
Targeting Inflammatory Pathways in Cardiovascular Disease: The Inflammasome, Interleukin-1, Interleukin-6 and Beyond

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cytokines
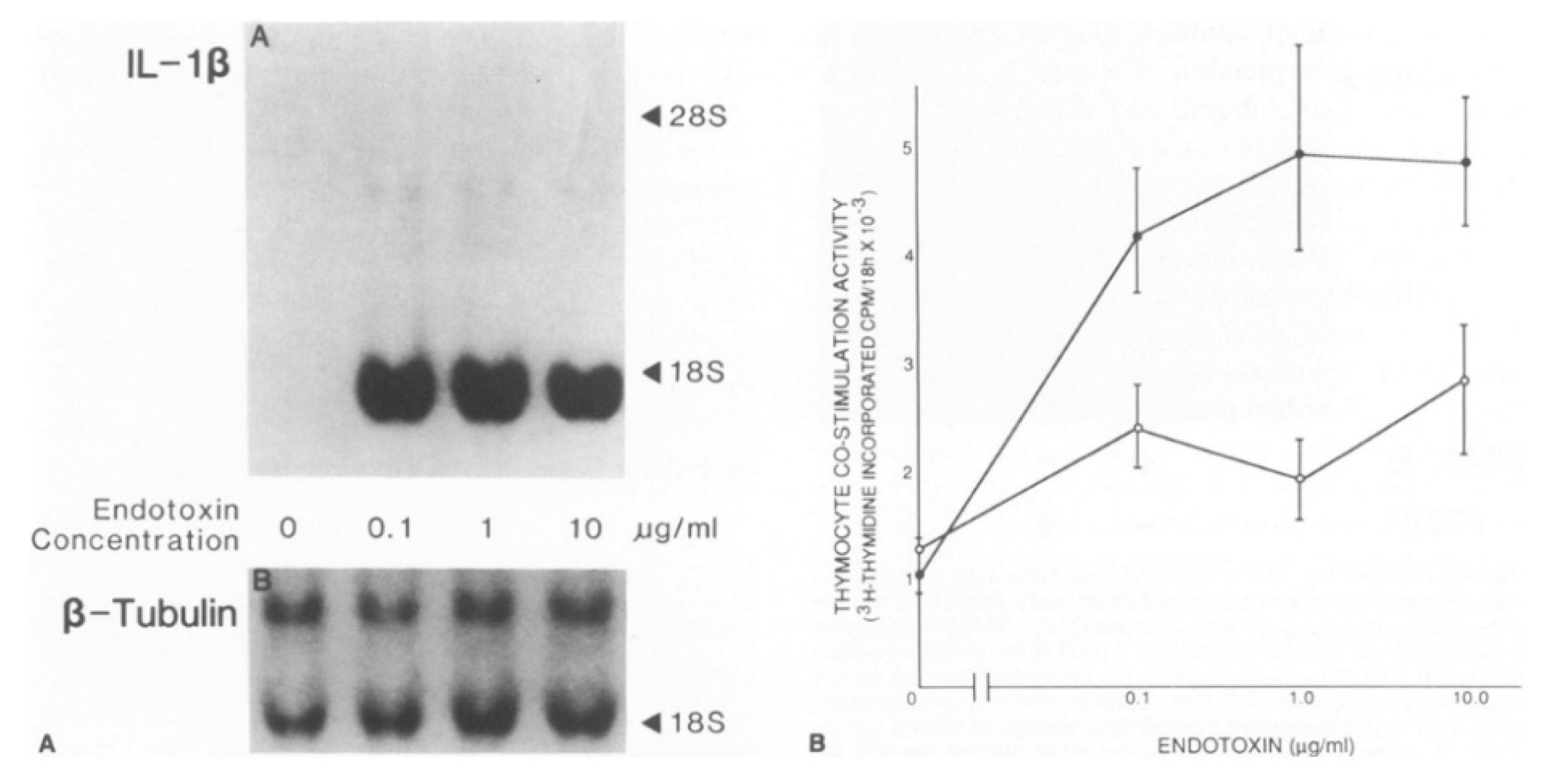
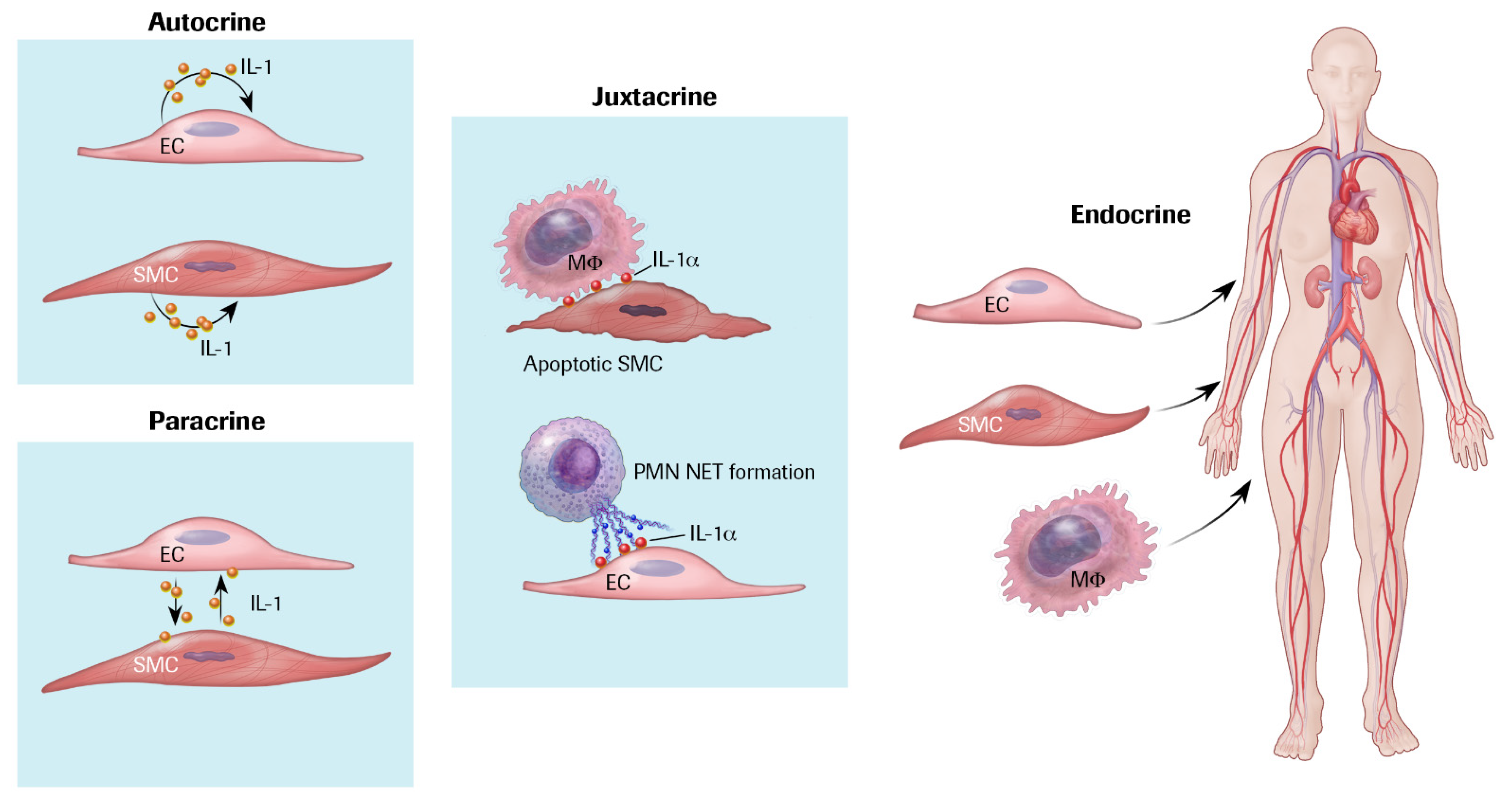
3. IL-1 Production and Responsiveness, and the Discovery of Arterial Autocrine/Paracrine Loops
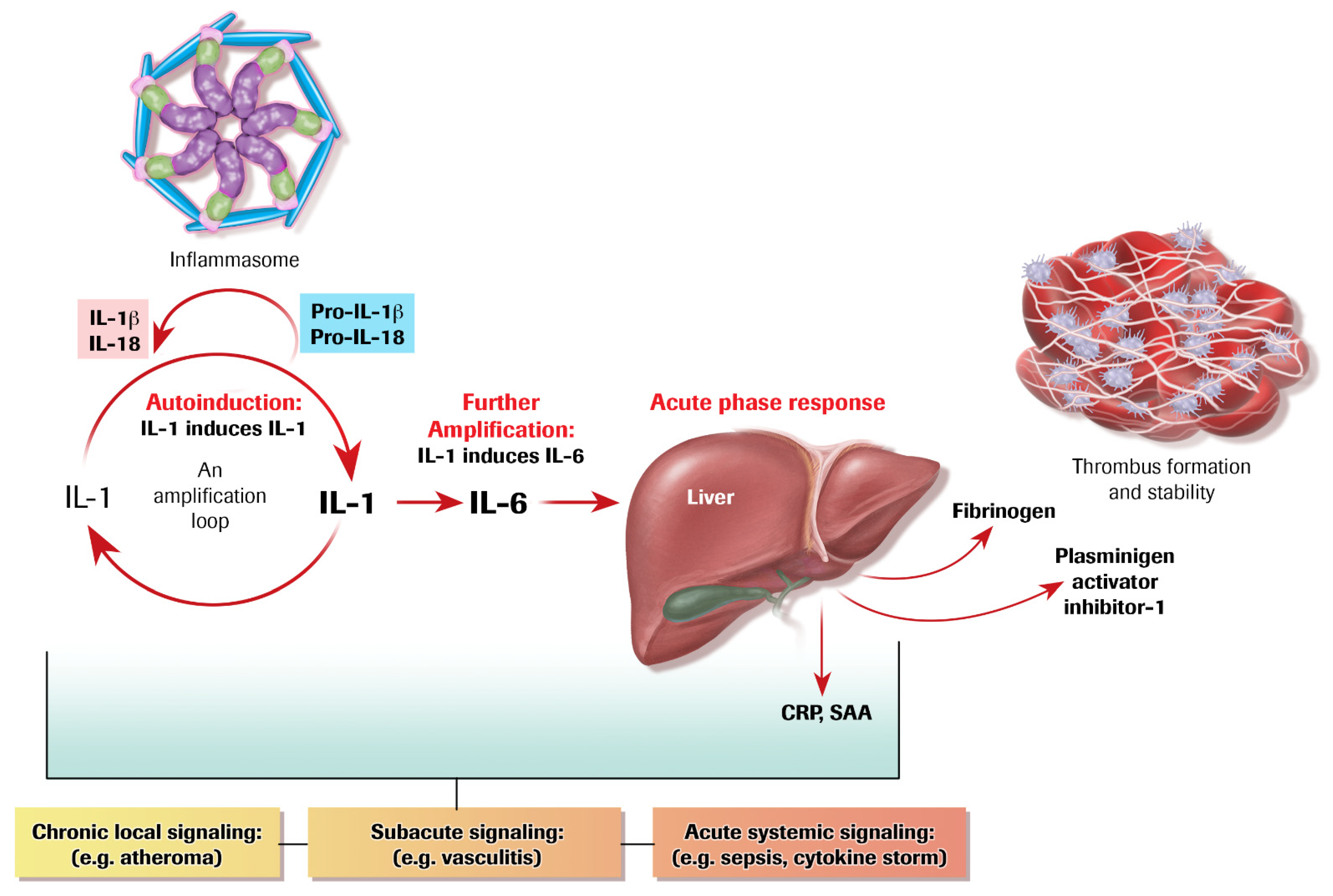
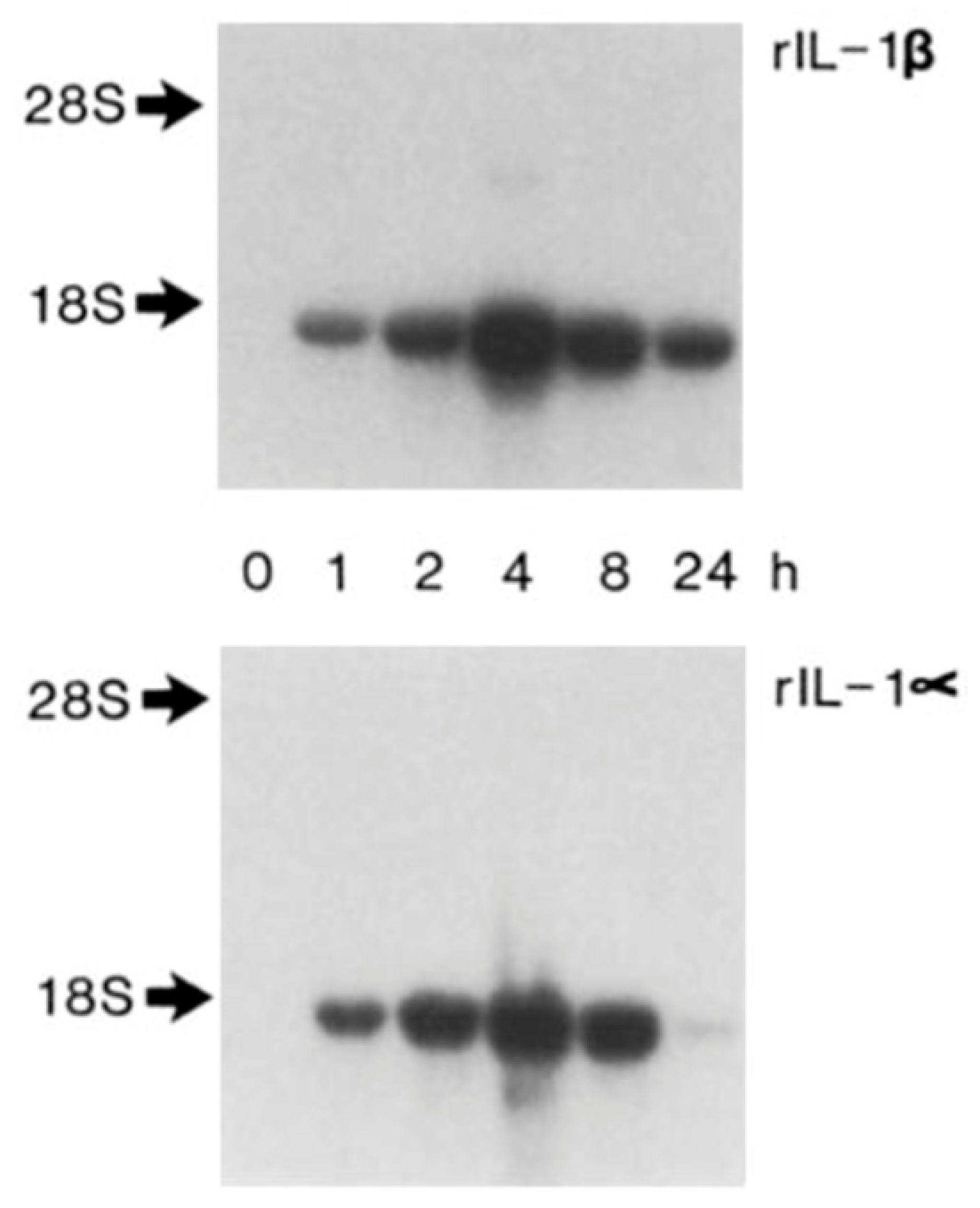
4. Autoinduction of IL-1: A Positive Feedback Loop
5. IL-1 Induces IL-6: Even More Amplification
6. The Acute Phase Response Lies Downstream of IL-1 and IL-6
7. The Inflammasome Family: Proximal Activators of Pro-Inflammatory Cytokines
8. Targeting Inflammasomes: Colchicine and Small Molecules
9. Targeting IL-1β
10. Targeting IL-1α
11. Targeting Interleukin-6
12. Targeting TNF
13. Targeting IL-18
14. Targeting Nuclear Factor-Kappa B (NF-κB)
15. Targeting Adaptive Immune Responses
16. Precision Medicine—A Path Forward?
17. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gencer, S.; Evans, B.; van der Vorst, E.; Döring, Y.; Weber, C. Inflammatory Chemokines in Atherosclerosis. Cells 2021, 10, 226. [Google Scholar] [CrossRef]
- Libby, P.; Ordovas, J.M.; Auger, K.R.; Robbins, A.H.; Birinyi, L.K.; Dinarello, C.A. Endotoxin and tumor necrosis factor induce interleukin-1 gene expression in adult human vascular endothelial cells. Am. J. Pathol. 1986, 124, 179–185. [Google Scholar] [PubMed]
- Libby, P.; Ordovas, J.M.; Birinyi, L.K.; Auger, K.R.; A Dinarello, C. Inducible interleukin-1 gene expression in human vascular smooth muscle cells. J. Clin. Investig. 1986, 78, 1432–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, P.; Fleet, J.; Salomon, R.; Li, H.; Loppnow, H.; Clinton, S. Possible roles of cytokines in atherogenesis. In Atherosclerosis IX Tel Aviv; Stein, O., Eisenberg, S., Stein, Y., Eds.; R&L Creative Communications: Tel Aviv, Isræl, 1992; pp. 339–350. [Google Scholar]
- Ait-Oufella, H.; Taleb, S.; Mallat, Z.; Tedgui, A. Recent Advances on the Role of Cytokines in Atherosclerosis. Arter. Thromb. Vasc. Biol. 2011, 31, 969–979. [Google Scholar] [CrossRef] [Green Version]
- Kusters, P.J.; Lutgens, E. Cytokines and Immune Responses in Murine Atherosclerosis. Methods Mol. Biol. 2015, 1339, 17–40. [Google Scholar] [PubMed]
- Warner, S.J.; Auger, K.R.; Libby, P. Human interleukin 1 induces interleukin 1 gene expression in human vascular smooth muscle cells. J. Exp. Med. 1987, 165, 1316–1331. [Google Scholar] [CrossRef]
- Warner, S.J.; Auger, K.R.; Libby, P. Interleukin 1 induces interleukin 1. II. Recombinant human interleukin 1 induces interleukin 1 production by adult human vascular endothelial cells. J. Immunol. 1987, 139, 1911–1917. [Google Scholar] [PubMed]
- Auron, P.E.; Webb, A.C.; Rosenwasser, L.J.; Mucci, S.F.; Rich, A.; Wolff, S.M.; Dinarello, C.A. Nucleotide sequence of human monocyte interleukin-1 precursor cDNA. Proc. Natl. Acad. Sci. USA 1984, 81, 7907–7911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Dinarello, C.; Ikejima, T.; Warner, S.J.; Orencole, S.F.; Lonnemann, G.; Cannon, J.G.; Libby, P. Interleukin 1 induces interleukin 1. I. Induction of circulating interleukin 1 in rabbits in vivo and in human mononuclear cells in vitro. J. Immunol. 1987, 139, 1902–1910. [Google Scholar] [PubMed]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 Family: Back to the Future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Lüscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef]
- Buckley, L.F.; Wohlford, G.F.; Ting, C.; Alahmed, A.; Van Tassell, B.W.; Abbate, A.; Devlin, J.W.; Libby, P. Role for Anti-Cytokine Therapies in Severe Coronavirus Disease 2019. Crit. Care Explor. 2020, 2, e0178. [Google Scholar] [CrossRef]
- Siddiqi, H.K.; Libby, P.; Ridker, P.M. COVID-19—A vascular disease. Trends Cardiovasc. Med. 2021, 31, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Falus, A.; Rokita, H.; Walcz, E.; Brozik, M.; Hidvegi, T.; Meretey, K. Purification, cloning, expression and biological characterization of an interleukin-1 receptor antagonist protein [see comments]. Nature 1990, 344, 633–638. [Google Scholar]
- Arend, W. Interleukin 1 receptor antagonist. J. Clin. Investig. 1991, 88, 1445–1451. [Google Scholar] [CrossRef]
- Cavalli, G.; Dinarello, C.A. Anakinra Therapy for Non-cancer Inflammatory Diseases. Front Pharmacol. 2018, 9, 1157. [Google Scholar] [CrossRef] [Green Version]
- Abbate, A.; Toldo, S.; Marchetti, C.; Kron, J.; Van Tassell, B.W.; Dinarello, C.A. Interleukin-1 and the Inflammasome as Therapeutic Targets in Cardiovascular Disease. Circ. Res. 2020, 126, 1260–1280. [Google Scholar] [CrossRef] [PubMed]
- Ronsein, G.E.; Vaisar, T. Inflammation, remodeling, and other factors affecting HDL cholesterol efflux. Curr. Opin. Lipidol. 2017, 28, 52–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M. A Test in Context: High-Sensitivity C-Reactive Protein. J. Am. Coll. Cardiol. 2016, 67, 712–723. [Google Scholar] [CrossRef]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.H.; Genest, J.; Gotto, A.M.; Kastelein, J.J.P.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to Prevent Vascular Events in Men and Women with Elevated C-Reactive Protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Y.J.; Libby, P. Evidence for apoptosis in advanced human atheroma. Colocalization with interleukin-1 beta-converting enzyme. Am. J. Pathol. 1995, 147, 251–266. [Google Scholar]
- Schoenbeck, U.; Mach, F.; Bonnefoy, J.Y.; Loppnow, H.; Flad, H.D.; Libby, P. Ligation of CD40 activates interleukin-1beta-converting enzyme (caspase-1) activity in vascular smooth muscle and endothelial cells and promotes elaboration of active interleukin-1 beta. J. Biol. Chem. 1997, 272, 19569–19574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pétrilli, V.; Dostert, C.; A Muruve, D.; Tschopp, J. The inflammasome: A danger sensing complex triggering innate immunity. Curr. Opin. Immunol. 2007, 19, 615–622. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; I Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nat. Cell Biol. 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [Green Version]
- Abe, J.; Berk, B.C. Atheroprone flow activation of the sterol regulatory element binding protein 2 and nod-like receptor protein 3 inflammasome mediates focal atherosclerosis. Circulation 2013, 128, 579–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folco, E.J.; Sukhova, G.K.; Quillard, T.; Libby, P. Moderate hypoxia potentiates interleukin-1beta production in activated human macrophages. Circ. Res. 2014, 115, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M. NLRP3 inflammasome as a key driver of vascular disease. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Zewinger, S.; Reiser, J.; Jankowski, V.; AlAnsary, D.; Hahm, E.; Triem, S.; Klug, M.; Schunk, S.J.; Schmit, D.; Kramann, R.; et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat. Immunol. 2019, 21, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Prochnicki, T.; Latz, E. Inflammasomes on the Crossroads of Innate Immune Recognition and Metabolic Control. Cell Metab. 2017, 26, 71–93. [Google Scholar] [CrossRef] [Green Version]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef]
- Buckley, L.F.; Libby, P. Inhibiting NLRP3 Inflammasome Activity in Acute Myocardial Infarction: A Review of Pharmacologic Agents and Clinical Outcomes. J. Cardiovasc. Pharmcol. 2019, 74, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, Y.; Puri, R.; Hammadah, M.; Duggal, B.; Uno, K.; Kapadia, S.R.; Tuzcu, E.M.; Nissen, S.E.; Nicholls, S.J. Cholesterol Crystals Associate with Coronary Plaque Vulnerability In Vivo. J. Am. Coll. Cardiol. 2015, 65, 630–632. [Google Scholar] [CrossRef] [PubMed]
- Schunk, S.J.; E Kleber, M.; März, W.; Pang, S.; Zewinger, S.; Triem, S.; Ege, P.; Reichert, M.C.; Krawczyk, M.; Weber, S.N.; et al. Genetically determined NLRP3 inflammasome activation associates with systemic inflammation and cardiovascular mortality. Eur. Heart J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Fidler, T.P.; Xue, C.; Yalcinkaya, M.; Hardaway, B.; Abramowicz, S.; Xiao, T.; Liu, W.; Thomas, D.G.; Hajebrahimi, M.A.; Pircher, J.; et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nat. Cell Biol. 2021, 1–6. [Google Scholar] [CrossRef]
- Toldo, S.; Mauro, A.G.; Cutter, Z.; Van Tassell, B.W.; Mezzaroma, E.; Del Buono, M.G.; Prestamburgo, A.; Potere, N.; Abbate, A. The NLRP3 Inflammasome Inhibitor, OLT1177 (Dapansutrile), Reduces Infarct Size and Preserves Contractile Function After Ischemia Reperfusion Injury in the Mouse. J. Cardiovasc. Pharmcol. 2019, 73, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Chusid, M.J.; Fauci, A.S.; Gallin, J.I.; Dale, D.C.; Wolff, S.M. Effect of prophylactic colchicine therapy on leukocyte function in patients with familial mediterranean fever. Arthritis Rheum. 1976, 19, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Imazio, M.; Brucato, A.; Cemin, R.; Ferrua, S.; Maggiolini, S.; Beqaraj, F.; Demarie, D.; Forno, D.; Ferro, S.; Maestroni, S.; et al. A Randomized Trial of Colchicine for Acute Pericarditis. N. Engl. J. Med. 2013, 369, 1522–1528. [Google Scholar] [CrossRef]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nat. Cell Biol. 2006, 440, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nidorf, S.M.; Eikelboom, J.W.; Budgeon, C.A.; Thompson, P.L. Low-Dose Colchicine for Secondary Prevention of Cardiovascular Disease. J. Am. Coll. Cardiol. 2013, 61, 404–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef]
- Opstal, T.S.J.; Hoogeveen, R.M.; Fiolet, A.T.L.; Silvis, M.J.M.; The, S.H.K.; Bax, W.A.; de Kleijn, D.P.V.; Mosterd, A.; Stroes, E.S.G.; Cornel, J.H. Colchicine Attenuates Inflammation Beyond the Inflammasome in Chronic Coronary Artery Disease: A LoDoCo2 Proteomic Substudy. Circulation 2020, 142, 1996–1998. [Google Scholar] [CrossRef] [PubMed]
- Morton, A.C.; Rothman, A.M.K.; Greenwood, J.P.; Gunn, J.; Chase, A.; Clarke, B.; Hall, A.S.; A A Fox, K.; Foley, C.; Banya, W.; et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: The MRC-ILA Heart Study. Eur. Heart J. 2015, 36, 377–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, A.L.; Imazio, M.; Cremer, P.; Brucato, A.; Abbate, A.; Fang, F.; Insalaco, A.; LeWinter, M.; Lewis, B.S.; Lin, D.; et al. Phase 3 Trial of Interleukin-1 Trap Rilonacept in Recurrent Pericarditis. N. Engl. J. Med. 2021, 384, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Thuren, T.; Zalewski, A.; Libby, P. Interleukin-1β inhibition and the prevention of recurrent cardiovascular events: Rationale and Design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Am. Heart J. 2011, 162, 597–605. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; Group, C.T. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Everett, B.M.; Libby, P.; Thuren, T.; Glynn, R.J.; Kastelein, J.; Koenig, W.; Genest, J.; Lorenzatti, A.; et al. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the CANTOS randomised controlled trial. Lancet 2018, 391, 319–328. [Google Scholar] [CrossRef]
- Loppnow, H.; Libby, P. Functional significance of human vascular smooth muscle cell-derived interleukin1 in paracrine and autocrine regulation pathways. Exp. Cell Res. 1992, 198, 283–290. [Google Scholar] [CrossRef]
- Kamari, Y.; Werman-Venkert, R.; Shaish, A.; Werman, A.; Harari, A.; Gonen, A.; Voronov, E.; Grosskopf, I.; Sharabi, Y.; Grossman, E.; et al. Differential role and tissue specificity of interleukin-1α gene expression in atherogenesis and lipid metabolism. Atherosclerosis 2007, 195, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Kamari, Y.; Shaish, A.; Shemesh, S.; Vax, E.; Grosskopf, I.; Dotan, S.; White, M.; Voronov, E.; Dinarello, C.A.; Apte, R.N.; et al. Reduced atherosclerosis and inflammatory cytokines in apolipoprotein-E-deficient mice lacking bone marrow-derived interleukin-1α. Biochem. Biophys. Res. Commun. 2011, 405, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Freigang, S.B.; Ampenberger, F.; Weiss, A.; Kanneganti, T.-D.; Iwakura, Y.; Hersberger, M.; Kopf, M. Fatty acid–induced mitochondrial uncoupling elicits inflammasome-independent IL-1α and sterile vascular inflammation in atherosclerosis. Nat. Immunol. 2013, 14, 1045–1053. [Google Scholar] [CrossRef]
- Folco, E.J.; Mawson, T.L.; Vromman, A.; Bernardes-Souza, B.; Franck, G.; Persson, O.; Nakamura, M.; Newton, G.; Luscinskas, F.W.; Libby, P. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production Through Interleukin-1α and Cathepsin G. Arter. Thromb. Vasc. Biol. 2018, 38, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Vromman, A.; Ruvkun, V.; Shvartz, E.; Wojtkiewicz, G.; Masson, G.S.; Tesmenitsky, Y.; Folco, E.; Gram, H.; Nahrendorf, M.; Swirski, F.K.; et al. Stage-dependent differential effects of interleukin-1 isoforms on experimental atherosclerosis. Eur. Heart J. 2019, 40, 2482–2491. [Google Scholar] [CrossRef]
- Liberale, L.; Bonetti Nicole, R.; Puspitasari Yustina, M.; Schwarz, L.; Akhmedov, A.; Montecucco, F.; Ruschitzka, F.; Beer Jürg, H.; Lüscher Thomas, F.; Simard, J.; et al. Postischemic Administration of IL-1α Neutralizing Antibody Reduces Brain Damage and Neurological Deficit in Experimental Stroke. Circulation 2020, 142, 187–189. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, P.; Rocha, V.Z. All roads lead to IL-6, A central hub of cardiometabolic signaling. Int. J. Cardiol. 2018, 259, 213–215. [Google Scholar] [CrossRef]
- Garbers, C.; Heink, S.; Korn, T.; Rose-John, S. Interleukin-6, designing specific therapeutics for a complex cytokine. Nat. Rev. Drug Discov. 2018, 17, 395–412. [Google Scholar] [CrossRef]
- Huber, S.A.; Sakkinen, P.; Conze, D.; Hardin, N.; Tracy, R. Interleukin-6 Exacerbates Early Atherosclerosis in Mice. Arter. Thromb. Vasc. Biol. 1999, 19, 2364–2367. [Google Scholar] [CrossRef] [Green Version]
- Schieffer, B.; Selle, T.; Hilfiker, A.; Hilfiker-Kleiner, D.; Grote, K.; Tietge, U.J.F.; Trautwein, C.; Luchtefeld, M.; Schmittkamp, C.; Heeneman, S.; et al. Impact of Interleukin-6 on Plaque Development and Morphology in Experimental Atherosclerosis. Circulation 2004, 110, 3493–3500. [Google Scholar] [CrossRef] [PubMed]
- Madan, M.; Bishayi, B.; Hoge, M.; Amar, S. Atheroprotective role of interleukin-6 in diet- and/or pathogen-associated atherosclerosis using an ApoE heterozygote murine model. Atherosclerosis 2008, 197, 504–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuett, H.; Oestreich, R.; Waetzig, G.H.; Annema, W.; Luchtefeld, M.; Hillmer, A.; Bavendiek, U.; Von Felden, J.; Divchev, D.; Kempf, T.; et al. Transsignaling of Interleukin-6 Crucially Contributes to Atherosclerosis in Mice. Arter. Thromb. Vasc. Biol. 2012, 32, 281–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luchtefeld, M.; Schunkert, H.; Stoll, M.; Selle, T.; Lorier, R.; Grote, K.; Sagebiel, C.; Jagavelu, K.; Tietge, U.J.; Assmus, U.; et al. Signal transducer of inflammation gp130 modulates atherosclerosis in mice and man. J. Exp. Med. 2007, 204, 1935–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuett, H.; Luchtefeld, M.; Grothusen, C.; Grote, K.; Schieffer, B. How much is too much? Interleukin-6 and its signalling in atherosclerosis. Thromb. Haemost. 2009, 102, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Reiss, A.B.; Siegart, N.M.; De Leon, J. Interleukin-6 in atherosclerosis: Atherogenic or atheroprotective? Clin. Lipidol. 2017, 12, 14–23. [Google Scholar]
- Kim, K.-W.; Ivanov, S.; Williams, J.W. Monocyte Recruitment, Specification, and Function in Atherosclerosis. Cells 2020, 10, 15. [Google Scholar] [CrossRef]
- I Swerdlow, D.; Holmes, M.V.; Kuchenbaecker, K.B.; Engmann, J.E.L.; Shah, T.; Sofat, R.; Guo, Y.; Chung, C.; Peasey, A.; Pfister, R.; et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: A mendelian randomisation analysis. Lancet 2012, 379, 1214–1224. [Google Scholar] [CrossRef] [Green Version]
- Sarwar, N.; Butterworth, A.S.; Freitag, D.F.; Gregson, J.; Willeit, P.; Gorman, D.N.; Gao, P.; Saleheen, D.; Rendon, A.; Nelson, C.P.; et al. IL6R Genetics Consortium Emerging Risk Factors Collaboration. Interleukin-6 receptor pathways in coronary heart disease: A collaborative meta-analysis of 82 studies. Lancet 2012, 379, 1205–1213. [Google Scholar]
- Ferrante, G.; Condorelli, G. Interleukin-6 trans-signalling and risk of future cardiovascular events: A new avenue for atheroprotection? Cardiovasc. Res. 2019, 115, 8–9. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rifai, N.; Stampfer, M.J.; Hennekens, C.H. Plasma Concentration of Interleukin-6 and the Risk of Future Myocardial Infarction Among Apparently Healthy Men. Circulation 2000, 101, 1767–1772. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Libby, P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1beta inhibition with canakinumab: Further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur. Heart J. 2020, 41, 2153–2163. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; MacFadyen, J.G.; Glynn, R.J.; Bradwin, G.; Hasan, A.A.; Rifai, N. Comparison of interleukin-6, C-reactive protein, and low-density lipoprotein cholesterol as biomarkers of residual risk in contemporary practice: Secondary analyses from the Cardiovascular Inflammation Reduction Trial. Eur. Heart J. 2020, 41, 2952–2961. [Google Scholar] [CrossRef] [Green Version]
- Biasucci, L.M.; Liuzzo, G.; Fantuzzi, G.; Caligiuri, G.; Rebuzzi, A.G.; Ginnetti, F.; Dinarello, C.A.; Maseri, A. Increasing Levels of Interleukin (IL)-1Ra and IL-6 During the First 2 Days of Hospitalization in Unstable Angina Are Associated with Increased Risk of In-Hospital Coronary Events. Circulation 1999, 99, 2079–2084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindmark, E.; Diderholm, E.; Wallentin, L.; Siegbahn, A. Relationship between interleukin 6 and mortality in patients with unstable coronary artery disease: Effects of an early invasive or noninvasive strategy. JAMA 2001, 286, 2107–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Held, C.; White, H.D.; Stewart, R.A.H.; Budaj, A.; Cannon, C.P.; Hochman, J.S.; Koenig, W.; Siegbahn, A.; Steg, P.G.; Soffer, J.; et al. Inflammatory Biomarkers Interleukin-6 and C-Reactive Protein and Outcomes in Stable Coronary Heart Disease: Experiences From the STABILITY (Stabilization of Atherosclerotic Plaque by Initiation of Darapladib Therapy) Trial. J. Am. Heart Assoc. 2017, 6, e005077. [Google Scholar] [CrossRef] [Green Version]
- Groot, H.E.; Al Ali, L.; van der Horst, I.C.C.; Schurer, R.A.J.; van der Werf, H.W.; Lipsic, E.; van Veldhuisen, D.J.; Karper, J.C.; van der Harst, P. Plasma interleukin 6 levels are associated with cardiac function after ST-elevation myocardial infarction. Clin. Res. Cardiol. 2019, 108, 612–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno Velásquez, I.; Golabkesh, Z.; Källberg, H.; Leander, K.; de Faire, U.; Gigante, B. Circulating levels of interleukin 6 soluble receptor and its natural antagonist, sgp130, and the risk of myocardial infarction. Atherosclerosis 2015, 240, 477–481. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, L.; Gajulapuri, A.; Frumento, P.; Bonomi, A.; Wallén, H.; De Faire, U.; Rose-John, S.; Gigante, B. Interleukin 6 trans-signalling and risk of future cardiovascular events. Cardiovasc. Res. 2018, 115, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bick, A.G.; Pirruccello, J.P.; Griffin, G.K.; Gupta, N.; Gabriel, S.; Saleheen, D.; Libby, P.; Kathiresan, S.; Natarajan, P. Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation 2020, 141, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Rohde, L.E.P.; Arroyo, L.H.; Rifai, N.; Creager, M.A.; Libby, P.; Ridker, P.M.; Lee, R.T. Plasma Concentrations of Interleukin-6 and Abdominal Aortic Diameter Among Subjects Without Aortic Dilatation. Arter. Thromb. Vasc. Biol. 1999, 19, 1695–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paige, E.; Clément, M.; Lareyre, F.; Sweeting, M.; Raffort, J.; Grenier, C.; Finigan, A.; Harrison, J.; Peters, J.E.; Sun, B.B.; et al. Interleukin-6 Receptor Signaling and Abdominal Aortic Aneurysm Growth Rates. Circ. Genom. Precis. Med. 2019, 12, e002413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleveland, O.; Kunszt, G.; Bratlie, M.; Ueland, T.; Broch, K.; Holte, E.; Michelsen, A.E.; Bendz, B.; Amundsen, B.H.; Espevik, T.; et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: A double-blind, randomized, placebo-controlled phase 2 trial. Eur. Heart J. 2016, 37, 2406–2413. [Google Scholar] [CrossRef] [PubMed]
- Kleveland, O.; Ueland, T.; Kunszt, G.; Bratlie, M.; Yndestad, A.; Broch, K.; Holte, E.; Ryan, L.; Amundsen, B.H.; Bendz, B.; et al. Interleukin-6 receptor inhibition with tocilizumab induces a selective and substantial increase in plasma IP-10 and MIP-1beta in non-ST-elevation myocardial infarction. Int. J. Cardiol. 2018, 271, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anstensrud, A.K.; Woxholt, S.; Sharma, K.; Broch, K.; Bendz, B.; Aakhus, S.; Ueland, T.; Amundsen, B.H.; Damås, J.K.; Hopp, E.; et al. Rationale for the ASSAIL-MI-trial: A randomised controlled trial designed to assess the effect of tocilizumab on myocardial salvage in patients with acute ST-elevation myocardial infarction (STEMI). Open. Heart 2019, 6, e001108. [Google Scholar] [CrossRef]
- Schiff, M.H.; Kremer, J.M.; Jahreis, A.; Vernon, E.; Isaacs, J.D.; Van Vollenhoven, R.F. Integrated safety in tocilizumab clinical trials. Arthritis Res. Ther. 2011, 13, R141. [Google Scholar] [CrossRef] [Green Version]
- Ait-Oufella, H.; Libby, P.; Tedgui, A. Anticytokine Immune Therapy and Atherothrombotic Cardiovascular Risk. Arter. Thromb. Vasc. Biol. 2019, 39, 1510–1519. [Google Scholar] [CrossRef]
- Old, L.J. Tumor Necrosis Factor. Sci. Am. 1988, 258, 59–75. [Google Scholar] [CrossRef]
- Beutler, B.; Cerami, A. Cachectin and tumour necrosis factor as two sides of the same biological coin. Nature 1986, 320, 584–588. [Google Scholar] [CrossRef]
- Sedger, L.M.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants—Past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, W.-M. Tumor necrosis factor. Cancer Lett. 2013, 328, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, S.M.; Naga Prasad, S.V. Tumor Necrosis Factor-alpha in Heart Failure: An Updated Review. Curr. Cardiol. Rep. 2018, 20, 117. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L. Innate immunity and the failing heart: The cytokine hypothesis revisited. Circ. Res. 2015, 116, 1254–1268. [Google Scholar] [CrossRef] [Green Version]
- Ait-Oufella, H.; Libby, P.; Tedgui, A. Antibody-based immunotherapy targeting cytokines and atherothrombotic cardiovascular diseases. Arch. Cardiovasc. Dis. 2020, 113, 5–8. [Google Scholar] [CrossRef]
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Besnard, S.; Lesèche, G.; Chvatchko, Y.; Tedgui, A. Expression of Interleukin-18 in Human Atherosclerotic Plaques and Relation to Plaque Instability. Circulation 2001, 104, 1598–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerdes, N.; Sukhova, G.K.; Libby, P.; Reynolds, R.S.; Young, J.L.; Schönbeck, U. Expression of Interleukin (IL)-18 and Functional IL-18 Receptor on Human Vascular Endothelial Cells, Smooth Muscle Cells, and Macrophages: Implication for atherogenesis. J. Exp. Med. 2002, 195, 245–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Sun, C.; Gerdes, N.; Liu, C.; Liao, M.; Liu, J.; Shi, M.A.; He, A.; Zhou, Y.; Sukhova, G.K.; et al. Interleukin 18 function in atherosclerosis is mediated by the interleukin 18 receptor and the Na-Cl co-transporter. Nat. Med. 2015, 21, 820–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltimore, D. NF-kappaB is 25. Nat. Immunol. 2011, 12, 683–685. [Google Scholar] [CrossRef]
- Brown, J.D.; Lin, C.Y.; Duan, Q.; Griffin, G.; Federation, A.; Paranal, R.M.; Bair, S.; Newton, G.; Lichtman, A.; Kung, A.; et al. NF-κB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell. 2014, 56, 219–231. [Google Scholar] [CrossRef] [Green Version]
- Gasparini, C.; Feldmann, M. NF-kappaB as a target for modulating inflammatory responses. Curr. Pharm. Des. 2012, 18, 5735–5745. [Google Scholar] [CrossRef]
- Zhao, T.X.; Mallat, Z. Targeting the Immune System in Atherosclerosis: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 1691–1706. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, S.; Zernecke, A. CD8+ T Cells in Atherosclerosis. Cells 2021, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.K.; Chyu, K.-Y.; Dimayuga, P.C.; Nilsson, J. Vaccine for Atherosclerosis. J. Am. Coll. Cardiol. 2014, 64, 2779–2791. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, J.; Hansson Göran, K. Vaccination Strategies and Immune Modulation of Atherosclerosis. Circ. Res. 2020, 126, 1281–1296. [Google Scholar] [CrossRef]
- Nettersheim, F.S.; De Vore, L.; Winkels, H. Vaccination in Atherosclerosis. Cells 2020, 9, 2560. [Google Scholar] [CrossRef]
- Ma, S.; Mussbacher, M.; Galkina, E. Functional Role of B Cells in Atherosclerosis. Cells 2021, 10, 270. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.X.; Ur-Rahman, M.A.; Sage, A.P.; Victor, S.; Kurian, R.; Fielding, S.; Ait-Oufella, H.; Chiu, Y.-D.; Binder, C.J.; Mckie, M.; et al. Rituximab in Patients with Acute ST-elevation Myocardial Infarction (RITA-MI): An Experimental Medicine Safety Study. Cardiovasc. Res. 2021, 106, A19–A21. [Google Scholar] [CrossRef]
- Ali, A.J.; Makings, J.; Ley, K. Regulatory T Cell Stability and Plasticity in Atherosclerosis. Cells 2020, 9, 2665. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.X.; Kostapanos, M.; Griffiths, C.; Arbon, E.L.; Hubsch, A.; Kaloyirou, F.; Helmy, J.; Hoole, S.P.; Rudd, J.H.F.; Wood, G.; et al. Low-dose interleukin-2 in patients with stable ischaemic heart disease and acute coronary syndromes (LILACS): Protocol and study rationale for a randomised, double-blind, placebo-controlled, phase I/II clinical trial. BMJ Open 2018, 8, e022452. [Google Scholar] [CrossRef]
- O’Brien, E.; Shi, C.; Deng, J.; Diao, C.; Clarkson, M.; Shrivastava, V.; Adijian, A.; Hu, A.; Chiu, M.H.; Gwilym, B.; et al. HSP27 immunization attenuates atherogenesis by markedly reducing plasma PCSK9 and cholesterol levels. Atherosclerosis 2018, 275, e152. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Braunwald, E.; White, H.D.; Lukas, M.A.; Tarka, E.; Steg, P.G.; Hochman, J.S.; Bode, C.; Maggioni, A.P.; Im, K.; et al. Effect of darapladib on major coronary events after an acute coronary syndrome: The SOLID-TIMI 52 randomized clinical trial. JAMA 2014, 312, 1006–1015. [Google Scholar] [CrossRef] [Green Version]
- White, H.D.; Held, C.; Stewart, R.; Tarka, E.; Brown, R.; Davies, R.Y.; Budaj, A.; Harrington, R.A.; Steg, P.G.; Ardissino, D.; et al. Darapladib for preventing ischemic events in stable coronary heart disease. N. Engl. J. Med. 2014, 370, 1702–1711. [Google Scholar] [PubMed] [Green Version]
- O’Donoghue, M.L.; Glaser, R.; Cavender, M.A.; Aylward, P.E.; Bonaca, M.P.; Budaj, A.; Davies, R.Y.; Dellborg, M.; Fox, K.A.; Gutierrez, J.A.; et al. Effect of Losmapimod on Cardiovascular Outcomes in Patients Hospitalized With Acute Myocardial Infarction: A Randomized Clinical Trial. JAMA 2016, 315, 1591–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henein, M.; Granåsen, G.; Wiklund, U.; Schmermund, A.; Guerci, A.; Erbel, R.; Raggi, P. High dose and long-term statin therapy accelerate coronary artery calcification. Int. J. Cardiol. 2015, 184, 581–586. [Google Scholar] [CrossRef]
- Puri, R.; Nicholls, S.J.; Shao, M.; Kataoka, Y.; Uno, K.; Kapadia, S.R.; Tuzcu, E.M.; Nissen, S.E. Impact of Statins on Serial Coronary Calcification During Atheroma Progression and Regression. J. Am. Coll. Cardiol. 2015, 65, 1273–1282. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.E.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.-L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [Green Version]
- Bick, A.G.; Weinstock, J.S.; Nandakumar, S.K.; Fulco, C.P.; Bao, E.L.; Zekavat, S.M.; Szeto, M.D.; Liao, X.; Leventhal, M.J.; Nasser, J.; et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 2020, 586, 763–768. [Google Scholar] [CrossRef]
- Svensson Eric, C.; Madar, A.; Campbell Catarina, D.; He, Y.; Sultan, M.; Healey Margaret, L.; D’Aco, K.; Fernandez, A.; Wache-Mainier, C.; Ridker Paul, M.; et al. TET2-Driven Clonal Hematopoiesis Predicts Enhanced Response to Canakinumab in the CANTOS Trial: An Exploratory Analysis. Circulation 2018, 138, A15111. [Google Scholar]
- Wang, W.; Liu, W.; Fidler, T.; Wang, Y.; Tang, Y.; Woods, B.; Welch, C.; Cai, B.; Silvestre-Roig, C.; Ai, D.; et al. Macrophage Inflammation, Erythrophagocytosis, and Accelerated Atherosclerosis in Jak2 V617F Mice. Circ. Res. 2018, 123, e35–e47. [Google Scholar] [CrossRef]
- Libby, P.; Tabas, I.; Fredman, G.; Fisher, E.A. Inflammation and its Resolution as Determinants of Acute Coronary Syndromes. Circ. Res. 2014, 114, 1867–1879. [Google Scholar] [CrossRef] [Green Version]
- Fredman, G.; Tabas, I. Boosting Inflammation Resolution in Atherosclerosis: The Next Frontier for Therapy. Am. J. Pathol. 2017, 187, 1211–1221. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N. Treating inflammation and infection in the 21st century: New hints from decoding resolution mediators and mechanisms. FASEB J. 2017, 31, 1273–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, P.; Hasan, A.A.; Nohria, A. Drugs Targeting Inflammation. In Opie’s Cardiovascular Drugs: A Companion to Braunwald’s Heart Disease, 9th ed.; Bhatt, D.L., Ed.; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- E Strandberg, T.; Libby, P.; Kovanen, P.T. A tale of two therapies lipid-lowering vs. anti-inflammatory therapy: A false dichotomy? Eur. Heart J. Cardiovasc. Pharmacother. 2020. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation and Atherosclerosis—The End of a Controversy. Circulation 2017, 136, 1875–1877. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in Atherosclerosis—No Longer a Theory. Clin. Chem. 2021, 67, 131–142. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Libby, P. Targeting Inflammatory Pathways in Cardiovascular Disease: The Inflammasome, Interleukin-1, Interleukin-6 and Beyond. Cells 2021, 10, 951. https://doi.org/10.3390/cells10040951
Libby P. Targeting Inflammatory Pathways in Cardiovascular Disease: The Inflammasome, Interleukin-1, Interleukin-6 and Beyond. Cells. 2021; 10(4):951. https://doi.org/10.3390/cells10040951
Chicago/Turabian StyleLibby, Peter. 2021. "Targeting Inflammatory Pathways in Cardiovascular Disease: The Inflammasome, Interleukin-1, Interleukin-6 and Beyond" Cells 10, no. 4: 951. https://doi.org/10.3390/cells10040951