
Tissue-Resident and Recruited Macrophages in Primary Tumor and Metastatic Microenvironments: Potential Targets in Cancer Therapy


Abstract
:1. Introduction
2. Macrophage Nomenclature and Identification
3. Monopoiesis and Macrophage Ontology
4. Tissue-Resident Macrophages
4.1. Tissue-Resident Macrophages in Cancer
4.2. Tissue-Resident Macrophages in Metastasis
5. Tumor-Associated Macrophages
Prognostic Significance of TAM Density across Different Tumor Types
6. Metastasis-Associated Macrophages (MAMs)
7. Macrophages and Therapeutic Efficacy
7.1. Macrophages Restrict T Cell Mobilization and Anti-Tumor Immune Responses
7.2. Macrophages Impair Responses to Chemotherapy and Radiotherapy
7.3. Macrophages Impair Immune Checkpoint Blockade Responses
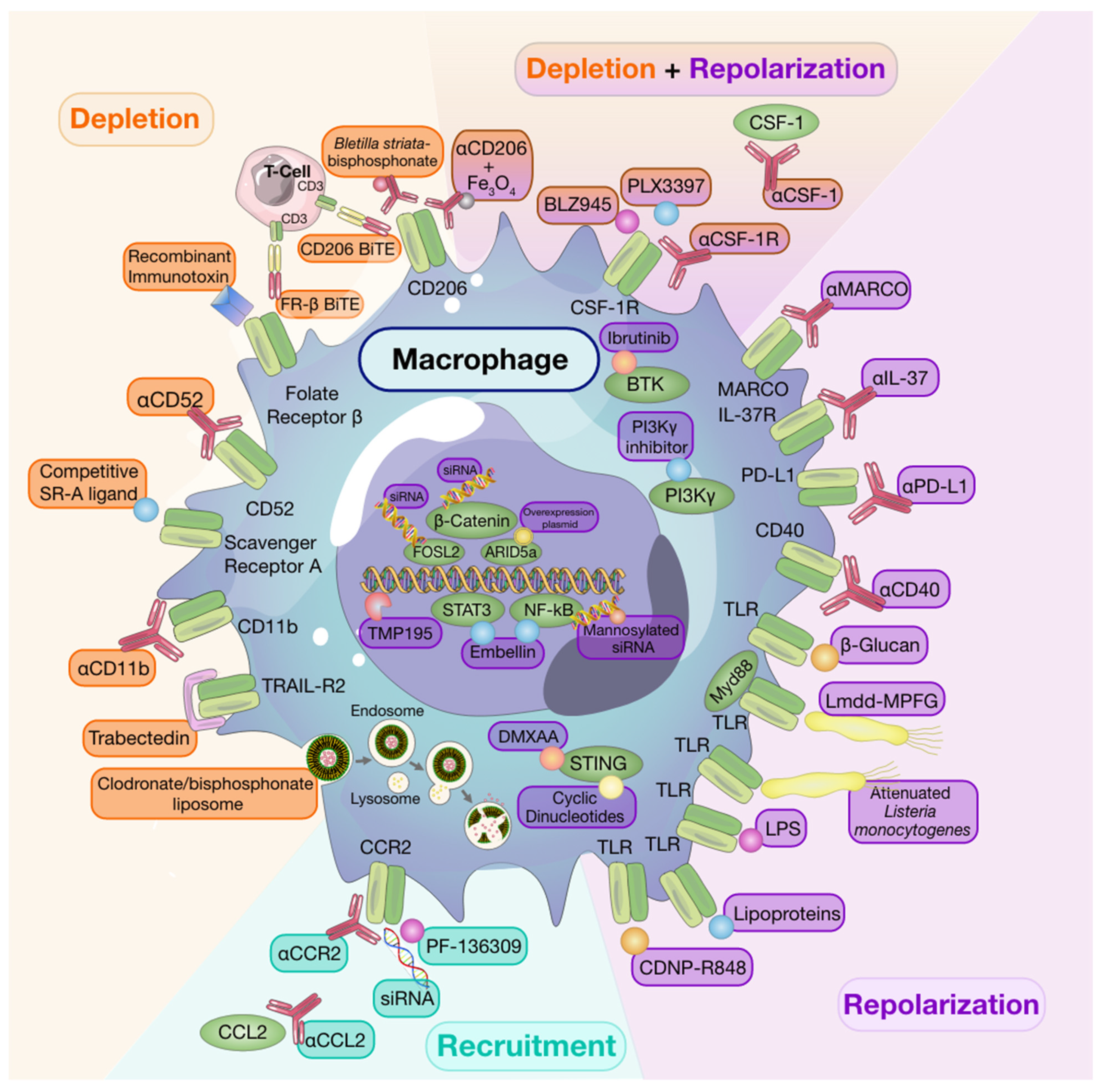
8. Targeting Macrophages to Potentiate Anti-Cancer Therapy
8.1. Macrophage Depletion
8.2. Recruitment
8.3. Repolarization
9. Exploiting Macrophages as Targeted Drug Delivery Systems
9.1. Chimeric Antigen Receptor Macrophage Therapy
9.2. Macrophage-Mediated Drug Delivery
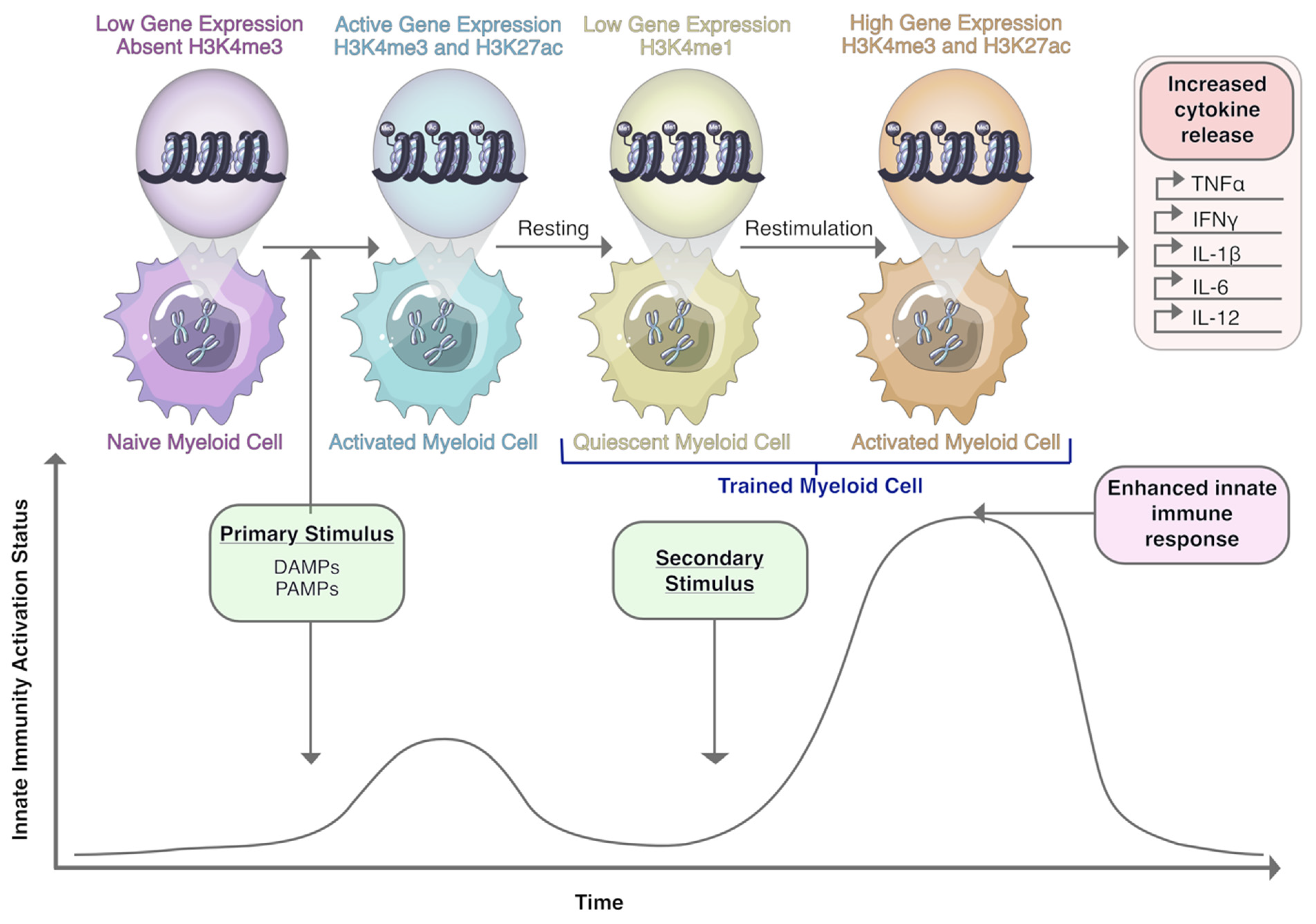
10. Macrophages and Trained Immunity: A Novel Therapeutic Target?
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Pawelek, J.; Chakraborty, A.; Lazova, R.; Yilmaz, Y.; Cooper, D.; Brash, D.; Handerson, T. Co-opting macrophage traits in cancer progression: A consequence of tumor cell fusion? Contrib. Microbiol. 2006, 13, 138–155. [Google Scholar]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Kitamura, T. Targeting Tumor-Associated Macrophages as a Potential Strategy to Enhance the Response to Immune Checkpoint Inhibitors. Front. Cell Dev. Biol. 2018, 6, 38. [Google Scholar] [CrossRef]
- Anfray, C.; Ummarino, A.; Andon, F.T.; Allavena, P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells 2019, 9, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Kowal, J.; Kornete, M.; Joyce, J.A. Re-education of macrophages as a therapeutic strategy in cancer. Immunotherapy 2019, 11, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.W.; Giannarelli, C.; Rahman, A.; Randolph, G.J.; Kovacic, J.C. Macrophage Biology, Classification, and Phenotype in Cardiovascular Disease: JACC Macrophage in CVD Series (Part 1). J. Am. Coll. Cardiol. 2018, 72, 2166–2180. [Google Scholar] [CrossRef] [PubMed]
- Perdiguero, E.G.; Geissmann, F. The development and maintenance of resident macrophages. Nat. Immunol. 2016, 17, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passlick, B.; Flieger, D.; Ziegler-Heitbrock, H.W. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood 1989, 74, 2527–2534. [Google Scholar] [CrossRef] [Green Version]
- Sunderkotter, C.; Nikolic, T.; Dillon, M.J.; Van Rooijen, N.; Stehling, M.; Drevets, D.A.; Leenen, P.J. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J. Immunol. 2004, 172, 4410–4417. [Google Scholar] [CrossRef]
- Lichanska, A.M.; Hume, D.A. Origins and functions of phagocytes in the embryo. Exp. Hematol. 2000, 28, 601–611. [Google Scholar] [CrossRef] [Green Version]
- Kratofil, R.M.; Kubes, P.; Deniset, J.F. Monocyte Conversion During Inflammation and Injury. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Jakubzick, C.V.; Randolph, G.J.; Henson, P.M. Monocyte differentiation and antigen-presenting functions. Nat. Rev. Immunol. 2017, 17, 349–362. [Google Scholar] [CrossRef]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Stanley, E.R.; Chitu, V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb. Perspect. Biol. 2014, 6, a021857. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, J.Y.; Jalil, A.; Klaine, M.; Jung, S.; Cumano, A.; Godin, I. Three pathways to mature macrophages in the early mouse yolk sac. Blood 2005, 106, 3004–3011. [Google Scholar] [CrossRef]
- Sheng, J.; Ruedl, C.; Karjalainen, K. Most Tissue-Resident Macrophages Except Microglia Are Derived from Fetal Hematopoietic Stem Cells. Immunity 2015, 43, 382–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdiguero, E.G.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2014. [Google Scholar] [CrossRef]
- Bain, C.C.; Bravo-Blas, A.; Scott, C.L.; Perdiguero, E.G.; Geissmann, F.; Henri, S.; Malissen, B.; Osborne, L.C.; Artis, D.; Mowat, A.M. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol. 2014, 15, 929–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, C.L.; Zheng, F.; De Baetselier, P.; Martens, L.; Saeys, Y.; De Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun. 2016, 7, 10321. [Google Scholar] [CrossRef] [PubMed]
- Molawi, K.; Wolf, Y.; Kandalla, P.K.; Favret, J.; Hagemeyer, N.; Frenzel, K.; Pinto, A.R.; Klapproth, K.; Henri, S.; Malissen, B.; et al. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 2014, 211, 2151–2158. [Google Scholar] [CrossRef]
- Van Hove, H.; Martens, L.; Scheyltjens, I.; De Vlaminck, K.; Pombo Antunes, A.R.; De Prijck, S.; Vandamme, N.; De Schepper, S.; Van Isterdael, G.; Scott, C.L.; et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 2019, 22, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Gu, Y.; Chakarov, S.; Bleriot, C.; Kwok, I.; Chen, X.; Shin, A.; Huang, W.; Dress, R.J.; Dutertre, C.A.; et al. Fate Mapping via Ms4a3-Expression History Traces Monocyte-Derived Cells. Cell 2019, 178, 1509–1525.e19. [Google Scholar] [CrossRef]
- T’Jonck, W.; Guilliams, M.; Bonnardel, J. Niche signals and transcription factors involved in tissue-resident macrophage development. Cell. Immunol. 2018, 330, 43–53. [Google Scholar] [CrossRef]
- Tan, S.Y.; Krasnow, M.A. Developmental origin of lung macrophage diversity. Development 2016, 143, 1318–1327. [Google Scholar] [CrossRef] [Green Version]
- De Schepper, S.; Verheijden, S.; Aguilera-Lizarraga, J.; Viola, M.F.; Boesmans, W.; Stakenborg, N.; Voytyuk, I.; Schmidt, I.; Boeckx, B.; Dierckx de Casterle, I.; et al. Self-Maintaining Gut Macrophages Are Essential for Intestinal Homeostasis. Cell 2018, 175, 400–415.e13. [Google Scholar] [CrossRef] [Green Version]
- Guilliams, M.; Svedberg, F.R. Does tissue imprinting restrict macrophage plasticity? Nat. Immunol. 2021, 22, 118–127. [Google Scholar] [CrossRef]
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef] [Green Version]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chaffee, T.S.; LaRue, R.S.; Huggins, D.N.; Witschen, P.M.; Ibrahim, A.M.; Nelson, A.C.; Machado, H.L.; Schwertfeger, K.L. Tissue-resident macrophages promote extracellular matrix homeostasis in the mammary gland stroma of nulliparous mice. Elife 2020, 9, e57438. [Google Scholar] [CrossRef]
- Jappinen, N.; Felix, I.; Lokka, E.; Tyystjarvi, S.; Pynttari, A.; Lahtela, T.; Gerke, H.; Elima, K.; Rantakari, P.; Salmi, M. Fetal-derived macrophages dominate in adult mammary glands. Nat. Commun. 2019, 10, 281. [Google Scholar] [CrossRef] [Green Version]
- Pollard, J.W.; Stanley, E.R. Pleiotropic roles for CSF-1 in development defined by the mouse mutation osteopetrotic (op). Adv. Dev. Biochem. 1996, 4, 153–193. [Google Scholar]
- Dai, X.; Ryan, G.R.; Hapel, A.J.; Dominguez, M.G.; Russell, R.G.; Kapp, S.; Sylvestre, V.; Stanley, E.R. Targeted disruption of the mouse CSF-1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primititive progenitor cell frequencies and reproductive defects. Blood 2002, 99, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Banaei-Bouchareb, L.; Gouon-Evans, V.; Samara-Boustani, D.; Castellotti, M.C.; Czernichow, P.; Pollard, J.W.; Polak, M. Insulin cell mass is altered in Csf1op/Csf1op macrophage-deficient mice. J. Leukoc. Biol. 2004, 76, 359–367. [Google Scholar] [CrossRef]
- Gouon-Evans, V.; Rothenberg, M.E.; Pollard, J.W. Postnatal mammary gland development requires macrophages and eosinophils. Development 2000, 127, 2269–2282. [Google Scholar]
- Pollard, J.W.; Hennighausen, L. Colony stimulating factor 1 is required for mammary gland development during pregnancy. Proc. Natl. Acad. Sci. USA 1994, 91, 9312–9316. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Lee, E.; Hestir, K.; Leo, C.; Huang, M.; Bosch, E.; Halenbeck, R.; Wu, G.; Zhou, A.; Behrens, D.; et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science 2008, 320, 807–811. [Google Scholar] [CrossRef] [Green Version]
- Chihara, T.; Suzu, S.; Hassan, R.; Chutiwitoonchai, N.; Hiyoshi, M.; Motoyoshi, K.; Kimura, F.; Okada, S. IL-34 and M-CSF share the receptor Fms but are not identical in biological activity and signal activation. Cell Death Differ. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulakirba, S.; Pfeifer, A.; Mhaidly, R.; Obba, S.; Goulard, M.; Schmitt, T.; Chaintreuil, P.; Calleja, A.; Furstoss, N.; Orange, F.; et al. IL-34 and CSF-1 display an equivalent macrophage differentiation ability but a different polarization potential. Sci. Rep. 2018, 8, 256. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; De Kleer, I.; Henri, S.; Post, S.; Vanhoutte, L.; De Prijck, S.; Deswarte, K.; Malissen, B.; Hammad, H.; Lambrecht, B.N. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 2013, 210, 1977–1992. [Google Scholar] [CrossRef] [Green Version]
- Stanley, E.; Lieschke, G.J.; Grail, D.; Metcalf, D.; Hodgson, G.; Gall, J.A.M.; Maher, D.W.; Cebon, J.; Sinickas, V.; Dunn, A.R. Granulocyte/Macrophage Colony-Stimulating Factor-Deficient Mice Show No Major Perturbation of Hematopoiesis but Develop a Characteristic Pulmonary Pathology. Proc. Natl. Acad. Sci. USA 1994, 91, 5592–5596. [Google Scholar] [CrossRef] [Green Version]
- Mu, X.; Li, Y.; Fan, G.C. Tissue-Resident Macrophages in the Control of Infection and Resolution of Inflammation. Shock 2021, 55, 14–23. [Google Scholar] [CrossRef]
- Taylor, P.R.; Martinez-Pomares, L.; Stacey, M.; Lin, H.H.; Brown, G.D.; Gordon, S. Macrophage receptors and immune recognition. Annu. Rev. Immunol. 2005, 23, 901–944. [Google Scholar] [CrossRef]
- Cailhier, J.F.; Partolina, M.; Vuthoori, S.; Wu, S.; Ko, K.; Watson, S.; Savill, J.; Hughes, J.; Lang, R.A. Conditional macrophage ablation demonstrates that resident macrophages initiate acute peritoneal inflammation. J. Immunol. 2005, 174, 2336–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balhara, J.; Gounni, A.S. The alveolar macrophages in asthma: A double-edged sword. Mucosal Immunol. 2012, 5, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Soroosh, P.; Doherty, T.A.; Duan, W.; Mehta, A.K.; Choi, H.; Adams, Y.F.; Mikulski, Z.; Khorram, N.; Rosenthal, P.; Broide, D.H.; et al. Lung-resident tissue macrophages generate Foxp3+ regulatory T cells and promote airway tolerance. J. Exp. Med. 2013, 210, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Kopf, M.; Schneider, C.; Nobs, S.P. The development and function of lung-resident macrophages and dendritic cells. Nat. Immunol 2015, 16, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Jakubzick, C.; Tacke, F.; Llodra, J.; van Rooijen, N.; Randolph, G.J. Modulation of dendritic cell trafficking to and from the airways. J. Immunol. 2006, 176, 3578–3584. [Google Scholar] [CrossRef] [Green Version]
- Holt, P.G.; Oliver, J.; Bilyk, N.; McMenamin, C.; McMenamin, P.G.; Kraal, G.; Thepen, T. Downregulation of the antigen presenting cell function(s) of pulmonary dendritic cells in vivo by resident alveolar macrophages. J. Exp. Med. 1993, 177, 397–407. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, S.J.; Ruckerl, D.; Cook, P.C.; Jones, L.H.; Finkelman, F.D.; van Rooijen, N.; MacDonald, A.S.; Allen, J.E. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 2011, 332, 1284–1288. [Google Scholar] [CrossRef] [Green Version]
- Loyher, P.L.; Hamon, P.; Laviron, M.; Meghraoui-Kheddar, A.; Goncalves, E.; Deng, Z.; Torstensson, S.; Bercovici, N.; Baudesson de Chanville, C.; Combadiere, B.; et al. Macrophages of distinct origins contribute to tumor development in the lung. J. Exp. Med. 2018, 215, 2536–2553. [Google Scholar] [CrossRef]
- Qian, B.; Deng, Y.; Im, J.H.; Muschel, R.J.; Zou, Y.; Li, J.; Lang, R.A.; Pollard, J.W. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS ONE 2009, 4, e6562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, K.P.; Palmer, J.S.; Cronau, S.; Seppanen, E.; Olver, S.; Raffelt, N.C.; Kuns, R.; Pettit, A.R.; Clouston, A.; Wainwright, B.; et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood 2010, 116, 3955–3963. [Google Scholar] [CrossRef] [Green Version]
- Pombo Antunes, A.R.; Scheyltjens, I.; Lodi, F.; Messiaen, J.; Antoranz, A.; Duerinck, J.; Kancheva, D.; Martens, L.; De Vlaminck, K.; Van Hove, H.; et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 2021, 24, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Zilionis, R.; Engblom, C.; Pfirschke, C.; Savova, V.; Zemmour, D.; Saatcioglu, H.D.; Krishnan, I.; Maroni, G.; Meyerovitz, C.V.; Kerwin, C.M.; et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 2019, 50, 1317–1334.e10. [Google Scholar] [CrossRef]
- Kitamura, T.; Doughty-Shenton, D.; Cassetta, L.; Fragkogianni, S.; Brownlie, D.; Kato, Y.; Carragher, N.; Pollard, J.W. Monocytes Differentiate to Immune Suppressive Precursors of Metastasis-Associated Macrophages in Mouse Models of Metastatic Breast Cancer. Front. Immunol. 2017, 8, 2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Etzerodt, A.; Moulin, M.; Doktor, T.K.; Delfini, M.; Mossadegh-Keller, N.; Bajenoff, M.; Sieweke, M.H.; Moestrup, S.K.; Auphan-Anezin, N.; Lawrence, T. Tissue-resident macrophages in omentum promote metastatic spread of ovarian cancer. J. Exp. Med. 2020, 217, e20191869. [Google Scholar] [CrossRef]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stange, G.; Van den Bossche, J.; Mack, M.; Pipeleers, D.; In’t Veld, P.; De Baetselier, P.; et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef] [Green Version]
- Wyckoff, J.; Wang, W.; Lin, E.Y.; Wang, Y.; Pixley, F.; Stanley, E.R.; Graf, T.; Pollard, J.W.; Segall, J.; Condeelis, J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004, 64, 7022–7029. [Google Scholar] [CrossRef] [Green Version]
- La Fleur, L.; Botling, J.; He, F.; Pelicano, C.; Zhou, C.; He, C.; Palano, G.; Mezheyeuski, A.; Micke, P.; Ravetch, J.V.; et al. Targeting MARCO and IL-37R on immunosuppressive macrophages in lung cancer blocks regulatory T cells and supports cytotoxic lymphocyte function. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linde, N.; Casanova-Acebes, M.; Sosa, M.S.; Mortha, A.; Rahman, A.; Farias, E.; Harper, K.; Tardio, E.; Reyes Torres, I.; Jones, J.; et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat. Commun. 2018, 9, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Lanzi, A. Immunoscore and its introduction in clinical practice. Q. J. Nucl. Med. Mol. Imaging 2020, 64, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Angell, H.K.; Bruni, D.; Barrett, J.C.; Herbst, R.; Galon, J. The Immunoscore: Colon Cancer and Beyond. Clin. Cancer Res. 2020, 26, 332–339. [Google Scholar] [CrossRef] [Green Version]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Mat Zin, A.A.; Ang, K.C.; Ch’ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2019, 9, 1512. [Google Scholar] [CrossRef] [Green Version]
- Ruffell, B.; Au, A.; Rugo, H.S.; Esserman, L.J.; Hwang, E.S.; Coussens, L.M. Leukocyte composition of human breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 2796–2801. [Google Scholar] [CrossRef] [Green Version]
- Giesen, C.; Wang, H.A.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schuffler, P.J.; Grolimund, D.; Buhmann, J.M.; Brandt, S.; et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 2014, 11, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, T.; Kumar, S.; Borkar, R.N.; Azimi, V.; Thibault, G.; Chang, Y.H.; Balter, A.; Kawashima, R.; Choe, G.; Sauer, D.; et al. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep. 2017, 19, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.; Thibault, G.; Luoh, S.W.; Gray, J.W.; Chang, Y.H.; Chin, K. Cyclic Multiplexed-Immunofluorescence (cmIF), a Highly Multiplexed Method for Single-Cell Analysis. Methods Mol. Biol. 2020, 2055, 521–562. [Google Scholar] [PubMed]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, H.; Liang, D.; Zhang, M.; Ren, D.; Chen, H.; Zhang, H.; Li, S.; Ding, G.; Zhang, C.; Ding, Z.; et al. The CD68+ macrophages to CD8+ T-cell ratio is associated with clinical outcomes in hepatitis B virus (HBV)-related hepatocellular carcinoma. HPB 2020. [Google Scholar] [CrossRef]
- Guo, F.; Dong, Y.S.; Tan, Q.Q.; Kong, J.; Yu, B. Tissue Infiltrating Immune Cells as Prognostic Biomarkers in Endometrial Cancer: A Meta-Analysis. Dis. Markers 2020, 2020, 1805764. [Google Scholar] [CrossRef] [Green Version]
- Maccio, A.; Gramignano, G.; Cherchi, M.C.; Tanca, L.; Melis, L.; Madeddu, C. Role of M1-polarized tumor-associated macrophages in the prognosis of advanced ovarian cancer patients. Sci. Rep. 2020, 10, 6096. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Chang, W.; Mao, Y.; He, G.; Zheng, P.; Tang, W.; Wei, Y.; Ren, L.; Zhu, D.; Ji, M.; et al. Tumor-associated Macrophages as Prognostic and Predictive Biomarkers for Postoperative Adjuvant Chemotherapy in Patients with Stage II Colon Cancer. Clin. Cancer Res. 2019, 25, 3896–3907. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [Green Version]
- Yin, S.; Huang, J.; Li, Z.; Zhang, J.; Luo, J.; Lu, C.; Xu, H.; Xu, H. The Prognostic and Clinicopathological Significance of Tumor-Associated Macrophages in Patients with Gastric Cancer: A Meta-Analysis. PLoS ONE 2017, 12, e0170042. [Google Scholar] [CrossRef] [PubMed]
- Hanada, T.; Nakagawa, M.; Emoto, A.; Nomura, T.; Nasu, N.; Nomura, Y. Prognostic value of tumor-associated macrophage count in human bladder cancer. Int. J. Urol. 2000, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Donadon, M.; Torzilli, G.; Cortese, N.; Soldani, C.; Di Tommaso, L.; Franceschini, B.; Carriero, R.; Barbagallo, M.; Rigamonti, A.; Anselmo, A.; et al. Macrophage morphology correlates with single-cell diversity and prognosis in colorectal liver metastasis. J. Exp. Med. 2020, 217, e20191847. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Zhang, C.; Wang, Z.; Wang, K.; Wang, J.; Qiu, X.; Jiang, T.; Yang, P. Prognostic value of a nine-gene signature in glioma patients based on tumor-associated macrophages expression profiling. Clin. Immunol. 2020, 216, 108430. [Google Scholar] [CrossRef]
- Cassetta, L.; Fragkogianni, S.; Sims, A.H.; Swierczak, A.; Forrester, L.M.; Zhang, H.; Soong, D.Y.H.; Cotechini, T.; Anur, P.; Lin, E.Y.; et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell 2019, 35, 588–602.e10. [Google Scholar] [CrossRef] [Green Version]
- Tuit, S.; Salvagno, C.; Kapellos, T.S.; Hau, C.S.; Seep, L.; Oestreich, M.; Klee, K.; de Visser, K.E.; Ulas, T.; Schultze, J.L. Transcriptional Signature Derived from Murine Tumor-Associated Macrophages Correlates with Poor Outcome in Breast Cancer Patients. Cell Rep. 2019, 29, 1221–1235.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [Green Version]
- Oshi, M.; Tokumaru, Y.; Asaoka, M.; Yan, L.; Satyananda, V.; Matsuyama, R.; Matsuhashi, N.; Futamura, M.; Ishikawa, T.; Yoshida, K.; et al. M1 Macrophage and M1/M2 ratio defined by transcriptomic signatures resemble only part of their conventional clinical characteristics in breast cancer. Sci. Rep. 2020, 10, 16554. [Google Scholar] [CrossRef]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017, 169, 736–749.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liudahl, S.M.; Betts, C.B.; Sivagnanam, S.; Morales-Oyarvide, V.; da Silva, A.; Yuan, C.; Hwang, S.; Grossblatt-Wait, A.; Leis, K.R.; Larson, W.; et al. Leukocyte Heterogeneity in Pancreatic Ductal Adenocarcinoma: Phenotypic and Spatial Features Associated with Clinical Outcome. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345.e18. [Google Scholar] [CrossRef] [Green Version]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, X.H.; Massague, J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell 2011, 20, 538–549. [Google Scholar] [CrossRef] [Green Version]
- Gast, C.E.; Silk, A.D.; Zarour, L.; Riegler, L.; Burkhart, J.G.; Gustafson, K.T.; Parappilly, M.S.; Roh-Johnson, M.; Goodman, J.R.; Olson, B.; et al. Cell fusion potentiates tumor heterogeneity and reveals circulating hybrid cells that correlate with stage and survival. Sci. Adv. 2018, 4, eaat7828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, B.Z.; Zhang, H.; Li, J.; He, T.; Yeo, E.J.; Soong, D.Y.; Carragher, N.O.; Munro, A.; Chang, A.; Bresnick, A.R.; et al. FLT1 signaling in metastasis-associated macrophages activates an inflammatory signature that promotes breast cancer metastasis. J. Exp. Med. 2015, 212, 1433–1448. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, T.; Qian, B.Z.; Soong, D.; Cassetta, L.; Noy, R.; Sugano, G.; Kato, Y.; Li, J.; Pollard, J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, R.Y.; Zhang, H.; Li, X.F.; Zhang, C.B.; Selli, C.; Tagliavini, G.; Lam, A.D.; Prost, S.; Sims, A.H.; Hu, H.Y.; et al. Monocyte-derived macrophages promote breast cancer bone metastasis outgrowth. J. Exp. Med. 2020, 217, e20191820. [Google Scholar] [CrossRef]
- Celus, W.; Di Conza, G.; Oliveira, A.I.; Ehling, M.; Costa, B.M.; Wenes, M.; Mazzone, M. Loss of Caveolin-1 in Metastasis-Associated Macrophages Drives Lung Metastatic Growth through Increased Angiogenesis. Cell Rep. 2017, 21, 2842–2854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freire Valls, A.; Knipper, K.; Giannakouri, E.; Sarachaga, V.; Hinterkopf, S.; Wuehrl, M.; Shen, Y.; Radhakrishnan, P.; Klose, J.; Ulrich, A.; et al. VEGFR1(+) Metastasis-Associated Macrophages Contribute to Metastatic Angiogenesis and Influence Colorectal Cancer Patient Outcome. Clin. Cancer Res. 2019, 25, 5674–5685. [Google Scholar] [CrossRef] [Green Version]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Meziani, L.; Deutsch, E.; Mondini, M. Macrophages in radiation injury: A new therapeutic target. Oncoimmunology 2018, 7, e1494488. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Weizman, N.; Krelin, Y.; Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.J.; Gil, Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 2014, 33, 3812–3819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Lou, D.; Yang, Z.; Shao, Z.; Jiang, Y.; Yu, X.; Guo, X.; Feng, Y. Transcriptome Profiling Reveals Inhibited Immune Responses Associated With Radiation Resistance In Triple-Negative Breast Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, e567. [Google Scholar] [CrossRef]
- Hughes, R.; Qian, B.-Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munn, D.H.; Mellor, A.L. Macrophages and the regulation of self-reactive T cells. Curr. Pharm. Des. 2003, 9, 257–264. [Google Scholar] [CrossRef]
- Matlack, R.; Yeh, K.; Rosini, L.; Gonzalez, D.; Taylor, J.; Silberman, D.; Pennello, A.; Riggs, J. Peritoneal macrophages suppress T-cell activation by amino acid catabolism. Immunology 2006, 117, 386–395. [Google Scholar] [CrossRef]
- Blumenthal, R.L.; Campbell, D.E.; Hwang, P.; DeKruyff, R.H.; Frankel, L.R.; Umetsu, D.T. Human alveolar macrophages induce functional inactivation in antigen-specific CD4 T cells. J. Allergy Clin. Immunol. 2001, 107, 258–264. [Google Scholar] [CrossRef]
- Hamilton, M.J.; Antignano, F.; von Rossum, A.; Boucher, J.L.; Bennewith, K.L.; Krystal, G. TLR agonists that induce IFN-beta abrogate resident macrophage suppression of T cells. J. Immunol. 2010, 185, 4545–4553. [Google Scholar] [CrossRef]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef]
- Kung, J.T.; Brooks, S.B.; Jakway, J.P.; Leonard, L.L.; Talmage, D.W. Suppression of in vitro cytotoxic response by macrophages due to induced arginase. J. Exp. Med. 1977, 146, 665–672. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Kryczek, I.; Chen, L.; Zou, W.; Welling, T.H. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Res. 2009, 69, 8067–8075. [Google Scholar] [CrossRef] [Green Version]
- Doedens, A.L.; Stockmann, C.; Rubinstein, M.P.; Liao, D.; Zhang, N.; DeNardo, D.G.; Coussens, L.M.; Karin, M.; Goldrath, A.W.; Johnson, R.S. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010, 70, 7465–7475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strachan, D.C.; Ruffell, B.; Oei, Y.; Bissell, M.J.; Coussens, L.M.; Pryer, N.; Daniel, D. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8 T cells. Oncoimmunology 2013, 2, e26968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guerin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, S.R.; Quaranta, V.; Linford, A.; Emeagi, P.; Rainer, C.; Santos, A.; Ireland, L.; Sakai, T.; Sakai, K.; Kim, Y.S.; et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat. Cell Biol. 2016, 18, 549–560. [Google Scholar] [CrossRef] [Green Version]
- Quaranta, V.; Rainer, C.; Nielsen, S.R.; Raymant, M.L.; Ahmed, M.S.; Engle, D.D.; Taylor, A.; Murray, T.; Campbell, F.; Palmer, D.H.; et al. Macrophage-Derived Granulin Drives Resistance to Immune Checkpoint Inhibition in Metastatic Pancreatic Cancer. Cancer Res. 2018, 78, 4253–4269. [Google Scholar] [CrossRef] [Green Version]
- Schuette, V.; Embgenbroich, M.; Ulas, T.; Welz, M.; Schulte-Schrepping, J.; Draffehn, A.M.; Quast, T.; Koch, K.; Nehring, M.; Konig, J.; et al. Mannose receptor induces T-cell tolerance via inhibition of CD45 and up-regulation of CTLA-4. Proc. Natl. Acad. Sci. USA 2016, 113, 10649–10654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeNardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009, 16, 91–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreu, P.; Johansson, M.; Affara, N.I.; Pucci, F.; Tan, T.; Junankar, S.; Korets, L.; Lam, J.; Tawfik, D.; DeNardo, D.G.; et al. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell 2010, 17, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Medler, T.R.; Murugan, D.; Horton, W.; Kumar, S.; Cotechini, T.; Forsyth, A.M.; Leyshock, P.; Leitenberger, J.J.; Kulesz-Martin, M.; Margolin, A.A.; et al. Complement C5a Fosters Squamous Carcinogenesis and Limits T Cell Response to Chemotherapy. Cancer Cell 2018, 34, 561–578.e6. [Google Scholar] [CrossRef] [Green Version]
- Shiao, S.L.; Ruffell, B.; DeNardo, D.G.; Faddegon, B.A.; Park, C.C.; Coussens, L.M. TH2-Polarized CD4+ T Cells and Macrophages Limit Efficacy of Radiotherapy. Cancer Immunol. Res. 2015, 3, 518–525. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.I.; Tiersma, J.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Buzzelli, J.; Im, J.H.; Muschel, R.J. Radiation combined with macrophage depletion promotes adaptive immunity and potentiates checkpoint blockade. EMBO Mol. Med. 2018, 10, e9342. [Google Scholar] [CrossRef]
- Neubert, N.J.; Schmittnaegel, M.; Bordry, N.; Nassiri, S.; Wald, N.; Martignier, C.; Tille, L.; Homicsko, K.; Damsky, W.; Maby-El Hajjami, H.; et al. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci. Transl. Med. 2018, 10, eaan3311. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, R.; Ijichi, H.; Sano, M.; Miyabayashi, K.; Mohri, D.; Kim, J.; Kimura, G.; Nakatsuka, T.; Fujiwara, H.; Yamamoto, K.; et al. Soluble VCAM-1 promotes gemcitabine resistance via macrophage infiltration and predicts therapeutic response in pancreatic cancer. Sci. Rep. 2020, 10, 21194. [Google Scholar] [CrossRef] [PubMed]
- Kalbasi, A.; Komar, C.; Tooker, G.M.; Liu, M.; Lee, J.W.; Gladney, W.L.; Ben-Josef, E.; Beatty, G.L. Tumor-Derived CCL2 Mediates Resistance to Radiotherapy in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2017, 23, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffell, B.; Coussens, L.M. Macrophages and Therapeutic Resistance in Cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Bulle, A.; Dekervel, J.; Deschuttere, L.; Nittner, D.; Libbrecht, L.; Janky, R.S.; Plaisance, S.; Topal, B.; Coosemans, A.; Lambrechts, D.; et al. Gemcitabine Recruits M2-Type Tumor-Associated Macrophages into the Stroma of Pancreatic Cancer. Transl. Oncol. 2020, 13, 100743. [Google Scholar] [CrossRef]
- Xu, X.; Ye, J.; Huang, C.; Yan, Y.; Li, J. M2 macrophage-derived IL6 mediates resistance of breast cancer cells to hedgehog inhibition. Toxicol. Appl. Pharmacol. 2019, 364, 77–82. [Google Scholar] [CrossRef]
- Song, E.; Su, S.; Zhao, J. Antibody-dependent Tumor Phagocytosis Enhances PD-L1 and IDOs Expression on Macrophages, Which Mediate Trastuzumab Resistance in Her2+ Breast Cancer. J. Immunol. 2017, 198, 204–220. [Google Scholar]
- Ben-Mordechai, T.; Lawrence, Y.; Symon, Z.; Shimoni-Sebag, A.; Appel, S.; Amit, U. CX3CR1 Expressing Macrophages Infiltrate the Tumor Microenvironment and Promote Radiation Resistance in a Mouse Model of Lung Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2019, 105, S125–S126. [Google Scholar] [CrossRef]
- Guan, W.; Hu, J.; Yang, L.; Tan, P.; Tang, Z.; West, B.L.; Bollag, G.; Xu, H.; Wu, L. Inhibition of TAMs improves the response to docetaxel in castration-resistant prostate cancer. Endocr. Relat. Cancer 2019, 26, 131–140. [Google Scholar] [CrossRef]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emami, F.; Pathak, S.; Nguyen, T.T.; Shrestha, P.; Maharjan, S.; Kim, J.O.; Jeong, J.-H.; Yook, S. Photoimmunotherapy with cetuximab-conjugated gold nanorods reduces drug resistance in triple negative breast cancer spheroids with enhanced infiltration of tumor-associated macrophages. J. Control. Release 2020, 329, 645–654. [Google Scholar] [CrossRef]
- Qiao, J.; Chen, Y.; Mi, Y.; Jin, H.; Wang, L.; Huang, T.; Li, H.; Song, Y.; Cao, J.; Wu, B.; et al. Macrophages confer resistance to BET inhibition in triple-negative breast cancer by upregulating IKBKE. Biochem. Pharmacol. 2020, 180, 114126. [Google Scholar] [CrossRef] [PubMed]
- Balmanno, K.; Cook, S.J. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 2009, 16, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, Y.Y.; Yang, X.F.; Shen, D.F.; Sun, H.Z.; Huang, K.Q.; Zheng, H.C. Effects and mechanism of STAT3 silencing on the growth and apoptosis of colorectal cancer cells. Oncol. Lett. 2018. [Google Scholar] [CrossRef]
- Olsson, M.; Zhivotovsky, B. Caspases and cancer. Cell Death Differ. 2011, 18, 1441–1449. [Google Scholar] [CrossRef] [Green Version]
- Castellaro, A.M.; Rodriguez-Baili, M.C.; Di Tada, C.E.; Gil, G.A. Tumor-Associated Macrophages Induce Endocrine Therapy Resistance in ER+ Breast Cancer Cells. Cancers 2019, 11, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, M.; Wicks, K.; Gardasevic, M.; Mace, K.A. Cx3CR1 Expression Identifies Distinct Macrophage Populations That Contribute Differentially to Inflammation and Repair. ImmunoHorizons 2019, 3, 262–273. [Google Scholar] [CrossRef]
- Goswami, S.; Sahai, E.; Wyckoff, J.B.; Cammer, M.; Cox, D.; Pixley, F.J.; Stanley, E.R.; Segall, J.E.; Condeelis, J.S. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 2005, 65, 5278–5283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Wang, W.; Wang, X.; Xu, C.; Zhang, N.; Di, W. Cisplatin-stimulated macrophages promote ovarian cancer migration via the CCL20-CCR6 axis. Cancer Lett. 2020, 472, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Y.; Hao, L.; Hou, A.; Chen, X.; Li, Y.; Wang, R.; Luo, P.; Ruan, Z.; Ou, J.; et al. Macrophages induce resistance to 5-fluorouracil chemotherapy in colorectal cancer through the release of putrescine. Cancer Lett. 2016, 381, 305–313. [Google Scholar] [CrossRef]
- Zheng, P.; Chen, L.; Yuan, X.; Luo, Q.; Liu, Y.; Xie, G.; Ma, Y.; Shen, L. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 53. [Google Scholar] [CrossRef] [Green Version]
- Au Yeung, C.L.; Co, N.-N.; Tsuruga, T.; Yeung, T.-L.; Kwan, S.-Y.; Leung, C.S.; Li, Y.; Lu, E.S.; Kwan, K.; Wong, K.-K.; et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat. Commun. 2016, 7, 11150. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.-H.; Wu, C.-L.; Tsao, C.-J.; Chang, J.-G.; Lu, P.-J.; Yeh, K.-T.; Uen, Y.-H.; Lee, J.-C.; Shiau, A.-L. Deregulated expression of sprouty2 and microRNA-21 in human colon cancer: Correlation with the clinical stage of the disease. Cancer Biol. Ther. 2011, 11, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci. Transl. Med. 2017, 9, eaal3604. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [Green Version]
- Fritz, J.M.; Tennis, M.A.; Orlicky, D.J.; Lin, H.; Ju, C.; Redente, E.F.; Choo, K.S.; Staab, T.A.; Bouchard, R.J.; Merrick, D.T.; et al. Depletion of tumor-associated macrophages slows the growth of chemically induced mouse lung adenocarcinomas. Front. Immunol. 2014, 5, 587. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, S.A.; Orf, J.; Skrzypczynska, K.M.; Tan, H.; Kim, J.; DeVoss, J.; Belmontes, B.; Egen, J.G. Activity of tumor-associated macrophage depletion by CSF1R blockade is highly dependent on the tumor model and timing of treatment. Cancer Immunol. Immunother. 2021. [Google Scholar] [CrossRef]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.M.; Ries, C.H.; Ruttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Tham, M.; Khoo, K.; Yeo, K.P.; Kato, M.; Prevost-Blondel, A.; Angeli, V.; Abastado, J.P. Macrophage depletion reduces postsurgical tumor recurrence and metastatic growth in a spontaneous murine model of melanoma. Oncotarget 2015, 6, 22857–22868. [Google Scholar] [CrossRef] [PubMed]
- Rogers, T.L.; Holen, I. Tumour macrophages as potential targets of bisphosphonates. J. Transl. Med. 2011, 9, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Acker, H.H.; Anguille, S.; Willemen, Y.; Smits, E.L.; Van Tendeloo, V.F. Bisphosphonates for cancer treatment: Mechanisms of action and lessons from clinical trials. Pharmacol. Ther. 2016, 158, 24–40. [Google Scholar] [CrossRef] [PubMed]
- Biewenga, J.; van der Ende, M.B.; Krist, L.F.; Borst, A.; Ghufron, M.; van Rooijen, N. Macrophage depletion in the rat after intraperitoneal administration of liposome-encapsulated clodronate: Depletion kinetics and accelerated repopulation of peritoneal and omental macrophages by administration of Freund’s adjuvant. Cell Tissue Res. 1995, 280, 189–196. [Google Scholar]
- Carron, E.C.; Homra, S.; Rosenberg, J.; Coffelt, S.B.; Kittrell, F.; Zhang, Y.; Creighton, C.J.; Fuqua, S.A.; Medina, D.; Machado, H.L. Macrophages promote the progression of premalignant mammary lesions to invasive cancer. Oncotarget 2017, 8, 50731–50746. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhu, X.D.; Sun, H.C.; Xiong, Y.Q.; Zhuang, P.Y.; Xu, H.X.; Kong, L.Q.; Wang, L.; Wu, W.Z.; Tang, Z.Y. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin. Cancer Res. 2010, 16, 3420–3430. [Google Scholar] [CrossRef] [Green Version]
- Priceman, S.J.; Sung, J.L.; Shaposhnik, Z.; Burton, J.B.; Torres-Collado, A.X.; Moughon, D.L.; Johnson, M.; Lusis, A.J.; Cohen, D.A.; Iruela-Arispe, M.L.; et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: Combating tumor evasion of antiangiogenic therapy. Blood 2010, 115, 1461–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberger, S.M.; Odermatt, B.; Marty, C.; Zehnder-Fjällman, A.H.M.; Ballmer-Hofer, K.; Schwendener, R.A. Clodronate-liposome-mediated depletion of tumour-associated macrophages: A new and highly effective antiangiogenic therapy approach. Br. J. Cancer 2006, 95, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of Macrophage Targeting in the Antitumor Activity of Trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Gordon, E.M.; Sankhala, K.K.; Chawla, N.; Chawla, S.P. Trabectedin for Soft Tissue Sarcoma: Current Status and Future Perspectives. Adv. Ther. 2016, 33, 1055–1071. [Google Scholar] [CrossRef] [Green Version]
- Allavena, P.; Signorelli, M.; Chieppa, M.; Erba, E.; Bianchi, G.; Marchesi, F.; Olimpio, C.O.; Bonardi, C.; Garbi, A.; Lissoni, A.; et al. Anti-inflammatory properties of the novel antitumor agent yondelis (trabectedin): Inhibition of macrophage differentiation and cytokine production. Cancer Res. 2005, 65, 2964–2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, J.P.; Gajdos, C.; Elias, A. Trabectedin: Novel insights in the treatment of advanced sarcoma. Curr. Oncol. Rep. 2014, 16, 387. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, D.; Kowal, J.; Akkari, L.; Schuhmacher, A.J.; Huse, J.T.; West, B.L.; Joyce, J.A. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene 2017, 36, 6049–6058. [Google Scholar] [CrossRef]
- Sawa-Wejksza, K.; Kandefer-Szerszeń, M. Tumor-Associated Macrophages as Target for Antitumor Therapy. Arch. Immunol. Ther. Exp. 2018, 66, 97–111. [Google Scholar] [CrossRef] [Green Version]
- Pulaski, H.L.; Spahlinger, G.; Silva, I.A.; McLean, K.; Kueck, A.S.; Reynolds, R.K.; Coukos, G.; Conejo-Garcia, J.R.; Buckanovich, R.J. Identifying alemtuzumab as an anti-myeloid cell antiangiogenic therapy for the treatment of ovarian cancer. J. Transl. Med. 2009, 7, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neyen, C.; Plüddemann, A.; Mukhopadhyay, S.; Maniati, E.; Bossard, M.; Gordon, S.; Hagemann, T. Macrophage Scavenger Receptor A Promotes Tumor Progression in Murine Models of Ovarian and Pancreatic Cancer. J. Immunol. 2013, 190, 3798–3805. [Google Scholar] [CrossRef] [Green Version]
- Puig-Kröger, A.; Sierra-Filardi, E.; Domínguez-Soto, A.; Samaniego, R.; Corcuera, M.T.; Gómez-Aguado, F.; Ratnam, M.; Sánchez-Mateos, P.; Corbí, A.L. Folate receptor beta is expressed by tumor-associated macrophages and constitutes a marker for M2 anti-inflammatory/regulatory macrophages. Cancer Res. 2009, 69, 9395–9403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Putt, K.S.; Visscher, D.W.; Murphy, L.; Cohen, C.; Singhal, S.; Sandusky, G.; Feng, Y.; Dimitrov, D.S.; Low, P.S. Assessment of folate receptor-β expression in human neoplastic tissues. Oncotarget 2015, 6, 14700–14709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagai, T.; Tanaka, M.; Tsuneyoshi, Y.; Xu, B.; Michie, S.A.; Hasui, K.; Hirano, H.; Arita, K.; Matsuyama, T. Targeting tumor-associated macrophages in an experimental glioma model with a recombinant immunotoxin to folate receptor beta. Cancer Immunol. Immunother. 2009, 58, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Jia, L.; Niu, Y.; Qi, H.; Chen, X.; Zhang, Q.; Zhang, J.; Wang, Y.; Dong, L.; Wang, C. Targeted depletion of tumour-associated macrophages by an alendronate-glucomannan conjugate for cancer immunotherapy. Biomaterials 2014, 35, 10046–10057. [Google Scholar] [CrossRef]
- Scott, E.M.; Jacobus, E.J.; Lyons, B.; Frost, S.; Freedman, J.D.; Dyer, A.; Khalique, H.; Taverner, W.K.; Carr, A.; Champion, B.R.; et al. Bi- and tri-valent T cell engagers deplete tumour-associated macrophages in cancer patient samples. J. Immunother. Cancer 2019, 7, 320. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 2016, 7, 28697–28710. [Google Scholar] [CrossRef] [Green Version]
- Argyle, D.; Kitamura, T. Targeting Macrophage-Recruiting Chemokines as a Novel Therapeutic Strategy to Prevent the Progression of Solid Tumors. Front. Immunol. 2018, 9, 2629. [Google Scholar] [CrossRef]
- Pienta, K.J.; Machiels, J.P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Investig. New Drugs 2013, 31, 760–768. [Google Scholar] [CrossRef]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.B.; Yao, M.; Brummer, G.; Acevedo, D.; Alhakamy, N.; Berkland, C.; Cheng, N. Targeted gene silencing of CCL2 inhibits triple negative breast cancer progression by blocking cancer stem cell renewal and M2 macrophage recruitment. Oncotarget 2016, 7, 49349–49367. [Google Scholar] [CrossRef]
- Tu, M.M.; Abdel-Hafiz, H.A.; Jones, R.T.; Jean, A.; Hoff, K.J.; Duex, J.E.; Chauca-Diaz, A.; Costello, J.C.; Dancik, G.M.; Tamburini, B.A.J.; et al. Inhibition of the CCL2 receptor, CCR2, enhances tumor response to immune checkpoint therapy. Commun. Biol. 2020, 3, 720. [Google Scholar] [CrossRef] [PubMed]
- van Dalen, F.J.; van Stevendaal, M.; Fennemann, F.L.; Verdoes, M.; Ilina, O. Molecular Repolarisation of Tumour-Associated Macrophages. Molecules 2018, 24, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidyarthi, A.; Khan, N.; Agnihotri, T.; Negi, S.; Das, D.K.; Aqdas, M.; Chatterjee, D.; Colegio, O.R.; Tewari, M.K.; Agrewala, J.N. TLR-3 Stimulation Skews M2 Macrophages to M1 Through IFN-alphabeta Signaling and Restricts Tumor Progression. Front. Immunol. 2018, 9, 1650. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.A.; Shah, M.; Kim, J.; Choi, S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med. Res. Rev. 2019, 39, 1053–1090. [Google Scholar] [CrossRef] [Green Version]
- Mullins, S.R.; Vasilakos, J.P.; Deschler, K.; Grigsby, I.; Gillis, P.; John, J.; Elder, M.J.; Swales, J.; Timosenko, E.; Cooper, Z.; et al. Intratumoral immunotherapy with TLR7/8 agonist MEDI9197 modulates the tumor microenvironment leading to enhanced activity when combined with other immunotherapies. J. Immunother. Cancer 2019, 7, 244. [Google Scholar] [CrossRef]
- Rodell, C.B.; Arlauckas, S.P.; Cuccarese, M.F.; Garris, C.S.; Li, R.; Ahmed, M.S.; Kohler, R.H.; Pittet, M.J.; Weissleder, R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018, 2, 578–588. [Google Scholar] [CrossRef]
- Downey, C.M.; Aghaei, M.; Schwendener, R.A.; Jirik, F.R. DMXAA causes tumor site-specific vascular disruption in murine non-small cell lung cancer, and like the endogenous non-canonical cyclic dinucleotide STING agonist, 2’3’-cGAMP, induces M2 macrophage repolarization. PLoS ONE 2014, 9, e99988. [Google Scholar] [CrossRef] [Green Version]
- Gadkaree, S.K.; Fu, J.; Sen, R.; Korrer, M.J.; Allen, C.; Kim, Y.J. Induction of tumor regression by intratumoral STING agonists combined with anti-programmed death-L1 blocking antibody in a preclinical squamous cell carcinoma model. Head Neck 2017, 39, 1086–1094. [Google Scholar] [CrossRef]
- Ao, J.-Y.; Zhu, X.-D.; Chai, Z.-T.; Cai, H.; Zhang, Y.-Y.; Zhang, K.-Z.; Kong, L.-Q.; Zhang, N.; Ye, B.-G.; Ma, D.-N.; et al. Colony-Stimulating Factor 1 Receptor Blockade Inhibits Tumor Growth by Altering the Polarization of Tumor-Associated Macrophages in Hepatocellular Carcinoma. Mol. Cancer Ther. 2017, 16, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Kaneda, M.M.; Cappello, P.; Nguyen, A.V.; Ralainirina, N.; Hardamon, C.R.; Foubert, P.; Schmid, M.C.; Sun, P.; Mose, E.; Bouvet, M.; et al. Macrophage PI3Kgamma Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discov. 2016, 6, 870–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truitt, M.; Olson, P.; et al. Bruton’s Tyrosine Kinase (BTK)-dependent immune cell crosstalk drives pancreas cancer. Cancer Discov. 2015, 6, 270–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luke, J.J.; Bao, R.; Sweis, R.F.; Spranger, S.; Gajewski, T.F. WNT/beta-catenin Pathway Activation Correlates with Immune Exclusion across Human Cancers. Clin. Cancer Res. 2019, 25, 3074–3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Sun, X.; Shi, S. MORC2 Enhances Tumor Growth by Promoting Angiogenesis and Tumor-Associated Macrophage Recruitment via Wnt/beta-Catenin in Lung Cancer. Cell Physiol. Biochem. 2018, 51, 1679–1694. [Google Scholar] [CrossRef]
- Sarode, P.; Zheng, X.; Giotopoulou, G.A.; Weigert, A.; Kuenne, C.; Gunther, S.; Friedrich, A.; Gattenlohner, S.; Stiewe, T.; Brune, B.; et al. Reprogramming of tumor-associated macrophages by targeting beta-catenin/FOSL2/ARID5A signaling: A potential treatment of lung cancer. Sci. Adv. 2020, 6, eaaz6105. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y.; Wang, W.; Teng, G.; Jiao, H.; Shuai, X.; Zhang, R.; Zhao, P.; Qiao, L. Macrophage targeting contributes to the inhibitory effects of embelin on colitis-associated cancer. Oncotarget 2016, 7, 19548–19558. [Google Scholar] [CrossRef] [Green Version]
- Zwar, T.D.; Read, S.; van Driel, I.R.; Gleeson, P.A. CD4+CD25+ regulatory T cells inhibit the antigen-dependent expansion of self-reactive T cells in vivo. J. Immunol. 2006, 176, 1609–1617. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Feng, D.; Yao, Y.; Li, P.; Sun, H.; Yang, H.; Li, C.; Jiang, R.; Sun, B.; Chen, Y. Listeria-based hepatocellular carcinoma vaccine facilitates anti-PD-1 therapy by regulating macrophage polarization. Oncogene 2020, 39, 1429–1444. [Google Scholar] [CrossRef] [PubMed]
- Lizotte, P.H.; Baird, J.R.; Stevens, C.A.; Lauer, P.; Green, W.R.; Brockstedt, D.G.; Fiering, S.N. Attenuated Listeria monocytogenes reprograms M2-polarized tumor-associated macrophages in ovarian cancer leading to iNOS-mediated tumor cell lysis. Oncoimmunology 2014, 3, e28926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Luo, F.; Ding, C.; Albeituni, S.; Hu, X.; Ma, Y.; Cai, Y.; McNally, L.; Sanders, M.A.; Jain, D.; et al. Dectin-1 Activation by a Natural Product beta-Glucan Converts Immunosuppressive Macrophages into an M1-like Phenotype. J. Immunol. 2015, 195, 5055–5065. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H.; Glennie, M.J. Agonistic CD40 antibodies and cancer therapy. Clin. Cancer Res. 2013, 19, 1035–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonderheide, R.H.; Bajor, D.L.; Winograd, R.; Evans, R.A.; Bayne, L.J.; Beatty, G.L. CD40 immunotherapy for pancreatic cancer. Cancer Immunol. Immunother. 2013, 62, 949–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosaka, A.; Ohkuri, T.; Okada, H. Combination of an agonistic anti-CD40 monoclonal antibody and the COX-2 inhibitor celecoxib induces anti-glioma effects by promotion of type-1 immunity in myeloid cells and T-cells. Cancer Immunol. Immunother. 2014, 63, 847–857. [Google Scholar] [CrossRef] [Green Version]
- Alderson, K.L.; Luangrath, M.; Elsenheimer, M.M.; Gillies, S.D.; Navid, F.; Rakhmilevich, A.L.; Sondel, P.M. Enhancement of the anti-melanoma response of Hu14.18K322A by alphaCD40 + CpG. Cancer Immunol. Immunother. 2013, 62, 665–675. [Google Scholar] [CrossRef] [Green Version]
- Hartley, G.P.; Chow, L.; Ammons, D.T.; Wheat, W.H.; Dow, S.W. Programmed Cell Death Ligand 1 (PD-L1) Signaling Regulates Macrophage Proliferation and Activation. Cancer Immunol. Res. 2018, 6, 1260–1273. [Google Scholar] [CrossRef] [Green Version]
- Georgoudaki, A.M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Ostling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Klevebring, D.; et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef] [Green Version]
- Zanganeh, S.; Hutter, G.; Spitler, R.; Lenkov, O.; Mahmoudi, M.; Shaw, A.; Pajarinen, J.S.; Nejadnik, H.; Goodman, S.; Moseley, M.; et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat. Nanotechnol. 2016, 11, 986–994. [Google Scholar] [CrossRef]
- Zhou, Y.; Que, K.T.; Tang, H.M.; Zhang, P.; Fu, Q.M.; Liu, Z.J. Anti-CD206 antibody-conjugated Fe3O4-based PLGA nanoparticles selectively promote tumor-associated macrophages to polarize to the pro-inflammatory subtype. Oncol. Lett. 2020, 20, 298. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, Y.X.; Qiao, S.L.; An, H.W.; Ma, Y.; Qiao, Z.Y.; Rajapaksha, R.P.; Wang, H. Polymeric nanoparticles promote macrophage reversal from M2 to M1 phenotypes in the tumor microenvironment. Biomaterials 2017, 112, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Gao, M.; Hu, Z.; Xu, T.; Chen, J.; Ma, Y.; Li, S.; Gu, Y. Synergistic ferroptosis and macrophage re-polarization using engineering exosome-mimic M1 nanovesicles for cancer metastasis suppression. Chem. Eng. J. 2021, 409, 128217. [Google Scholar] [CrossRef]
- Ramesh, A.; Kumar, S.; Nandi, D.; Kulkarni, A. CSF1R- and SHP2-Inhibitor-Loaded Nanoparticles Enhance Cytotoxic Activity and Phagocytosis in Tumor-Associated Macrophages. Adv. Mater. 2019, 31, e1904364. [Google Scholar] [CrossRef]
- Qi, Y.; Yan, X.; Xia, T.; Liu, S. Use of macrophage as a Trojan horse for cancer nanotheranostics. Mater. Des. 2021, 198, 15028–15044. [Google Scholar] [CrossRef]
- Poh, A.R.; Ernst, M. Targeting Macrophages in Cancer: From Bench to Bedside. Front. Oncol. 2018, 8, 49. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Xie, Z.; Kim, G.B.; Dong, C.; Yang, J. Design Strategies and Applications of Circulating Cell-Mediated Drug Delivery Systems. ACS Biomater. Sci. Eng. 2015, 1, 201–217. [Google Scholar] [CrossRef] [Green Version]
- Andón, F.T.; Digifico, E.; Maeda, A.; Erreni, M.; Mantovani, A.; Alonso, M.J.; Allavena, P. Targeting tumor associated macrophages: The new challenge for nanomedicine. Semin. Immunol. 2017, 34, 103–113. [Google Scholar] [CrossRef]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Patel, S.; Burga, R.A.; Powell, A.B.; Chorvinsky, E.A.; Hoq, N.; McCormack, S.E.; Van Pelt, S.N.; Hanley, P.J.; Cruz, C.R.Y. Beyond CAR T Cells: Other Cell-Based Immunotherapeutic Strategies Against Cancer. Front. Oncol. 2019, 9, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesley, J.D.; Whitmore, J.; Trager, J.; Sheikh, N. An overview of sipuleucel-T: Autologous cellular immunotherapy for prostate cancer. Hum. Vaccines Immunother. 2012, 8, 520–527. [Google Scholar] [CrossRef] [Green Version]
- Xia, A.L.; Wang, X.C.; Lu, Y.J.; Lu, X.J.; Sun, B. Chimeric-antigen receptor T (CAR-T) cell therapy for solid tumors: Challenges and opportunities. Oncotarget 2017, 8, 90521–90531. [Google Scholar] [CrossRef] [Green Version]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, L.; Su, H.; Liu, Q.; Shen, J.; Dai, H.; Zheng, W.; Lu, Y.; Zhang, W.; Bei, Y.; et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br. J. Cancer 2019, 121, 837–845. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Cai, Z.; Jiang, W.; Liu, J.; Tai, Z.; Li, G.; Gong, C.; Gao, S.; Gao, Y. A novel macrophage-mediated biomimetic delivery system with NIR-triggered release for prostate cancer therapy. J. Nanobiotechnol. 2019, 17, 83. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wang, D.; Mei, D.; Zhang, H.; Wang, Z.; He, B.; Dai, W.; Zhang, H.; Wang, X.; Zhang, Q. Macrophage mediated biomimetic delivery system for the treatment of lung metastasis of breast cancer. J. Control. Release 2015, 204, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Kim, H.Y.; Ju, E.J.; Jung, J.; Park, J.; Chung, H.K.; Lee, J.S.; Lee, J.S.; Park, H.J.; Song, S.Y.; et al. Use of macrophages to deliver therapeutic and imaging contrast agents to tumors. Biomaterials 2012, 33, 4195–4203. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.-R.; Bardhan, R.; Stanton-Maxey, K.J.; Badve, S.; Nakshatri, H.; Stantz, K.M.; Cao, N.; Halas, N.J.; Clare, S.E. Delivery of nanoparticles to brain metastases of breast cancer using a cellular Trojan horse. Cancer Nanotechnol. 2012, 3, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Bolli, E.; D’Huyvetter, M.; Murgaski, A.; Berus, D.; Stange, G.; Clappaert, E.J.; Arnouk, S.; Pombo Antunes, A.R.; Krasniqi, A.; Lahoutte, T.; et al. Stromal-targeting radioimmunotherapy mitigates the progression of therapy-resistant tumors. J. Control. Release 2019, 314, 1–11. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.B.; Latz, E.; Mills, K.H.G.; Natoli, G.; Stunnenberg, H.G.; Oneill, L.A.J.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Dominguez-Andres, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [Green Version]
- Blok, B.A.; Arts, R.J.W.; Van Crevel, R.; Benn, C.S.; Netea, M.G. Trained innate immunity as underlying mechanism for the long-term, nonspecific effects of vaccines. J. Leukoc. Biol. 2015, 98, 347–356. [Google Scholar] [CrossRef]
- Wout, J.W.; Poell, R.; Furth, R. The Role of BCG/PPD-Activated Macrophages in Resistance against Systemic Candidiasis in Mice. Scand. J. Immunol. 1992, 36, 713–720. [Google Scholar] [CrossRef]
- Morales, A.; Eidinger, D.; Bruce, A.W. Intracavitary Bacillus Calmette-Guerin in the treatment of superficial bladder tumors. J. Urol. 1976, 116, 180–183. [Google Scholar] [CrossRef]
- Packiam, V.T.; Johnson, S.C.; Steinberg, G.D. Non-muscle-invasive bladder cancer: Intravesical treatments beyond Bacille Calmette-Guérin. Cancer 2017, 123, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Kamat, A.M.; Briggman, J.; Urbauer, D.L.; Svatek, R.; Nogueras González, G.M.; Anderson, R.; Grossman, H.B.; Prat, F.; Dinney, C.P. Cytokine Panel for Response to Intravesical Therapy (CyPRIT): Nomogram of Changes in Urinary Cytokine Levels Predicts Patient Response to Bacillus Calmette-Guérin. Eur. Urol. 2016, 69, 197–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buffen, K.; Oosting, M.; Quintin, J.; Ng, A.; Kleinnijenhuis, J.; Kumar, V.; Van De Vosse, E.; Wijmenga, C.; Van Crevel, R.; Oosterwijk, E.; et al. Autophagy Controls BCG-Induced Trained Immunity and the Response to Intravesical BCG Therapy for Bladder Cancer. PLoS Pathog. 2014, 10, e1004485. [Google Scholar] [CrossRef]
- Graham, C.H.; Pare, J.F.; Cotechini, T.; Hopman, W.; Hindmarch, C.C.T.; Ghaffari, A.; Marginean, D.; Chenard, S.; Koti, M.; Siemens, D.R. Innate immune memory is associated with increased disease-free survival in bladder cancer patients treated with bacillus Calmette-Guerin. Can. Urol. Assoc. J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kalafati, L.; Kourtzelis, I.; Schulte-Schrepping, J.; Li, X.; Hatzioannou, A.; Grinenko, T.; Hagag, E.; Sinha, A.; Has, C.; Dietz, S.; et al. Innate Immune Training of Granulopoiesis Promotes Anti-tumor Activity. Cell 2020, 183, 771–785.e12. [Google Scholar] [CrossRef]
- Priem, B.; Van Leent, M.M.T.; Teunissen, A.J.P.; Sofias, A.M.; Mourits, V.P.; Willemsen, L.; Klein, E.D.; Oosterwijk, R.S.; Meerwaldt, A.E.; Munitz, J.; et al. Trained Immunity-Promoting Nanobiologic Therapy Suppresses Tumor Growth and Potentiates Checkpoint Inhibition. Cell 2020, 183, 786–801.e19. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Saeed, S.; Quintin, J.; Kerstens, H.H.D.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.C.; Ratter, J.; Berentsen, K.; Van Der Ent, M.A.; et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Heijden, C.D.C.C.; Noz, M.P.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P.; Keating, S.T. Epigenetics and Trained Immunity. Antioxid. Redox Signal. 2018, 29, 1023–1040. [Google Scholar] [CrossRef] [PubMed]
- Aegerter, H.; Kulikauskaite, J.; Crotta, S.; Patel, H.; Kelly, G.; Hessel, E.M.; Mack, M.; Beinke, S.; Wack, A. Influenza-induced monocyte-derived alveolar macrophages confer prolonged antibacterial protection. Nat. Immunol. 2020, 21, 145–157. [Google Scholar] [CrossRef]
- Caslini, C.; Hong, S.; Ban, Y.J.; Chen, X.S.; Ince, T.A. HDAC7 regulates histone 3 lysine 27 acetylation and transcriptional activity at super-enhancer-associated genes in breast cancer stem cells. Oncogene 2019, 38, 6599–6614. [Google Scholar] [CrossRef] [Green Version]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| NCT | Phase | Tumour Type | Therapy Name | Primary Outcome |
|---|---|---|---|---|
| NCT00857025 | I | Lung Cancer | β-glucan MM-10-001 | Safety, MTD, and toxicity |
| NCT00682032 | N/A | Non-Small Cell Lung Cancer | β-glucan dietary supplement | Ability of β-glucan to prime neutrophils complement receptor 3, neutrophil cytotoxicity, and macrophage phenotype |
| NCT04710290 | II and III | Metastatic Cancer | β-glucan dietary supplement | Total white blood cells, neutrophils, and lymphocytes at various times |
| NCT00533364 | I and II | Breast Cancer | Soluble β-glucan | Safety of SBG in combination with standard antibody and chemotherapy treatment |
| NCT04387682 | N/A | Squamous Cell Carcinoma of Oral Cavity | β-glucan dietary supplement | Recurrence-free survival or overall survival rate |
| NCT00089258 | II | Neuroblastoma | β-glucan, isotretinoin, sargramostim, and 3F8 mAb | Disease response and efficacy |
| NCT00087009 | 1 | Leukemia, Lymphoma, Lymphoproliferative Disorder | β-glucan and rituximab | MTD and safety |
| NCT04513028 | N/A | Melanoma Stage III and IV | β-glucan dietary supplement and mAb 3F8 | Lymphocyte cell surface expression markers |
| NCT00037011 | I | Neuroblastoma | β-glucan and mAb 3F8 | MTD and toxicity |
| NCT00492167 | I | Neuroblastoma | β-glucan and mAb 3F8 | Toxicity |
| NCT00290407 | II | CLL, Small Lymphocytic Lymphoma | Rituximab and dietary supplement β-glucan | Clinical effect |
| NCT01269385 | I and II | CLL | Alemtuzumab, rituximab, β-glucan (combination) | MTD |
| NCT01829373 | I | Lung Cancer | Vaccine 1650-G and oral β-glucan | Immunological response to vaccine |
| NCT00911560 | I and II | Neuroblastoma | Adjuvant OPT-821 in a vaccine containing two antigens (GD2L and GD3L) covalently linked to KLH and oral β-glucan | MTD and adjuvant effect of β-glucan. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cotechini, T.; Atallah, A.; Grossman, A. Tissue-Resident and Recruited Macrophages in Primary Tumor and Metastatic Microenvironments: Potential Targets in Cancer Therapy. Cells 2021, 10, 960. https://doi.org/10.3390/cells10040960
Cotechini T, Atallah A, Grossman A. Tissue-Resident and Recruited Macrophages in Primary Tumor and Metastatic Microenvironments: Potential Targets in Cancer Therapy. Cells. 2021; 10(4):960. https://doi.org/10.3390/cells10040960
Chicago/Turabian StyleCotechini, Tiziana, Aline Atallah, and Arielle Grossman. 2021. "Tissue-Resident and Recruited Macrophages in Primary Tumor and Metastatic Microenvironments: Potential Targets in Cancer Therapy" Cells 10, no. 4: 960. https://doi.org/10.3390/cells10040960