, S.; Kushida, Y.; Tatsumi, K.; Kitada, M.; Abe, T.; Niizuma, K.; Tominaga, T.; Kushimoto, S.; Dezawa, M. A Novel Type of Stem Cells Double-Positive for SSEA-3 and CD45 in Human Peripheral Blood. Cell Transplant. 2020, 29, 963689720923574. [Google Scholar] [CrossRef] [PubMed]
- Weigert, A.; Olesch, C.; Brüne, B. Sphingosine-1-Phosphate and Macrophage Biology—How the Sphinx Tames the Big Eater. Front. Immunol. 2019, 10, 1706. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Wakao, S.; Kushida, Y.; Minatoguchi, S.; Mikami, A.; Higashi, K.; Baba, S.; Shigemoto, T.; Kuroda, Y.; Kanamori, H.; et al. S1P–S1PR2 Axis Mediates Homing of Muse Cells Into Damaged Heart for Long-Lasting Tissue Repair and Functional Recovery After Acute Myocardial Infarction. Circ. Res. 2018, 122, 1069–1083. [Google Scholar] [CrossRef] [PubMed]
- Hori, E.; Hayakawa, Y.; Hayashi, T.; Hori, S.; Okamoto, S.; Shibata, T.; Kubo, M.; Horie, Y.; Sasahara, M.; Kuroda, S. Mobilization of Pluripotent Multilineage-Differentiating Stress-Enduring Cells in Ischemic Stroke. J. Stroke Cerebrovasc. Dis. 2016, 25, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Dezawa, M. Muse Cells Provide the Pluripotency of Mesenchymal Stem Cells: Direct Contribution of Muse Cells to Tissue Regeneration. Cell Transplant. 2016, 25, 849–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minatoguchi, S.; Mikami, A.; Tanaka, T.; Minatoguchi, S.; Yamada, Y. Acute Myocardial Infarction, Cardioprotection, and Muse Cells. Adv. Exp. Med. Biol. 2018, 1103, 153–166. [Google Scholar] [CrossRef]
- Noda, T.; Nishigaki, K.; Minatoguchi, S. Safety and Efficacy of Human Muse Cell-Based Product for Acute Myocardial Infarction in a First-in-Human Trial. Circ. J. 2020, 84, 1189–1192. [Google Scholar] [CrossRef]
- Fujita, Y.; Nohara, T.; Takashima, S.; Natsuga, K.; Adachi, M.; Yoshida, K.; Shinkuma, S.; Takeichi, T.; Nakamura, H.; Wada, O.; et al. Intravenous allogeneic multilineage-differentiating stress-enduring cells in adults with dystrophic epidermolysis bullosa: A phase 1/2 open-label study. J. Eur. Acad. Dermatol. Venereol. 2021. [Google Scholar] [CrossRef]
- Shevinsky, L.H.; Knowles, B.B.; Damjanov, I.; Solter, D. Monoclonal antibody to murine embryos defines a stage-specific embryonic antigen expressed on mouse embryos and human teratocarcinoma cells. Cell 1982, 30, 697–705. [Google Scholar] [CrossRef]
- Kannagi, R.; Cochran, N.A.; Ishigami, F.; Hakomori, S.; Andrews, P.W.; Knowles, B.B.; Solter, D. Stage-specific em-bryonic antigens (SSEA-3 and -4) are epitopes of a unique globo-series ganglioside isolated from human teratocarcino-ma cells. EMBO J. 1983, 2, 2355–2361. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liu, J.; Liu, H.; Qiu, M.; Liu, Q.; Zheng, L.; Pang, M.; Quan, F.; Zhang, Y. Isolation and Characterization of SSEA3+ Stem Cells Derived from Goat Skin Fibroblasts. Cell. Reprogr. 2013, 15, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Nitobe, Y.; Nagaoki, T.; Kumagai, G.; Sasaki, A.; Liu, X.; Fujita, T.; Fukutoku, T.; Wada, K.; Tanaka, T.; Kudo, H.; et al. Neurotrophic Factor Secretion and Neural Differentiation Potential of Multilineage-differentiating Stress-enduring (Muse) Cells Derived from Mouse Adipose Tissue. Cell Transplant. 2019, 28, 1132–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iseki, M.; Mizuma, M.; Wakao, S.; Kushida, Y.; Kudo, K.; Fukase, M.; Ishida, M.; Ono, T.; Shimura, M.; Ise, I.; et al. The evaluation of the safety and efficacy of intravenously administered allogeneic multilineage-differentiating stress-enduring cells in a swine hepatectomy model. Surg. Today 2021, 51, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Yang, L.; Cao, H.; Shen, Z.; Song, H. Study of the protective effect on damaged intestinal epithelial cells of rat multilineage-differentiating stress-enduring (Muse) cells. Cell Biol. Int. 2020, 44, 549–559. [Google Scholar] [CrossRef] [Green Version]
- Wakao, S.; Kushida, Y.; Dezawa, M. Correction to: Basic Characteristics of Muse Cells. In Muse Cells; Springer: Tokyo, Japan, 2019; p. C1. [Google Scholar]
- Leng, Z.; Sun, D.; Huang, Z.; Tadmori, I.; Chiang, N.; Kethidi, N.; Sabra, A.; Kushida, Y.; Fu, Y.-S.; Dezawa, M.; et al. Quantitative Analysis of SSEA3+ Cells from Human Umbilical Cord after Magnetic Sorting. Cell Transplant. 2019, 28, 907–923. [Google Scholar] [CrossRef]
- Rompolas, P.; Greco, V. Stem cell dynamics in the hair follicle niche. Semin. Cell Dev. Biol. 2014, 25–26, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Boulais, P.E.; Frenette, P.S. Making sense of hematopoietic stem cell niches. Blood 2015, 125, 2621–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakao, S.; Kitada, M.; Kuroda, Y.; Shigemoto, T.; Matsuse, D.; Akashi, H.; Tanimura, Y.; Tsuchiyama, K.; Kikuchi, T.; Goda, M.; et al. Multilineage-differentiating stress-enduring (Muse) cells are a primary source of induced pluripotent stem cells in human fibroblasts. Proc. Natl. Acad. Sci. USA 2011, 108, 9875–9880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiyama, K.; Wakao, S.; Kuroda, Y.; Ogura, F.; Nojima, M.; Sawaya, N.; Yamasaki, K.; Aiba, S.; Dezawa, M. Functional Melanocytes Are Readily Reprogrammable from Multilineage-Differentiating Stress-Enduring (Muse) Cells, Distinct Stem Cells in Human Fibroblasts. J. Investig. Dermatol. 2013, 133, 2425–2435. [Google Scholar] [CrossRef] [Green Version]
- Amin, M.; Kushida, Y.; Wakao, S.; Kitada, M.; Tatsumi, K.; Dezawa, M. Cardiotrophic Growth Factor–Driven Induction of Human Muse Cells into Cardiomyocyte-Like Phenotype. Cell Transplant. 2018, 27, 285–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, Y.; Wakao, S.; Kitada, M.; Murakami, T.; Nojima, M.; Dezawa, M. Isolation, culture and evaluation of multilineage-differentiating stress-enduring (Muse) cells. Nat. Protoc. 2013, 8, 1391–1415. [Google Scholar] [CrossRef]
- Ogura, F.; Wakao, S.; Kuroda, Y.; Tsuchiyama, K.; Bagheri, M.; Heneidi, S.; Chazenbalk, G.; Aiba, S.; Dezawa, M. Human Adipose Tissue Possesses a Unique Population of Pluripotent Stem Cells with Nontumorigenic and Low Telomerase Activities: Potential Implications in Regenerative Medicine. Stem Cells Dev. 2014, 23, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Iseki, M.; Kushida, Y.; Wakao, S.; Akimoto, T.; Mizuma, M.; Motoi, F.; Asada, R.; Shimizu, S.; Unno, M.; Chazenbalk, G.; et al. Human Muse Cells, Nontumorigenic Phiripotent-Like Stem Cells, Have Liver Regeneration Capacity through Specific Homing and Cell Replacement in a Mouse Model of Liver Fibrosis. Cell Transplant. 2017, 26, 821–840. [Google Scholar] [CrossRef]
- Alessio, N.; Özcan, S.; Tatsumi, K.; Murat, A.; Peluso, G.; Dezawa, M.; Galderisi, U. The secretome of MUSE cells contains factors that may play a role in regulation of stemness, apoptosis and immunomodulation. Cell Cycle 2017, 16, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Alessio, N.; Squillaro, T.; Özcan, S.; Di Bernardo, G.; Venditti, M.; Melone, M.; Peluso, G.; Galderisi, U. Stress and stem cells: Adult Muse cells tolerate extensive genotoxic stimuli better than mesenchymal stromal cells. Oncotarget 2018, 9, 19328–19341. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, M.L.; Fuertes, F.; Tabarrozzi, A.E.B.; Attorressi, A.I.; Cucchiani, R.; Corrales, L.; Oliveira, T.C.; Sogayar, M.C.; Labriola, L.; Dewey, R.A.; et al. Pluripotent Nontumorigenic Adipose Tissue-Derived Muse Cells have Immunomodulatory Capacity Mediated by Transforming Growth Factor-β1. Stem Cells Transl. Med. 2016, 6, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Milstien, S.; Spiegel, S. Generation and metabolism of bioactive sphingosine-1-phosphate. J. Cell. Biochem. 2004, 92, 882–899. [Google Scholar] [CrossRef]
- De Becker, A.; Van Riet, I. Homing and migration of mesenchymal stromal cells: How to improve the efficacy of cell therapy? World J. Stem Cells 2016, 8, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Komatsu, M.; Lee, S.E.; Kushida, Y.; Nakayama-Nishimura, C.; Matsumura, W.; Takashima, S.; Shinkuma, S.; Nomura, T.; Masutomi, N.; et al. Intravenous Injection of Muse Cells as a Potential Therapeutic Approach for Epidermolysis Bullosa. J. Investig. Dermatol. 2021, 141, 198–202.e6. [Google Scholar] [CrossRef]
- Uchida, H.; Niizuma, K.; Kushida, Y.; Wakao, S.; Tominaga, T.; Borlongan, C.V.; Dezawa, M. Human Muse Cells Reconstruct Neuronal Circuitry in Subacute Lacunar Stroke Model. Stroke 2017, 48, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Uchida, N.; Kushida, Y.; Kitada, M.; Wakao, S.; Kumagai, N.; Kuroda, Y.; Kondo, Y.; Hirohara, Y.; Kure, S.; Chazenbalk, G.; et al. Beneficial Effects of Systemically Administered Human Muse Cells in Adriamycin Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 2946–2960. [Google Scholar] [CrossRef]
- Yamashita, T.; Kushida, Y.; Wakao, S.; Tadokoro, K.; Nomura, E.; Omote, Y.; Takemoto, M.; Hishikawa, N.; Ohta, Y.; Dezawa, M.; et al. Therapeutic benefit of Muse cells in a mouse model of amyotrophic lateral sclerosis. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Ozuru, R.; Wakao, S.; Tsuji, T.; Ohara, N.; Matsuba, T.; Amuran, M.Y.; Isobe, J.; Iino, M.; Nishida, N.; Matsumoto, S.; et al. Rescue from Stx2-Producing E. coli-Associated Encephalopathy by Intravenous Injection of Muse Cells in NOD-SCID Mice. Mol. Ther. 2020, 28, 100–118. [Google Scholar] [CrossRef] [Green Version]
- Uchida, H.; Morita, T.; Niizuma, K.; Kushida, Y.; Kuroda, Y.; Wakao, S.; Sakata, H.; Matsuzaka, Y.; Mushiake, H.; Tominaga, T.; et al. Transplantation of Unique Subpopulation of Fibroblasts, Muse Cells, Ameliorates Experimental Stroke Possibly via Robust Neuronal Differentiation. Stem Cells 2016, 34, 160–173. [Google Scholar] [CrossRef]
- Suzuki, T.; Sato, Y.; Kushida, Y.; Tsuji, M.; Wakao, S.; Ueda, K.; Imai, K.; Iitani, Y.; Shimizu, S.; Hida, H.; et al. Intra-venously delivered multilineage-differentiating stress enduring cells dampen excessive glutamate metabolism and mi-croglial activation in experimental perinatal hypoxic ischemic encephalopathy. J. Cereb. Blood Flow 2020, 0271678X20972656. [Google Scholar] [CrossRef]
- Shimamura, N.; Kakuta, K.; Wang, L.; Naraoka, M.; Uchida, H.; Wakao, S.; Dezawa, M.; Ohkuma, H. Neuro-regeneration therapy using human Muse cells is highly effective in a mouse intracerebral hemorrhage model. Exp. Brain Res. 2017, 235, 565–572. [Google Scholar] [CrossRef]
- Katagiri, H.; Kushida, Y.; Nojima, M.; Kuroda, Y.; Wakao, S.; Ishida, K.; Endo, F.; Kume, K.; Takahara, T.; Nitta, H.; et al. A Distinct Subpopulation of Bone Marrow Mesenchymal Stem Cells, Muse Cells, Directly Commit to the Replacement of Liver Components. Arab. Archaeol. Epigr. 2016, 16, 468–483. [Google Scholar] [CrossRef]
- Hosoyama, K.; Wakao, S.; Kushida, Y.; Ogura, F.; Maeda, K.; Adachi, O.; Kawamoto, S.; Dezawa, M.; Saiki, Y. Intravenously injected human multilineage-differentiating stress-enduring cells selectively engraft into mouse aortic aneurysms and attenuate dilatation by differentiating into multiple cell types. J. Thorac. Cardiovasc. Surg. 2018, 155, 2301–2313.e4. [Google Scholar] [CrossRef]
- Ankrum, J.A.; Ong, J.F.; Karp, J.M. Mesenchymal stem cells: Immune evasive, not immune privileged. Nat. Biotechnol. 2014, 32, 252–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Chen, S.; Yang, P.; Cao, H.; Li, L. The role of mesenchymal stem cells in hematopoietic stem cell transplantation: Prevention and treatment of graft-versus-host disease. Stem Cell Res. Ther. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Najima, Y.; Ohashi, K. Mesenchymal Stem Cells: The First Approved Stem Cell Drug in Japan. J. Hematop. Cell Transplant. 2017, 6, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Loustau, M.; Anna, F.; Dréan, R.; LeComte, M.; Langlade-Demoyen, P.; Caumartin, J. HLA-G Neo-Expression on Tumors. Front. Immunol. 2020, 11, 1685. [Google Scholar] [CrossRef] [PubMed]
- Yabuki, H.; Wakao, S.; Kushida, Y.; Dezawa, M.; Okada, Y. Human Multilineage-differentiating Stress-Enduring Cells Exert Pleiotropic Effects to Ameliorate Acute Lung Ischemia–Reperfusion Injury in a Rat Model. Cell Transplant. 2018, 27, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Shono, Y.; Kushida, Y.; Wakao, S.; Kuroda, Y.; Unno, M.; Kamei, T.; Miyagi, S.; Dezawa, M. Protection of liver sinusoids by intravenous administration of human Muse cells in a rat extra-small partial liver transplantation model. Arab. Archaeol. Epigr. 2020. [Google Scholar] [CrossRef]
- Surgucheva, I.; Chidambaram, K.; Willoughby, D.A.; Surguchov, A. Matrix metalloproteinase 9 expression: New regulatory elements. J. Ocul. Biol. Dis. Inform. 2010, 3, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Ratajczak, M.Z.; Shin, D.-M.; Liu, R.; Mierzejewska, K.; Ratajczak, J.; Kucia, M.; Zuba-Surma, E.K. Very small embryonic/epiblast-like stem cells (VSELs) and their potential role in aging and organ rejuvenation—An update and comparison to other primitive small stem cells isolated from adult tissues. Aging 2012, 4, 235–246. [Google Scholar] [CrossRef]
- Jiang, Y.; Vaessen, B.; Lenvik, T.; Blackstad, M.; Reyes, M.; Verfaillie, C.M. Multipotent progenitor cells can be isolated from postnatal murine bone marrow, muscle, and brain. Exp. Hematol. 2002, 30, 896–904. [Google Scholar] [CrossRef]
- Nichols, J.; Smith, A. Naive and Primed Pluripotent States. Cell Stem Cell 2009, 4, 487–492. [Google Scholar] [CrossRef] [Green Version]
- Ying, Q.-L.; Nichols, J.; Chambers, I.; Smith, A. BMP Induction of Id Proteins Suppresses Differentiation and Sustains Embryonic Stem Cell Self-Renewal in Collaboration with STAT3. Cell 2003, 115, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Tesar, P.J.; Chenoweth, J.G.; Brook, F.A.; Davies, T.J.; Evans, E.P.; Mack, D.L.; Gardner, R.L.; McKay, R.D.G. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nat. Cell Biol. 2007, 448, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef]
- Ogasawara, M.; Matsubara, Y.; Narisawa, K.; Aoki, M.; Nakamura, S.; Itoyama, Y.; Abe, K. Mild ALS in Japan associated with novel SOD mutation. Nat. Genet. 1993, 5, 323–324. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Paré, B.; Lehmann, M.; Beaudin, M.; Nordström, U.; Saikali, S.; Julien, J.-P.; Gilthorpe, J.D.; Marklund, S.L.; Cashman, N.R.; Andersen, P.M.; et al. Misfolded SOD1 pathology in sporadic Amyotrophic Lateral Sclerosis. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of Increased Oxidative Damage in Both Sporadic and Familial Amyotrophic Lateral Sclerosis. J. Neurochem. 2002, 69, 2064–2074. [Google Scholar] [CrossRef]
- Warita, H.; Hayashi, T.; Murakami, T.; Manabe, Y.; Abe, K. Oxidative damage to mitochondrial DNA in spinal motoneurons of transgenic ALS mice. Mol. Brain Res. 2001, 89, 147–152. [Google Scholar] [CrossRef]
- Miyazaki, K.; Ohta, Y.; Nagai, M.; Morimoto, N.; Kurata, T.; Takehisa, Y.; Ikeda, Y.; Matsuura, T.; Abe, K. Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J. Neurosci. Res. 2011, 89, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Itoyama, Y.; Sobue, G.; Tsuji, S.; Aoki, M.; Doyu, M.; Hamada, C.; Kondo, K.; Yoneoka, T.; Akimoto, M.; et al. Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 610–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, K.; Aoki, M.; Tsuji, S.; Itoyama, Y.; Sobue, G.; Togo, M.; Hamada, C.; Tanaka, M.; Akimoto, M.; Nakamura, K.; et al. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Abe, T.; Aburakawa, D.; Niizuma, K.; Iwabuchi, N.; Kajitani, T.; Wakao, S.; Kushida, Y.; Dezawa, M.; Borlongan, C.V.; Tominaga, T. Intravenously Transplanted Human Multilineage-Differentiating Stress-Enduring Cells Afford Brain Repair in a Mouse Lacunar Stroke Model. Stroke 2020, 51, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage Potential of Adult Human Mesenchymal Stem Cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Karbaat, L.; Wu, L.; Leijten, J.; Both, S.K.; Karperien, M. Trophic Effects of Mesenchymal Stem Cells in Tissue Regeneration. Tissue Eng. Part B Rev. 2017, 23, 515–528. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamashita, T.; Kushida, Y.; Abe, K.; Dezawa, M. Non-Tumorigenic Pluripotent Reparative Muse Cells Provide a New Therapeutic Approach for Neurologic Diseases. Cells 2021, 10, 961. https://doi.org/10.3390/cells10040961
Yamashita T, Kushida Y, Abe K, Dezawa M. Non-Tumorigenic Pluripotent Reparative Muse Cells Provide a New Therapeutic Approach for Neurologic Diseases. Cells. 2021; 10(4):961. https://doi.org/10.3390/cells10040961
Chicago/Turabian StyleYamashita, Toru, Yoshihiro Kushida, Koji Abe, and Mari Dezawa. 2021. "Non-Tumorigenic Pluripotent Reparative Muse Cells Provide a New Therapeutic Approach for Neurologic Diseases" Cells 10, no. 4: 961. https://doi.org/10.3390/cells10040961