
The Potential of FGF-2 in Craniofacial Bone Tissue Engineering: A Review


Abstract
:1. Introduction
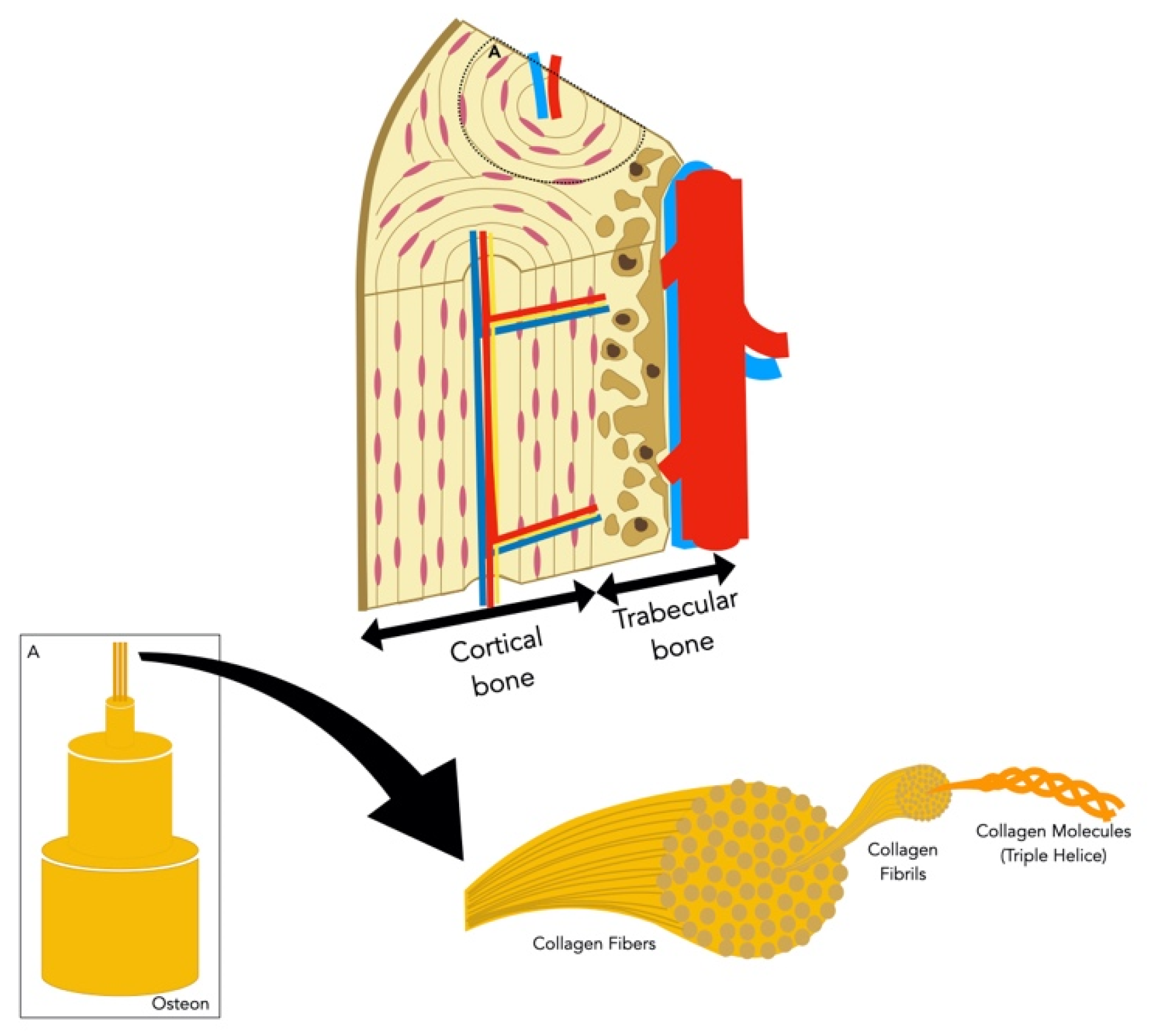
2. Background on Bone Physiology
2.1. Bone Composition
2.1.1. Bone Cells
2.1.2. Bone Extracellular Matrix
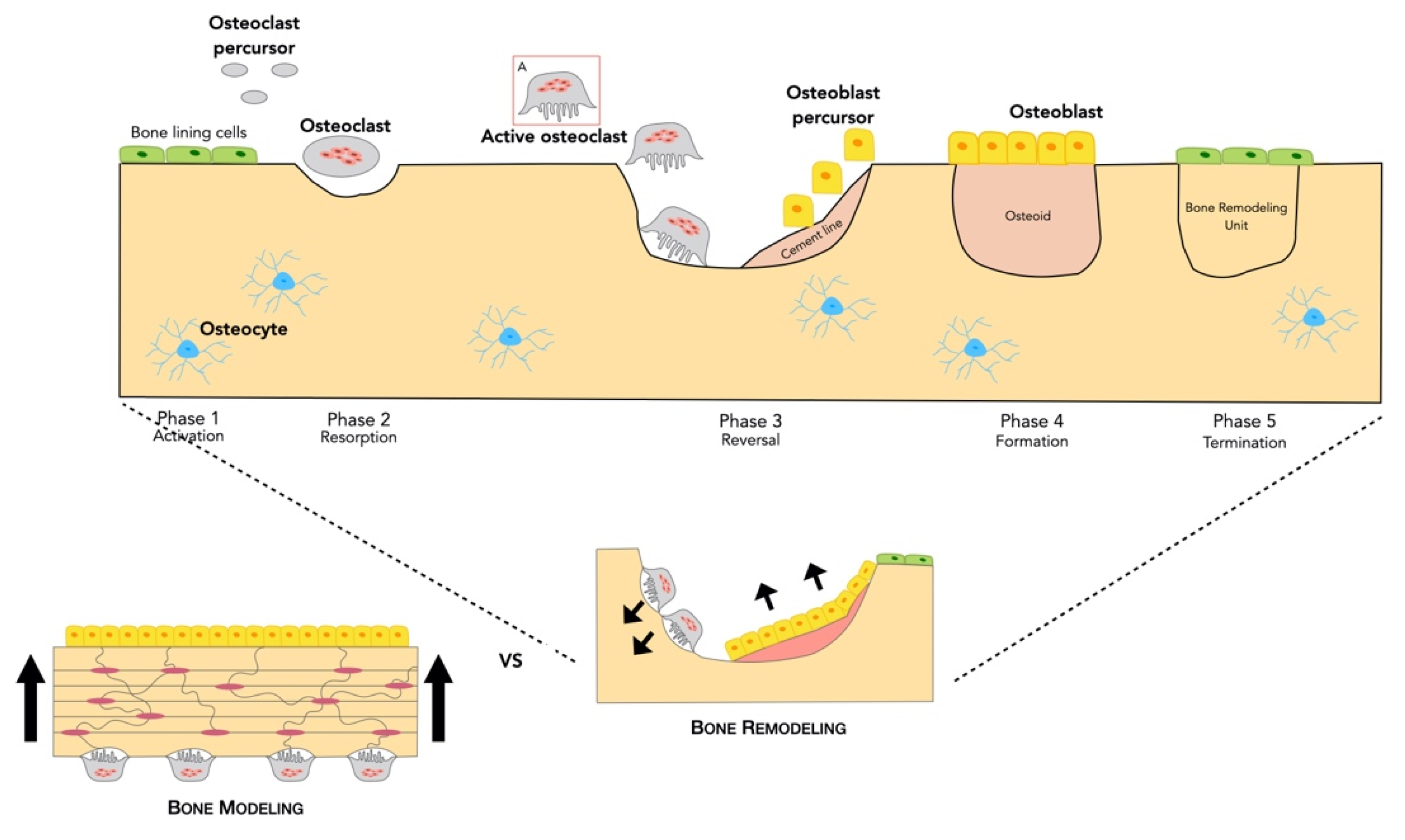
2.2. Bone Formation
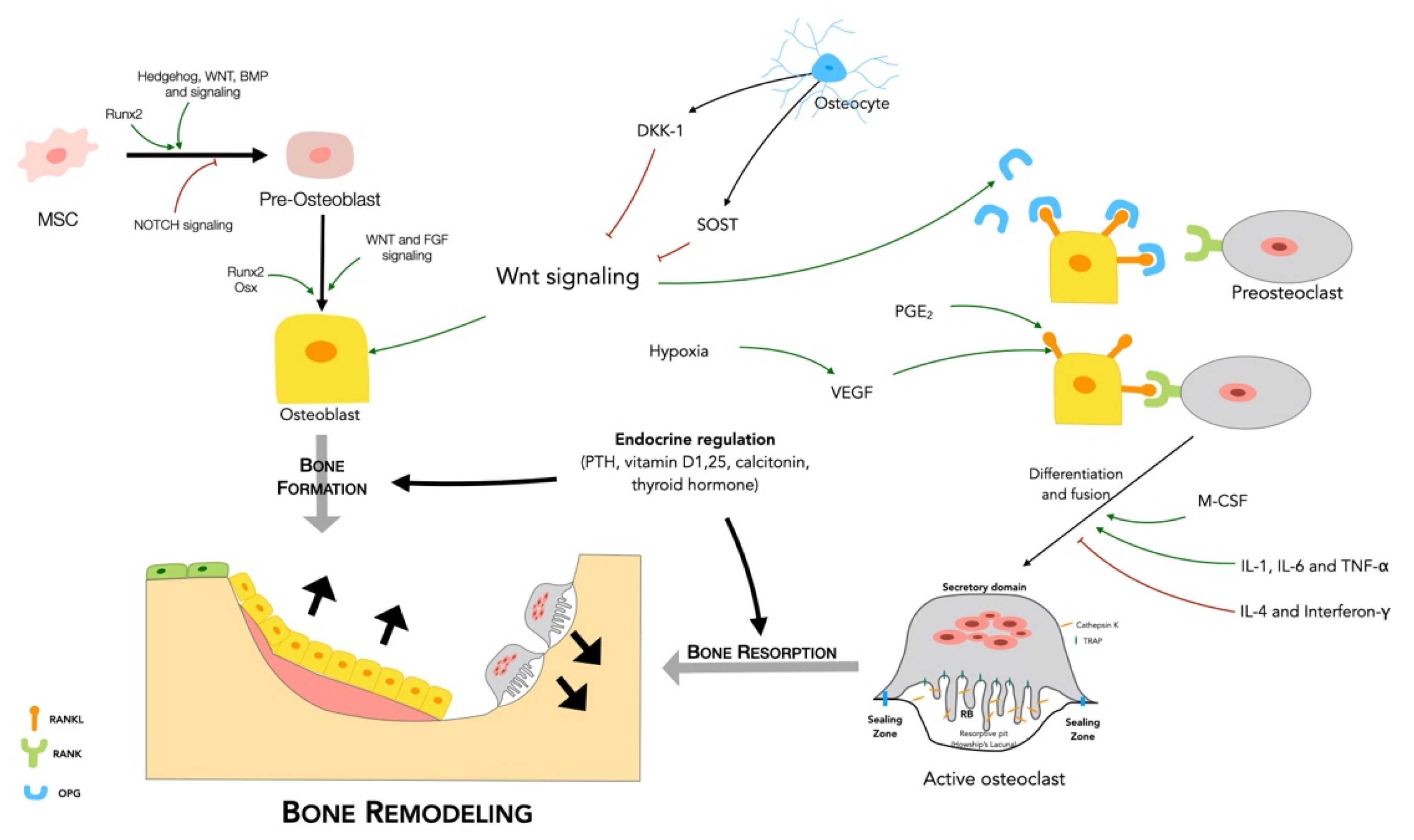
2.2.1. Bone Remodeling Process
2.2.2. Regulation of Bone Remodeling
- Major signaling pathways
- RANKL/RANK/OPG
- 2.
- Wnt signaling
- Endocrine regulation
- Paracrine regulation
2.3. Bone Vascularization
3. FGF-2 in Bone Homeostasis
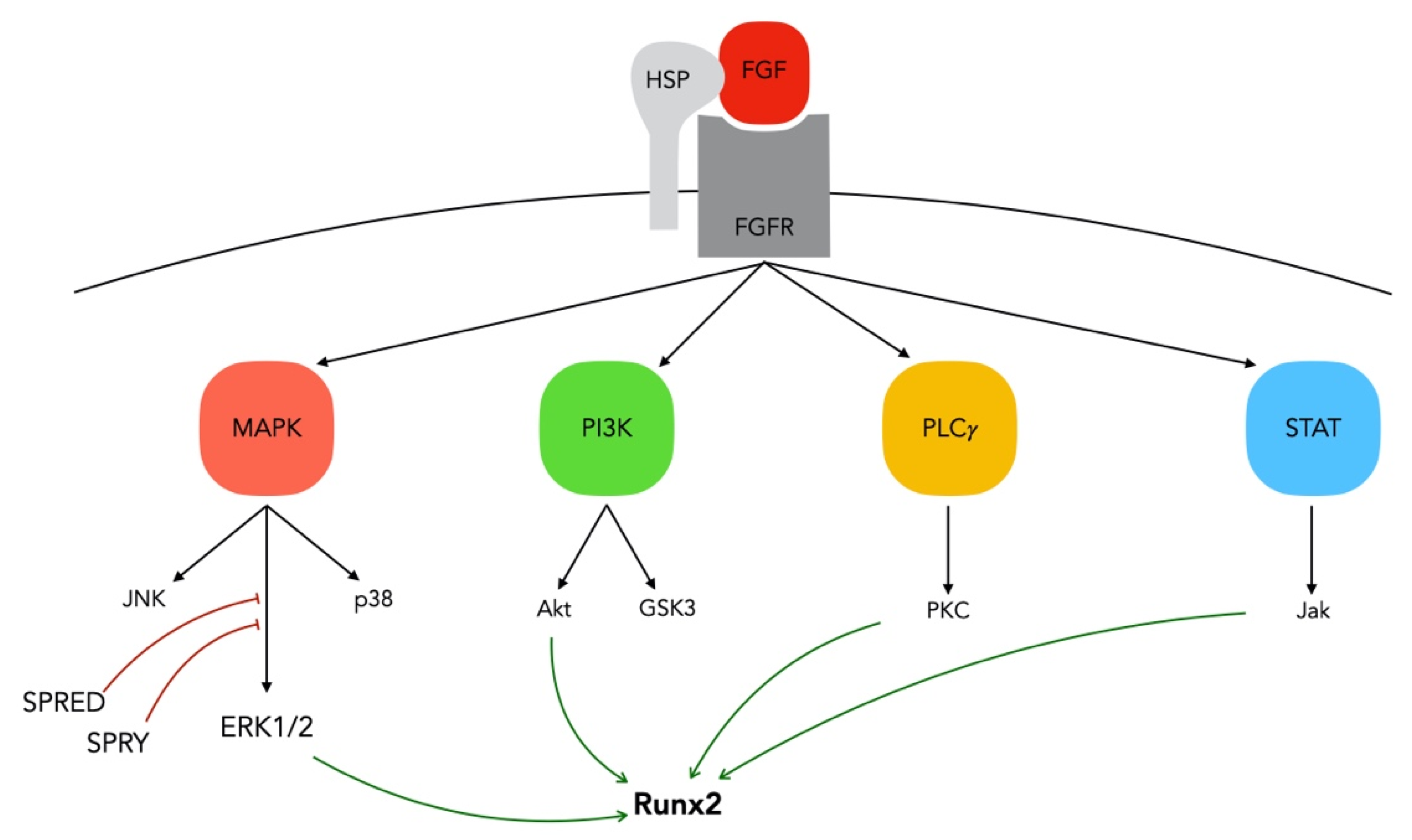
3.1. FGF/FGFR Signaling in Bone
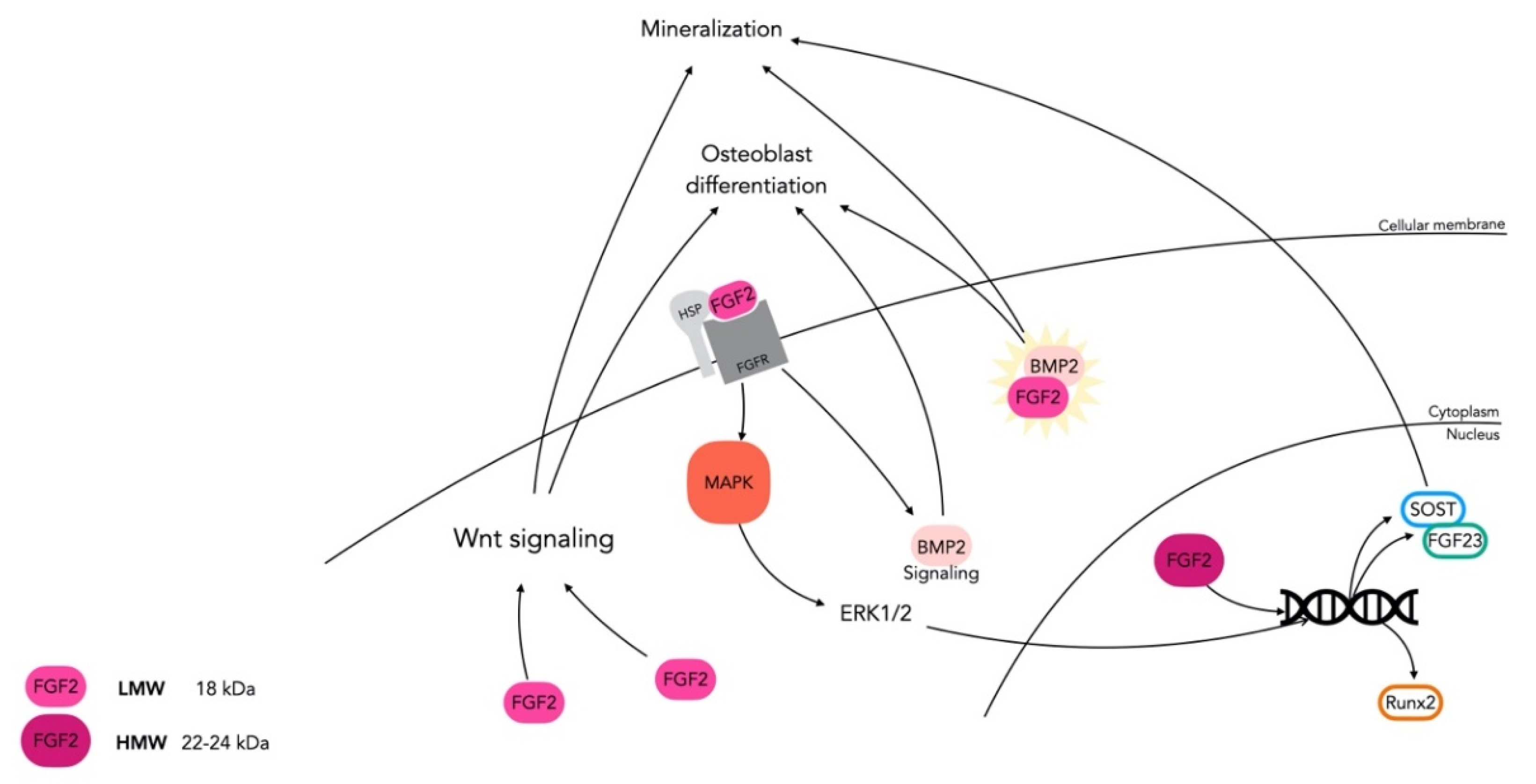
3.2. FGF-2: An Essential Regulator in Skeletal Tissue
4. FGF-2 in Craniofacial Bone Tissue Engineering
4.1. FGF-2: An Exogenous Factor In Vitro
4.2. FGF-2: An Exogenous Factor In Vivo
4.3. Human Clinical Applications of FGF-2 in the Craniofacial Area
4.4. FGF-2 as an Exogenous Factor: A Synthesis of Current Knowledge
4.4.1. Dosage (In Vitro/In Vivo)
4.4.2. Scaffolding and Stabilization by Administration Mode
4.4.3. BMP-2: An Interesting Cytokine for Combined Treatments
5. Discussion, Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Endochondral Ossification | Intramembranous Ossification | |
|---|---|---|
| Process | Replacement of cartilage with bone | Direct conversion of mesenchyme to bone |
| Site | Mainly long bones | Mainly flat bones |
| Cellular embryonic origin | Neural crest-derived mesenchymal cells | Mesoderm-derived mesenchymal cells |
| Functional cells | Chondrocytes that secrete ECM to form cartilage | Osteoblasts that secrete osteoid matrix |
| Angiogenic Effect | Osteogenic Effect | Function | |
|---|---|---|---|
| BMP | Yes (indirectly) [241] | Yes [242] | Differentiation of osteoblast-like cells chemoattractant for neighboring endothelial cells (ECs) [241,243,244]; proliferation and differentiation of mesenchymal osteoprogenitors [245]; BMP-2 and -7 enhance bone formation and repair, through induction of vascular endothelial growth factor (VEGF) expression and angiogenesis stimulation [246,247] |
| TGF-β | Yes, but role not well understood [248] | Yes [247] | Chemoattractant for mesenchymal stem cells, differentiation of osteoblasts [249,250,251], chondroblast, and osteoprogenitor cells and matrix production stimulation in healing process [252,253] |
| PDGF | Yes | Yes | Chemoattractant for and mitogenic stimulation of osteoblasts [251,254] |
| MMP | Yes | Yes | Matrix metallopeptidase 9 (MMP9) induces vascularization in bone formation by the release of VEGF from ECM [255]; while matrix metallopeptidase 13 (MMP13) induces osteogenesis [256] |
| Notch signaling | Yes | Yes | Notch pathway modulates the angiogenic effect of VEGF in ECs. Activation of Notch signaling in bone ECs promotes local angiogenesis and osteogenesis [257]. |
References
- Clarke, B. Normal Bone Anatomy and Physiology. Clin. J. Am. Soc. Nephrol. 2008, 3, S131–S139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiGirolamo, D.J.; Clemens, T.L.; Kousteni, S. The skeleton as an endocrine organ. Nat. Rev. Rheumatol. 2012, 8, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Oldknow, K.J.; Macrae, V.E.; Farquharson, C. Endocrine role of bone: Recent and emerging perspectives beyond osteocalcin. J. Endocrinol. 2015, 225, R1–R19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenkre, J.S.; Bassett, J.H. The bone remodelling cycle. Ann. Clin. Biochem. Int. J. Lab. Med. 2018, 55, 308–327. [Google Scholar] [CrossRef] [PubMed]
- Hadjidakis, D.J.; Androulakis, I.I. Bone remodeling. Ann. N. Y. Acad. Sci. 2006, 1092, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Alford, A.I.; Kozloff, K.M.; Hankenson, K.D. Extracellular matrix networks in bone remodeling. Int. J. Biochem. Cell Biol. 2015, 65, 20–31. [Google Scholar] [CrossRef]
- Yang, G.; Zhu, L.; Hou, N.; Lan, Y.; Wu, X.-M.; Zhou, B.; Teng, Y.; Yang, X. Osteogenic fate of hypertrophic chondrocytes. Cell Res. 2014, 24, 1266–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matic, I.; Matthews, B.G.; Wang, X.; Dyment, N.A.; Worthley, D.L.; Rowe, D.W.; Grcevic, D.; Kalajzic, I. Quiescent Bone Lining Cells Are a Major Source of Osteoblasts During Adulthood. Stem Cells 2016, 34, 2930–2942. [Google Scholar] [CrossRef] [Green Version]
- Murshed, M.; Harmey, D.; Millán, J.L.; McKee, M.D.; Karsenty, G. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 2005, 19, 1093–1104. [Google Scholar] [CrossRef] [Green Version]
- Katsimbri, P. The biology of normal bone remodelling. Eur. J. Cancer Care 2017, 26, e12740. [Google Scholar] [CrossRef]
- Franz-Odendaal, T.A.; Hall, B.K.; Witten, P.E. Buried alive: How osteoblasts become osteocytes. Dev. Dyn. 2005, 235, 176–190. [Google Scholar] [CrossRef]
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, Y.; Pacios, S.; Li, S.; Graves, D.T. Cellular and Molecular Aspects of Bone Remodeling. Craniofac. Sutures 2015, 18, 9–16. [Google Scholar] [CrossRef]
- Bonewald, L.F.; Johnson, M.L. Osteocytes, mechanosensing and Wnt signaling. Bone 2008, 42, 606–615. [Google Scholar] [CrossRef] [Green Version]
- Dallas, S.L.; Prideaux, M.; Bonewald, L.F. The Osteocyte: An Endocrine Cell … and More. Endocr. Rev. 2013, 34, 658–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanyon, L.E. Osteocytes, strain detection, bone modeling and remodeling. Calcif. Tissue Int. 1993, 53, S102–S107. [Google Scholar] [CrossRef]
- Sims, A.N.; Martin, T.J. Coupling the activities of bone formation and resorption: A multitude of signals within the basic multicellular unit. Bonekey Rep. 2014, 3, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teitelbaum, S.L. Bone Resorption by Osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Mackie, E.J.; Tatarczuch, L.; Mirams, M. The skeleton: A multi-functional complex organ. The growth plate chondrocyte and endochondral ossification. J. Endocrinol. 2011, 211, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, B.; Persikov, A.V. Molecular Structure of the Collagen Triple Helix. Adv. Protein Chem. 2005, 70, 301–339. [Google Scholar] [CrossRef] [PubMed]
- Ducy, P.; Desbois, C.; Boyce, B.; Pinero, G.; Story, B.; Dunstan, C.; Smith, E.; Bonadio, J.; Goldstein, S.; Gundberg, C.; et al. Increased bone formation in osteocalcin-deficient mice. Nat. Cell Biol. 1996, 382, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nat. Cell Biol. 1997, 386, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Burr, D.; Schaffler, M.; Yang, K.; Wu, D.; Lukoschek, M.; Kandzari, D.; Sivaneri, N.; Blaha, J.; Radin, E. The effects of altered strain environments on bone tissue kinetics. Bone 1989, 10, 215–221. [Google Scholar] [CrossRef]
- Krahl, H.; Michaelis, U.; Pieper, H.G.; Quack, G.; Montag, M. Stimulation of bone growth through sports. A radiologic investigation of the upper extremities in professional tennis players. Am. J. Sports Med. 1994, 22, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; McDonald, J.M. Disorders of Bone Remodeling. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 121–145. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Takahashi, H.; Ito, A.; Saito, N.; Nawata, M.; Horiuchi, H.; Ohta, H.; Iorio, R.; Yamamoto, N.; Takaoka, K. Trabecular minimodeling in human iliac bone. Bone 2003, 32, 163–169. [Google Scholar] [CrossRef]
- Frost, H.M. Skeletal structural adaptations to mechanical usage (SATMU): Mechanical influences on intact fibrous tissues. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 1990, 226, 433–439. [Google Scholar] [CrossRef]
- Manolagas, S.C. Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 2000, 21, 115–137. [Google Scholar] [PubMed] [Green Version]
- Takayanagi, H. Osteoimmunology: Shared mechanisms and crosstalk between the immune and bone systems. Nat. Rev. Immunol. 2007, 7, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nat. Cell Biol. 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.; Lacey, D.L.; Dunstan, C.R.; Solovyev, I.; Colombero, A.; Timms, E.; Tan, H.-L.; Elliott, G.; Kelley, M.J.; Sarosi, I.; et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc. Natl. Acad. Sci. USA 1999, 96, 3540–3545. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.-Y.; Yoshida, H.; Sarosi, I.; Tan, H.-L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-Dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nat. Cell Biol. 1999, 397, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Belibasakis, G.N.; Bostanci, N. The RANKL-OPG system in clinical periodontology. J. Clin. Periodontol. 2012, 39, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Simonet, W.; Lacey, D.; Dunstan, C.; Kelley, M.; Chang, M.-S.; Lüthy, R.; Nguyen, H.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A Novel Secreted Protein Involved in the Regulation of Bone Density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Hofbauer, L.C.; Khosla, S.; Dunstan, C.R.; Lacey, D.L.; Boyle, W.J.; Riggs, B.L. The Roles of Osteoprotegerin and Osteoprotegerin Ligand in the Paracrine Regulation of Bone Resorption. J. Bone Miner. Res. 2000, 15, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Maeda, K.; Ishihara, A.; Uehara, S.; Kobayashi, Y. Regulatory mechanism of osteoclastogenesis by RANKL and Wnt signals. Front. Biosci. 2011, 16, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, T.F.; Guo, X.; Garrett-Beal, L.; Yang, Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 2005, 8, 739–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, D.A., II.; Bialek, P.; Ahn, J.D.; Starbuck, M.; Patel, M.S.; Clevers, H.; Taketo, M.M.; Long, F.; McMahon, A.P.; Lang, R.A.; et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 2005, 8, 751–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daoussis, D.; Andonopoulos, A.P. The Emerging Role of Dickkopf-1 in Bone Biology: Is It the Main Switch Controlling Bone and Joint Remodeling? Semin. Arthritis Rheum. 2011, 41, 170–177. [Google Scholar] [CrossRef]
- Caetano-Lopes, J.; Canhão, H.; Fonseca, J.E. Osteoblasts and bone formation. Acta Reumatol. Port. 2007, 32, 103–110. [Google Scholar]
- Carter, P.H.; Schipan, E. The roles of parathyroid hormone and calcitonin in bone remodeling: Prospects for novel therapeutics. Endocr. Metab. Immune Disord. Drug Targets 2006, 6, 59–76. [Google Scholar] [CrossRef]
- Bassett, J.H.D.; Williams, G.R. Role of Thyroid Hormones in Skeletal Development and Bone Maintenance. Endocr. Rev. 2016, 37, 135–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iglesias, L.; Yeh, J.K.; Castro-Magana, M.; Aloia, J.F. Effects of growth hormone on bone modeling and remodeling in hypophysectomized young female rats: A bone histomorphometric study. J. Bone Miner. Metab. 2010, 29, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, R.S.; Jilka, R.L.; Parfitt, A.M.; Manolagas, S.C. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J. Clin. Investig. 1998, 102, 274–282. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Imai, Y.; Matsumoto, T.; Sato, S.; Takeuchi, K.; Igarashi, K.; Harada, Y.; Azuma, Y.; Krust, A.; Yamamoto, Y.; et al. Estrogen Prevents Bone Loss via Estrogen Receptor α and Induction of Fas Ligand in Osteoclasts. Cell 2007, 130, 811–823. [Google Scholar] [CrossRef]
- Roodman, G.D. Role of cytokines in the regulation of bone resorption. Calcif. Tissue Int. 1993, 53, S94–S98. [Google Scholar] [CrossRef] [PubMed]
- Graves, D.T.; Oates, T.; Garlet, G.P. Review of osteoimmunology and the host response in endodontic and periodontal lesions. J. Oral Microbiol. 2011, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Raisz, L. Prostaglandins and bone: Physiology and pathophysiology. Osteoarthr. Cartil. 1999, 7, 419–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandi, M.L.; Collin-Osdoby, P. Vascular Biology and the Skeleton. J. Bone Miner. Res. 2006, 21, 183–192. [Google Scholar] [CrossRef]
- Fang, T.D.; Salim, A.; Xia, W.; Nacamuli, R.P.; Guccione, S.; Song, H.M.; Carano, R.A.; Filvaroff, E.H.; Bednarski, M.D.; Giaccia, A.J.; et al. Angiogenesis Is Required for Successful Bone Induction During Distraction Osteogenesis. J. Bone Miner. Res. 2005, 20, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Seebach, C.; Henrich, D.; Wilhelm, K.; Barker, J.H.; Marzi, I. Endothelial Progenitor Cells Improve Directly and Indirectly Early Vascularization of Mesenchymal Stem Cell-Driven Bone Regeneration in a Critical Bone Defect in Rats. Cell Transplant. 2012, 21, 1667–1677. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, R.; Einhorn, T.; Lehmann, W.; Edgar, C.; Al-Yamani, A.; Apazidis, A.; Pacicca, D.; Clemens, T.; Gerstenfeld, L. The role of angiogenesis in a murine tibial model of distraction osteogenesis. Bone 2004, 34, 849–861. [Google Scholar] [CrossRef]
- Percival, C.J.; Richtsmeier, J.T. Angiogenesis and intramembranous osteogenesis. Dev. Dyn. 2013, 242, 909–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, C.; Clemens, T.L. Angiogenic–osteogenic coupling: The endothelial perspective. Bonekey Rep. 2014, 3, 578. [Google Scholar] [CrossRef] [Green Version]
- Schipani, E.; Maes, C.; Carmeliet, G.; Semenza, G.L. Regulation of Osteogenesis-Angiogenesis Coupling by HIFs and VEGF. J. Bone Miner. Res. 2009, 24, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Sivaraj, K.K.; Adams, R.H. Blood vessel formation and function in bone. Development 2016, 143, 2706–2715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jośko, J.; Gwóźdź, B.; Jedrzejowska-Szypułka, H.; Hendryk, S. Vascular endothelial growth factor (VEGF) and its effect on angiogenesis. Med. Sci. Monit. 2001, 6, 1047–1052. [Google Scholar]
- Bluteau, G.; Julien, M.; Magne, D.; Mallein-Gerin, F.; Weiss, P.; Daculsi, G.; Guicheux, J. VEGF and VEGF receptors are differentially expressed in chondrocytes. Bone 2007, 40, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Olsen, B.R. Osteoblast-derived VEGF regulates osteoblast differentiation and bone formation during bone repair. J. Clin. Investig. 2016, 126, 509–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouletreau, P.J.; Warren, S.M.; Spector, J.A.; Peled, Z.M.; Gerrets, R.P.; Greenwald, J.A.; Longaker, M.T. Hypoxia and VEGF Up-Regulate BMP-2 mRNA and Protein Expression in Microvascular Endothelial Cells: Implications for Fracture Healing. Plast. Reconstr. Surg. 2002, 109, 2384–2397. [Google Scholar] [CrossRef] [PubMed]
- Mayr-Wohlfart, U.; Waltenberger, J.; Hausser, H.; Kessler, S.; Günther, K.-P.; Dehio, C.; Puhl, W.; Brenner, R. Vascular endothelial growth factor stimulates chemotactic migration of primary human osteoblasts. Bone 2002, 30, 472–477. [Google Scholar] [CrossRef]
- Henriksen, K.; Karsdal, M.; Delaissé, J.M.; Engsig, M.T. RANKL and vascular endothelial growth factor (VEGF) induce osteoclast chemotaxis through an ERK1/2-dependent mechanism. J. Biol. Chem. 2003, 278, 48745–48753. [Google Scholar] [CrossRef] [Green Version]
- Carlevaro, M.F.; Cermelli, S.; Cancedda, R.; Cancedda, F.D. Vascular endothelial growth factor (VEGF) in cartilage neovascularization and chondrocyte differentiation: Auto-paracrine role during endochondral bone formation. J. Cell Sci. 2000, 113, 59–69. [Google Scholar]
- Street, J.; Bao, M.; DeGuzman, L.; Bunting, S.; Peale, F.V., Jr.; Ferrara, N.; Steinmetz, H.; Hoeffel, J.; Cleland, J.L.; Daugherty, A.; et al. Vascular endothelial growth factor stimulates bone repair by promoting angiogenesis and bone turnover. Proc. Natl. Acad. Sci. USA 2002, 99, 9656–9661. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-M.; Lee, Y.M.; Kim, H.-S.; Kim, J.D.; Choi, Y.; Kim, K.-W.; Lee, S.-Y.; Kwon, Y.-G. TNF-related Activation-induced Cytokine (TRANCE) Induces Angiogenesis through the Activation of Src and Phospholipase C (PLC) in Human Endothelial Cells. J. Biol. Chem. 2002, 277, 6799–6805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, J.K.; Kim, Y.M.; Kim, Y.M.; Kim, E.C.; Gho, Y.S.; Kang, I.J.; Lee, S.Y.; Kong, Y.Y.; Kwon, Y.G. Vascular endothelial growth factor up-regulates expression of receptor activator of NF-kappa B (RANK) in endothelial cells. Concomitant increase of angiogenic responses to RANK ligand. J. Biol. Chem. 2003, 278, 39548–39557. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-H.; Shin, H.S.; Kwak, H.J.; Ahn, K.Y.; Kim, J.-H.; Lee, H.J.; Lee, M.-S.; Lee, Z.H.; Koh, G.Y. RANKL regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. FASEB J. 2003, 17, 1–17. [Google Scholar] [CrossRef]
- Ulici, V.; Hoenselaar, K.D.; Agoston, H.; McErlain, D.D.; Umoh, J.; Chakrabarti, S.; Holdsworth, D.W.; Beier, F. The role of Akt1 in terminal stages of endochondral bone formation: Angiogenesis and ossification. Bone 2009, 45, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Aragonés, J.; Fraisl, P.; Baes, M.; Carmeliet, P. Oxygen Sensors at the Crossroad of Metabolism. Cell Metab. 2009, 9, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wan, C.; Deng, L.; Liu, X.; Cao, X.; Gilbert, S.R.; Bouxsein, M.L.; Faugere, M.-C.; Guldberg, R.E.; Gerstenfeld, L.C.; et al. The hypoxia-inducible factor α pathway couples angiogenesis to osteogenesis during skeletal development. J. Clin. Investig. 2007, 117, 1616–1626. [Google Scholar] [CrossRef]
- Riddle, R.C.; Khatri, R.; Schipani, E.; Clemens, T.L. Role of hypoxia-inducible factor-1α in angiogenic–osteogenic coupling. J. Mol. Med. 2009, 87, 583–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ornitz, D.M. FGFs, heparan sulfate and FGFRs: Complex interactions essential for development. BioEssays 2000, 22, 108–112. [Google Scholar] [CrossRef]
- Collin-Osdoby, P.; Rothe, L.; Bekker, S.; Anderson, F.; Huang, Y.; Osdoby, P. Basic fibroblast growth factor stimulates osteoclast recruitment, development, and bone pit resorption in association with angiogenesis in vivo on the chick chorioallantoic membrane and activates isolated avian osteoclast resorption in vitro. J. Bone Miner. Res. 2002, 17, 1859–1871. [Google Scholar] [CrossRef]
- De Moerlooze, L.; Spencer-Dene, B.; Revest, J.M.; Hajihosseini, M.; Rosewell, I.; Dickson, C. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Dev. 2000, 127, 483–492. [Google Scholar]
- Miraoui, H.; Marie, P.J. Fibroblast Growth Factor Receptor Signaling Crosstalk in Skeletogenesis. Sci. Signal. 2010, 3, re9. [Google Scholar] [CrossRef]
- Marie, P.J.; Miraoui, H.; Severe, N. FGF/FGFR signaling in bone formation: Progress and perspectives. Growth Factors 2012, 30, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, J.-C.; Kim, S.-H.; Im, G.-I.; Kim, B.-S.; Lee, J.-B.; Choi, E.-Y.; Song, J.-S.; Cho, K.-S.; Kim, C.-S. Treatment of FGF-2 on stem cells from inflamed dental pulp tissue from human deciduous teeth. Oral Dis. 2014, 20, 191–204. [Google Scholar] [CrossRef]
- Hatch, N.E. FGF Signaling in Craniofacial Biological Control and Pathological Craniofacial Development. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 295–311. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Eswarakumar, V.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef]
- Li, X.; Wang, C.; Xiao, J.; McKeehan, W.L.; Wang, F. Fibroblast growth factors, old kids on the new block. Semin. Cell Dev. Biol. 2016, 53, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Potthoff, J.M.; Kliewer, S.A.; Mangelsdorf, D.J. Endocrine fibroblast growth factors 15/19 and 21: From feast to famine. Genes Dev. 2012, 26, 312–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlinson, E.; Fu, L.; John, L.; Hultgren, B.; Huang, X.; Renz, M.; Stephan, J.P.; Tsai, S.P.; Powell-Braxton, L.; French, D.; et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology 2002, 143, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; McMahon, L.P.; Holt, S.G. Fibroblast growth factor. Ann. Clin. Biochem. Int. J. Lab. Med. 2014, 51, 203–227. [Google Scholar] [CrossRef]
- Belov, A.A.; Mohammadi, M. Molecular Mechanisms of Fibroblast Growth Factor Signaling in Physiology and Pathology. Cold Spring Harb. Perspect. Biol. 2013, 5, a015958. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Marie, P.J. Fibroblast growth factor signaling in skeletal development and disease. Genes Dev. 2015, 29, 1463–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yayon, A.; Klagsbrun, M.; Esko, J.D.; Leder, P.; Ornitz, D.M. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell 1991, 64, 841–848. [Google Scholar] [CrossRef]
- Rapraeger, C.A.; Krufka, A.; Olwin, B.B. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science 1991, 252, 1705–1708. [Google Scholar] [CrossRef] [Green Version]
- Ornitz, D.M.; Yayon, A.; Flanagan, J.G.; Svahn, C.M.; Levi, E.; Leder, P. Heparin is required for cell-free binding of basic fibroblast growth factor to a soluble receptor and for mitogenesis in whole cells. Mol. Cell. Biol. 1992, 12, 240–247. [Google Scholar] [CrossRef] [Green Version]
- Spivak-Kroizman, T.; Lemmon, M.; Dikic, I.; Ladbury, J.; Pinchasi, D.; Huang, J.; Jaye, M.; Crumley, G.; Schlessinger, J.; Lax, I. Heparin-induced oligomerization of FGF molecules is responsible for FGF receptor dimerization, activation, and cell proliferation. Cell 1994, 79, 1015–1024. [Google Scholar] [CrossRef]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, I.; Kimura-Yoshida, C. Extracellular modulation of Fibroblast Growth Factor signaling through heparan sulfate proteoglycans in mammalian development. Curr. Opin. Genet. Dev. 2013, 23, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Ori, A.; Rudd, T.R.; Uniewicz, K.A.; Ahmed, Y.A.; Guimond, S.E.; Skidmore, M.A.; Siligardi, G.; Yates, E.A.; Fernig, D.G. Diversification of the structural determinants of fibroblast growth factor-heparin interactions: Implications for binding specificity. J. Biol. Chem. 2012, 287, 40061–40073. [Google Scholar] [CrossRef] [Green Version]
- Goetz, R.; Mohammadi, M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell Biol. 2013, 14, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Hart, K.C.; Robertson, S.C.; Kanemitsu, M.Y.; Meyer, A.N.; Tynan, J.A.; Donoghue, D.J. Transformation and Stat activation by derivatives of FGFR1, FGFR3, and FGFR4. Oncogene 2000, 19, 3309–3320. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Lazar, M.A. Modulating nuclear receptor function: May the phos be with you. J. Clin. Investig. 1999, 103, 1617–1618. [Google Scholar] [CrossRef] [Green Version]
- Yun, Y.-R.; Won, J.E.; Jeon, E.; Lee, S.; Kang, W.; Jo, H.; Jang, J.H.; Shin, U.S.; Kim, H.W. Fibroblast Growth Factors: Biology, Function, and Application for Tissue Regeneration. J. Tissue Eng. 2010, 2010, 218142. [Google Scholar] [CrossRef] [PubMed]
- Nugent, M.A.; Iozzo, R.V. Fibroblast growth factor-2. Int. J. Biochem. Cell Biol. 2000, 32, 115–120. [Google Scholar] [CrossRef]
- Johnson, L.G.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taketomi, T.; Onimura, T.; Yoshiga, D.; Muratsu, D.; Sanui, T.; Fukuda, T.; Kusukawa, J.; Nakamura, S. Sprouty2 is involved in the control of osteoblast proliferation and differentiation through the FGF and BMP signaling pathways. Cell Biol. Int. 2017, 42, 1106–1114. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.; Ladage, D.; Schinköthe, T.; Klausmann, U.; Ulrichs, C.; Klinz, F.-J.; Brixius, K.; Arnhold, S.; Desai, B.; Mehlhorn, U.; et al. Basic Fibroblast Growth Factor Controls Migration in Human Mesenchymal Stem Cells. Stem Cells 2006, 24, 1750–1758. [Google Scholar] [CrossRef]
- Jin, L.; Hu, X.; Feng, L. NT3 inhibits FGF2-induced neural progenitor cell proliferation via the PI3K/GSK3 pathway. J. Neurochem. 2005, 93, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, S.; Fujiwara, T.; Matsuzaki, S.; Shingaki, K.; Taniguchi, M.; Miyata, S.; Tohyama, M.; Sakai, Y.; Yano, K.; Hosokawa, K.; et al. bFGF Regulates PI3-Kinase-Rac1-JNK Pathway and Promotes Fibroblast Migration in Wound Healing. PLoS ONE 2010, 5, e12228. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.C.; Kim, S.J.; Choi, J.H.; Park, C.Y.; Shim, W.J.; Lim, D.S. Fibroblast growth factor-2 and -4 promote the proliferation of bone marrow mesenchymal stem cells by the activation of the PI3K-Akt and ERK1/2 signaling pathways. Stem Cells Dev. 2008, 17, 725–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, H.; Kim, H.-L.; Chun, Y.-S.; Shin, D.H.; Lee, K.-H.; Shin, C.S.; Lee, D.Y.; Kim, H.-H.; Lee, Z.H.; Ryoo, H.-M.; et al. NAA10 controls osteoblast differentiation and bone formation as a feedback regulator of Runx2. Nat. Commun. 2014, 5, 5176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.-F.; Chaudhary, L.; Fausto, A.; Halstead, L.R.; Ory, D.S.; Avioli, L.V.; Cheng, S.-L. Erk Is Essential for Growth, Differentiation, Integrin Expression, and Cell Function in Human Osteoblastic Cells. J. Biol. Chem. 2001, 276, 14443–14450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marie, P. Fibroblast growth factor signaling controlling osteoblast differentiation. Gene 2003, 316, 23–32. [Google Scholar] [CrossRef]
- Chaudhary, L.R.; Avioli, L.V. Extracellular-signal regulated kinase signaling pathway mediates downregulation of type I procollagen gene expression by FGF-2, PDGF-BB, and okadaic acid in osteoblastic cells. J. Cell. Biochem. 2000, 76, 354–359. [Google Scholar] [CrossRef]
- Ge, C.; Xiao, G.; Jiang, D.; Franceschi, R.T. Critical role of the extracellular signal–regulated kinase–MAPK pathway in osteoblast differentiation and skeletal development. J. Cell Biol. 2007, 176, 709–718. [Google Scholar] [CrossRef]
- Marshak, D.R.; Jaiswal, R.K.; Jaiswal, N.; Bruder, S.P.; Mbalaviele, G.; Pittenger, M.F. Adult Human Mesenchymal Stem Cell Differentiation to the Osteogenic or Adipogenic Lineage Is Regulated by Mitogen-activated Protein Kinase. J. Biol. Chem. 2000, 275, 9645–9652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratchmarova, I.; Blagoev, B.; Haack-Sorensen, M.; Kassem, M.; Mann, M. Mechanism of Divergent Growth Factor Effects in Mesenchymal Stem Cell Differentiation. Science 2005, 308, 1472–1477. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, T.; Chan, Y.Y.; Kawanami, A.; Balmes, G.; Landreth, G.E.; Murakami, S. Extracellular Signal-Regulated Kinase 1 (ERK1) and ERK2 Play Essential Roles in Osteoblast Differentiation and in Supporting Osteoclastogenesis. Mol. Cell. Biol. 2009, 29, 5843–5857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.-S.; Lind, A.-K.; Dahm-Kähler, P.; Brännström, M.; Carletti, M.Z.; Christenson, L.K.; Curry, T.E.; Jo, M. RUNX2 Transcription Factor Regulates Gene Expression in Luteinizing Granulosa Cells of Rat Ovaries. Mol. Endocrinol. 2010, 24, 846–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bundschu, K.; Knobeloch, K.-P.; Ullrich, M.; Schinke, T.; Amling, M.; Engelhardt, C.M.; Renné, T.; Walter, U.; Schuh, K. Gene Disruption of Spred-2 Causes Dwarfism. J. Biol. Chem. 2005, 280, 28572–28580. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, K.; Ayada, T.; Ichiyama, K.; Kohno, R.-I.; Yonemitsu, Y.; Minami, Y.; Kikuchi, A.; Maehara, Y.; Yoshimura, A. Sprouty2 and Sprouty4 are essential for embryonic morphogenesis and regulation of FGF signaling. Biochem. Biophys. Res. Commun. 2007, 352, 896–902. [Google Scholar] [CrossRef]
- Ozaki, K.-I.; Miyazaki, S.; Tanimura, S.; Kohno, M. Efficient suppression of FGF-2-induced ERK activation by the cooperative interaction among mammalian Sprouty isoforms. J. Cell Sci. 2005, 118, 5861–5871. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, A. Regulation of Cytokine Signaling by the SOCS and Spred Family Proteins. Keio J. Med. 2009, 58, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.C.; Swindell, E.C.; Sierralta, W.D.; Eichele, G.; Thaller, C. Evidence for a role of protein kinase C in FGF signal transduction in the developing chick limb bud. Development 2001, 128, 2451–2460. [Google Scholar]
- Mohammadi, M.; Schlessinger, J.; Hubbard, S.R. Structure of the FGF Receptor Tyrosine Kinase Domain Reveals a Novel Autoinhibitory Mechanism. Cell 1996, 86, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-J.; Kim, J.-H.; Bae, S.-C.; Choi, J.-Y.; Ryoo, H.-M. The Protein Kinase C Pathway Plays a Central Role in the Fibroblast Growth Factor-stimulated Expression and Transactivation Activity of Runx2. J. Biol. Chem. 2003, 278, 319–326. [Google Scholar] [CrossRef] [Green Version]
- Osathanon, T.; Nowwarote, N.; Pavasant, P. Basic fibroblast growth factor inhibits mineralization but induces neuronal differentiation by human dental pulp stem cells through a FGFR and PLCγ signaling pathway. J. Cell. Biochem. 2011, 112, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Spivak-Kroizman, T.; Mohammadi, M.; Hu, P.; Jaye, M.; Schlessinger, J.; Lax, I. Point mutation in the fibroblast growth factor receptor eliminates phosphatidylinositol hydrolysis without affecting neuronal differentiation of PC12 cells. J. Biol. Chem. 1994, 269, 14419–14423. [Google Scholar] [CrossRef]
- Kang, S.; Elf, S.; Dong, S.; Hitosugi, T.; Lythgoe, K.; Guo, A.; Ruan, H.; Lonial, S.; Khoury, H.J.; Williams, I.R.; et al. Fibroblast Growth Factor Receptor 3 Associates with and Tyrosine Phosphorylates p90 RSK2, Leading to RSK2 Activation That Mediates Hematopoietic Transformation. Mol. Cell. Biol. 2009, 29, 2105–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dailey, L.; Ambrosetti, D.; Mansukhani, A.; Basilico, C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 2005, 16, 233–247. [Google Scholar] [CrossRef]
- Marie, P.; Coffin, J.; Hurley, M. FGF and FGFR signaling in chondrodysplasias and craniosynostosis. J. Cell. Biochem. 2005, 96, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Muenke, M.; Schell, U. Fibroblast-growth-factor receptor mutations in human skeletal disorders. Trends Genet. 1995, 11, 308–313. [Google Scholar] [CrossRef]
- Ornitz, M.D.; Marie, P.J. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002, 16, 1446–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ornitz, D.M.; Itoh, N. Fibroblast growth factors. Genome Biol. 2001, 2, 3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffin, J.D.; Homer-Bouthiette, C.; Hurley, M.M. Fibroblast Growth Factor 2 and Its Receptors in Bone Biology and Disease. J. Endocr. Soc. 2018, 2, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Du, E.; Xiao, L.; Hurley, M.M. FGF23 Neutralizing Antibody Ameliorates Hypophosphatemia and Impaired FGF Receptor Signaling in Kidneys of HMWFGF2 Transgenic Mice. J. Cell. Physiol. 2016, 232, 610–616. [Google Scholar] [CrossRef]
- Yang, J.; Meyer, M.; Müller, A.-K.; Böhm, F.; Grose, R.; Dauwalder, T.; Verrey, F.; Kopf, M.; Partanen, J.; Bloch, W.; et al. Fibroblast growth factor receptors 1 and 2 in keratinocytes control the epidermal barrier and cutaneous homeostasis. J. Cell Biol. 2010, 188, 935–952. [Google Scholar] [CrossRef] [Green Version]
- Sufen, G.; Xianghong, Y.; Yongxia, C.; Qian, P. bFGF and PDGF-BB have a synergistic effect on the proliferation, migration and VEGF release of endothelial progenitor cells. Cell Biol. Int. 2011, 35, 545–551. [Google Scholar] [CrossRef]
- Hill, P.A.; Tumber, A.; Meikle, M.C. Multiple Extracellular Signals Promote Osteoblast Survival and Apoptosis. Endocrinology 1997, 138, 3849–3858. [Google Scholar] [CrossRef]
- Nowwarote, N.; Sukarawan, W.; Pavasant, P.; Osathanon, T. Basic Fibroblast Growth Factor Regulates REX1 Expression Via IL-6 In Stem Cells Isolated From Human Exfoliated Deciduous Teeth. J. Cell. Biochem. 2017, 118, 1480–1488. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, H.; Zhao, Y.; Li, J.; Cai, J.; Wang, P.; Meng, S.; Feng, J.; Miao, C.; Ding, M.; et al. Noggin and bFGF cooperate to maintain the pluripotency of human embryonic stem cells in the absence of feeder layers. Biochem. Biophys. Res. Commun. 2005, 330, 934–942. [Google Scholar] [CrossRef]
- Kawaguchi, H.; Kurokawa, T.; Hanada, K.; Hiyama, Y.; Tamura, M.; Ogata, E.; Matsumoto, T. Stimulation of fracture repair by recombinant human basic fibroblast growth factor in normal and streptozotocin-diabetic rats. Endocrinology 1994, 135, 774–781. [Google Scholar] [CrossRef]
- Liang, H.; Pun, S.; Wronski, T.J. Bone anabolic effects of basic fibroblast growth factor in ovariectomized rats. Endocrinology 1999, 140, 5780–5788. [Google Scholar] [CrossRef]
- Yamaguchi, T.P.; Rossant, J. Fibroblast growth factors in mammalian development. Curr. Opin. Genet. Dev. 1995, 5, 485–491. [Google Scholar] [CrossRef]
- Kimelman, D.; Kirschner, M. Synergistic induction of mesoderm by FGF and TGF-beta and the identification of an mRNA coding for FGF in the early Xenopus embryo. Cell 1987, 51, 869–877. [Google Scholar] [CrossRef]
- Peters, K.; Ornitz, D.; Werner, S.; Williams, L. Unique Expression Pattern of the FGF Receptor 3 Gene during Mouse Organogenesis. Dev. Biol. 1993, 155, 423–430. [Google Scholar] [CrossRef]
- Hurley, M.M.; Abreu, C.; Gronowicz, G.; Kawaguchi, H.; Lorenzo, J. Expression and regulation of basic fibroblast growth factor mRNA levels in mouse osteoblastic MC3T3-E1 cells. J. Biol. Chem. 1994, 269, 9392–9396. [Google Scholar] [CrossRef]
- Vagner, S.; Gensac, M.C.; Maret, A.; Bayard, F.; Amalric, F.; Prats, H.; Prats, A.C. Alternative translation of human fibroblast growth factor 2 mRNA occurs by internal entry of ribosomes. Mol. Cell. Biol. 1995, 15, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, V.M.; Ling, L.; Natarajan, S.; Yap, M.G.; Cool, S.M.; Choo, A.B. FGF-2 modulates Wnt signaling in undifferentiated hESC and iPS cells through activated PI3-K/GSK3beta signaling. J. Cell Physiol. 2010, 225, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Fakhry, A.; Ratisoontorn, C.; Vedhachalam, C.; Salhab, I.; Koyama, E.; Leboy, P.; Pacifici, M.; Kirschner, R.E.; Nah, H.D. Effects of FGF-2/-9 in calvarial bone cell cultures: Differentiation stage-dependent mitogenic effect, inverse regulation of BMP-2 and noggin, and enhancement of osteogenic potential. Bone 2005, 36, 254–266. [Google Scholar] [CrossRef]
- Homer-Bouthiette, C.; Doetschman, T.; Xiao, L.; Hurley, M.M. Knockout of Nuclear High Molecular Weight FGF2 Isoforms in Mice Modulates Bone and Phosphate Homeostasis. J. Biol. Chem. 2014, 289, 36303–36314. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Xu, J.; Liu, Z.; Sosic, D.; Shao, J.; Olson, E.N.; Towler, D.A.; Ornitz, D.M. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 2003, 130, 3063–3074. [Google Scholar] [CrossRef] [Green Version]
- Jacob, A.L.; Smith, C.; Partanen, J.; Ornitz, D.M. Fibroblast growth factor receptor 1 signaling in the osteo-chondrogenic cell lineage regulates sequential steps of osteoblast maturation. Dev. Biol. 2006, 296, 315–328. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-H.; Su, S.-Y.; Sittampalam, K.; Chen, P.C.-H.; Petersson, F.; Kao, Y.-C.; Carpenter, T.O.; Hsieh, T.-H.; Konishi, E.; Tsai, J.-W.; et al. Frequent overexpression of klotho in fusion-negative phosphaturic mesenchymal tumors with tumorigenic implications. Mod. Pathol. 2019, 33, 858–870. [Google Scholar] [CrossRef]
- Fei, Y.; Xiao, L.; Doetschman, T.; Coffin, D.J.; Hurley, M.M. Fibroblast Growth Factor 2 Stimulation of Osteoblast Differentiation and Bone Formation Is Mediated by Modulation of the Wnt Signaling Pathway. J. Biol. Chem. 2011, 286, 40575–40583. [Google Scholar] [CrossRef] [Green Version]
- Naganawa, T.; Xiao, L.; Abogunde, E.; Sobue, T.; Kalajzic, I.; Sabbieti, M.; Agas, D.; Hurley, M. In vivo and in vitro comparison of the effects of FGF-2 null and haplo-insufficiency on bone formation in mice. Biochem. Biophys. Res. Commun. 2006, 339, 490–498. [Google Scholar] [CrossRef]
- Mansukhani, A.; Bellosta, P.; Sahni, M.; Basilico, C. Signaling by Fibroblast Growth Factors (Fgf) and Fibroblast Growth Factor Receptor 2 (Fgfr2)–Activating Mutations Blocks Mineralization and Induces Apoptosis in Osteoblasts. J. Cell Biol. 2000, 149, 1297–1308. [Google Scholar] [CrossRef]
- Mansukhani, A.; Ambrosetti, D.; Holmes, G.; Cornivelli, L.; Basilico, C. Sox2 induction by FGF and FGFR2 activating mutations inhibits Wnt signaling and osteoblast differentiation. J. Cell Biol. 2005, 168, 1065–1076. [Google Scholar] [CrossRef] [Green Version]
- Miraoui, H.; Oudina, K.; Petite, H.; Tanimoto, Y.; Moriyama, K.; Marie, P.J. Fibroblast growth factor receptor 2 promotes osteogenic differentiation in mesenchymal cells via ERK1/2 and protein kinase C signaling. J. Biol. Chem. 2009, 284, 4897–4904. [Google Scholar] [CrossRef] [Green Version]
- Naganawa, T.; Xiao, L.; Coffin, J.; Doetschman, T.; Sabbieti, M.G.; Agas, D.; Hurley, M.M. Reduced expression and function of bone morphogenetic protein-2 in bones of Fgf2 null mice. J. Cell. Biochem. 2008, 103, 1975–1988. [Google Scholar] [CrossRef]
- Xiao, G.; Jiang, D.; Gopalakrishnan, R.; Franceschi, R.T. Fibroblast Growth Factor 2 Induction of the Osteocalcin Gene Requires MAPK Activity and Phosphorylation of the Osteoblast Transcription Factor, Cbfa1/Runx2. J. Biol. Chem. 2002, 277, 36181–36187. [Google Scholar] [CrossRef] [Green Version]
- Ai-Aql, Z.S.; Alagl, A.S.; Graves, D.T.; Gerstenfeld, L.C.; Einhorn, T.A. Molecular mechanisms controlling bone formation during fracture healing and distraction osteogenesis. J. Dent Res. 2008, 87, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Lemonnier, J.; Haÿ, E.; Delannoy, P.; Lomri, A.; Modrowski, D.; Caverzasio, J.; Marie, P.J. Role of N-Cadherin and Protein Kinase C in Osteoblast Gene Activation Induced by the S252W Fibroblast Growth Factor Receptor 2 Mutation in Apert Craniosynostosis. J. Bone Miner. Res. 2001, 16, 832–845. [Google Scholar] [CrossRef] [PubMed]
- Debiais, F.; Lefèvre, G.; Lemonnier, J.; Le Mée, S.; Lasmoles, F.; Mascarelli, F.; Marie, P. Fibroblast growth factor-2 induces osteoblast survival through a phosphatidylinositol 3-kinase-dependent, -β-catenin-independent signaling pathway. Exp. Cell Res. 2004, 297, 235–246. [Google Scholar] [CrossRef]
- Montero, A.; Okada, Y.; Tomita, M.; Ito, M.; Tsurukami, H.; Nakamura, T.; Doetschman, T.; Coffin, J.D.; Hurley, M.M. Disruption of the fibroblast growth factor-2 gene results in decreased bone mass and bone formation. J. Clin. Investig. 2000, 105, 1085–1093. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Sobue, T.; Esliger, A.; Kronenberg, M.S.; Coffin, J.D.; Doetschman, T.; Hurley, M.M. Disruption of the Fgf2 gene activates the adipogenic and suppresses the osteogenic program in mesenchymal marrow stromal stem cells. Bone 2010, 47, 360–370. [Google Scholar] [CrossRef] [Green Version]
- Coffin, J.D.; Florkiewicz, R.Z.; Neumann, J.; Mort-Hopkins, T.; Dorn, G.W.; Lightfoot, P.; German, R.; Howles, P.N.; Kier, A.; O’Toole, B.A. Abnormal bone growth and selective translational regulation in basic fibroblast growth factor (FGF-2) transgenic mice. Mol. Biol. Cell 1995, 6, 1861–1873. [Google Scholar] [CrossRef]
- Sobue, T.; Naganawa, T.; Xiao, L.; Okada, Y.; Tanaka, Y.; Ito, M.; Okimoto, N.; Nakamura, T.; Coffin, J.; Hurley, M. Over-expression of fibroblast growth factor-2 causes defective bone mineralization and osteopenia in transgenic mice. J. Cell. Biochem. 2005, 95, 83–94. [Google Scholar] [CrossRef]
- Berthiaume, F.; Maguire, T.J.; Yarmush, M.L. Tissue Engineering and Regenerative Medicine: History, Progress, and Challenges. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 403–430. [Google Scholar] [CrossRef]
- Langer, R.; Vacanti, J.P. Tissue engineering. Science 1993, 260, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Black, C.R.M.; Goriainov, V.; Gibbs, D.; Kanczler, J.M.; Tare, R.S.; Oreffo, R.O.C. Bone Tissue Engineering. Curr. Mol. Biol. Rep. 2015, 1, 132–140. [Google Scholar] [CrossRef]
- Porter, J.R.; Ruckh, T.T.; Popat, K.C. Bone tissue engineering: A review in bone biomimetics and drug delivery strategies. Biotechnol. Prog. 2009, 25, 1539–1560. [Google Scholar] [CrossRef]
- McCarthy, T.L.; Centrella, M.; Canalis, E. Effects of Fibroblast Growth Factors on Deoxyribonucleic Acid and Collagen Synthesis in Rat Parietal Bone Cells. Endocrinology 1989, 125, 2118–2126. [Google Scholar] [CrossRef]
- Martin, I.; Muraglia, A.; Campanile, G.; Cancedda, R.; Quarto, R. Fibroblast growth factor-2 supports ex vivo expansion and maintenance of osteogenic precursors from human bone marrow. Endocrinology 1997, 138, 4456–4462. [Google Scholar] [CrossRef]
- Gorin, C.; Rochefort, G.Y.; Bascetin, R.; Ying, H.; Lesieur, J.; Sadoine, J.; Beckouche, N.; Berndt, S.; Novais, A.; Lesage, M.; et al. Priming Dental Pulp Stem Cells With Fibroblast Growth Factor-2 Increases Angiogenesis of Implanted Tissue-Engineered Constructs Through Hepatocyte Growth Factor and Vascular Endothelial Growth Factor Secretion. Stem Cells Transl. Med. 2016, 5, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Chosa, N.; Asakawa, T.; Yoshimura, Y.; Asakawa, A.; Ishisaki, A.; Tanaka, M. Effect of fibroblast growth factor-2 on periodontal ligament cells derived from human deciduous teeth in vitro. Exp. Ther. Med. 2010, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Huang, G.T.-J.; He, W.; Wang, P.; Tong, Z.; Jia, Q.; Dong, L.; Niu, Z.; Ni, L. Basic Fibroblast Growth Factor Enhances Stemness of Human Stem Cells from the Apical Papilla. J. Endod. 2012, 38, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Novais, A.; Lesieur, J.; Sadoine, J.; Slimani, L.; Baroukh, B.; Saubaméa, B.; Schmitt, A.; Vital, S.; Poliard, A.; Hélary, C.; et al. Priming Dental Pulp Stem Cells from Human Exfoliated Deciduous Teeth with Fibroblast Growth Factor-2 Enhances Mineralization Within Tissue-Engineered Constructs Implanted in Craniofacial Bone Defects. Stem Cells Transl. Med. 2019, 8, 844–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debiais, F.; Hott, M.; Graulet, A.M.; Marie, P.J. The Effects of Fibroblast Growth Factor-2 on Human Neonatal Calvaria Osteoblastic Cells Are Differentiation Stage Specific. J. Bone Miner. Res. 1998, 13, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Hurley, M.M.; Adams, U.J.; Wang, L.; Jiang, X.; Burt, P.M.; Du, E.; Xiao, L. Accelerated fracture healing in transgenic mice overexpressing an anabolic isoform of fibroblast growth factor 2. J. Cell. Biochem. 2016, 117, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.M.; Quarto, N.; Warren, S.M.; Salim, A.; Longaker, M.T. Age-related changes in the biomolecular mechanisms of calvarial osteoblast biology affect fibroblast growth factor-2 signaling and osteogenesis. J. Biol. Chem. 2003, 278, 45040. [Google Scholar] [CrossRef]
- Shimabukuro, Y.; Ueda, M.; Ozasa, M.; Anzai, J.; Takedachi, M.; Yanagita, M.; Ito, M.; Hashikawa, T.; Yamada, S.; Murakami, S. Fibroblast Growth Factor–2 Regulates the Cell Function of Human Dental Pulp Cells. J. Endod. 2009, 35, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Yu, J.; Liu, Y.; Lu, S.; Liu, H.; Shi, J.; Jin, Y. Effects of FGF2 and TGFbeta1 on the differentiation of human dental pulp stem cells in vitro. Cell Biol. Int. 2008, 32, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Nakao, K.; Itoh, M.; Tomita, Y.; Tomooka, Y.; Tsuji, T. FGF-2 potently induces both proliferation and DSP expression in collagen type I gel cultures of adult incisor immature pulp cells. Biochem. Biophys. Res. Commun. 2004, 325, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Hakki, S.S.; Hakki, E.E.; Nohutcu, R.M. Regulation of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases by basic fibroblast growth factor and dexamethasone in periodontal ligament cells. J. Periodontal Res. 2009, 44, 794–802. [Google Scholar] [CrossRef]
- Shimabukuro, Y.; Ueda, M.; Ichikawa, T.; Terashi, Y.; Yamada, S.; Kusumoto, Y.; Takedachi, M.; Terakura, M.; Kohya, A.; Hashikawa, T.; et al. Fibroblast Growth Factor-2 Stimulates Hyaluronan Production by Human Dental Pulp Cells. J. Endod. 2005, 31, 805–808. [Google Scholar] [CrossRef]
- Nauman, E.A.; Sakata, T.; Keaveny, T.M.; Halloran, B.P.; Bikle, D.D. bFGF Administration Lowers the Phosphate Threshold for Mineralization in Bone Marrow Stromal Cells. Calcif. Tissue Int. 2003, 73, 147–152. [Google Scholar] [CrossRef]
- Nakamura, S.; Nambu, M.; Ishizuka, T.; Hattori, H.; Kanatani, Y.; Takase, B.; Kishimoto, S.; Amano, Y.; Aoki, H.; Kiyosawa, T.; et al. Effect of controlled release of fibroblast growth factor-2 from chitosan/fucoidan micro complex-hydrogel onin vitro andin vivo vascularization. J. Biomed. Mater. Res. Part. A 2007, 85, 619–627. [Google Scholar] [CrossRef]
- Tabata, Y.; Yamada, K.; Hong, L.; Miyamoto, S.; Hashimoto, N.; Ikada, Y. Skull bone regeneration in primates in response to basic fibroblast growth factor. J. Neurosurg. 1999, 91, 851–856. [Google Scholar] [CrossRef]
- Kwan, M.D.; Sellmyer, M.A.; Quarto, N.; Ho, A.M.; Wandless, T.J.; Longaker, M.T. Chemical Control of FGF-2 Release for Promoting Calvarial Healing with Adipose Stem Cells. J. Biol. Chem. 2011, 286, 11307–11313. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, T.; Nakamura, T.; Kobayashi, E.; Inoue, M.; Shigeno, K.; Tabata, Y.; Eto, K.; Shimizu, Y. Novel Approach to Regeneration of Periodontal Tissues Based on in Situ Tissue Engineering: Effects of Controlled Release of Basic Fibroblast Growth Factor from a Sandwich Membrane. Tissue Eng. 2003, 9, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Anzai, J.; Nagayasu-Tanaka, T.; Terashima, A.; Asano, T.; Yamada, S.; Nozaki, T.; Kitamura, M.; Murakami, S. Long-term Observation of Regenerated Periodontium Induced by FGF-2 in the Beagle Dog 2-Wall Periodontal Defect Model. PLoS ONE 2016, 11, e0158485. [Google Scholar] [CrossRef]
- Lee, J.-H.; Kang, K.-J.; Jang, Y.-J. Functional efficacy of human recombinant FGF-2s tagged with (His) 6 and (His-Asn) 6 at the N- and C-termini in human gingival fibroblast and periodontal ligament-derived cells. Protein Expr. Purif. 2017, 135, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Charles, L.F.; Woodman, J.L.; Ueno, D.; Gronowicz, G.; Hurley, M.M.; Kuhn, L.T. Effects of low dose FGF-2 and BMP-2 on healing of calvarial defects in old mice. Exp. Gerontol. 2015, 64, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Kigami, R.; Sato, S.; Tsuchiya, N.; Yoshimakai, T.; Arai, Y.; Ito, K. FGF-2 Angiogenesis in Bone Regeneration Within Critical-Sized Bone Defects in Rat Calvaria. Implant. Dent. 2013, 22, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Rao, N.-J.; Chen, M.-L.; Huang, Z.-J.; Cao, Y.-G. Enhanced bone regeneration with sequential delivery of basic fibroblast growth factor and sonic hedgehog. Injury 2011, 42, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Mastrogiacomo, S.; Yang, F.; Shao, J.; Ong, M.M.A.; Chanchareonsook, N.; Jansen, J.A.; Walboomers, X.F.; Yu, N. Application of BMP-Bone Cement and FGF-Gel on Periodontal Tissue Regeneration in Nonhuman Primates. Tissue Eng. Part C Methods 2019, 25, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Collignon, A.-M.; Lesieur, J.; Anizan, N.; Ben Azzouna, R.; Poliard, A.; Gorin, C.; Letourneur, D.; Chaussain, C.; Rouzet, F.; Rochefort, G.Y. Early angiogenesis detected by PET imaging with 64Cu-NODAGA-RGD is predictive of bone critical defect repair. Acta Biomater. 2018, 82, 111–121. [Google Scholar] [CrossRef]
- Grosso, A.; Burger, M.G.; Lunger, A.; Schaefer, D.J.; Banfi, A.; Di Maggio, N. It Takes Two to Tango: Coupling of Angiogenesis and Osteogenesis for Bone Regeneration. Front. Bioeng. Biotechnol. 2017, 5, 68. [Google Scholar] [CrossRef]
- Atlas, Y.; Gorin, C.; Novais, A.; Marchand, M.F.; Chatzopoulou, E.; Lesieur, J.; Bascetin, R.; Binet-Moussy, C.; Sadoine, J.; Lesage, M.; et al. Microvascular maturation by mesenchymal stem cells in vitro improves blood perfusion in implanted tissue constructs. Biomaterials 2021, 268, 120594. [Google Scholar] [CrossRef]
- Kitamura, M.; Akamatsu, M.; Machigashira, M.; Hara, Y.; Sakagami, R.; Hirofuji, T.; Hamachi, T.; Maeda, K.; Yokota, M.; Kido, J.; et al. FGF-2 stimulates periodontal regeneration: Results of a multi-center randomized clinical trial. J. Dent. Res. 2011, 90, 35–40. [Google Scholar] [CrossRef]
- Santana, D.B.R.; de Santana, C.M. Human intrabony defect regeneration with rhFGF-2 and hyaluronic acid—A randomized controlled clinical trial. J. Clin. Periodontol. 2015, 42, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Cochran, D.; Oh, T.-J.; Mills, M.; Clem, D.; McClain, P.; Schallhorn, R.; McGuire, M.; Scheyer, E.; Giannobile, W.; Reddy, M.; et al. A Randomized Clinical Trial Evaluating rh-FGF-2/β-TCP in Periodontal Defects. J. Dent. Res. 2016, 95, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Khoshkam, V.; Chan, H.L.; Lin, G.H.; Mailoa, J.; Giannobile, W.V.; Wang, H.L.; Oh, T.J. Outcomes of regenerative treatment with rhPDGF-BB and rhFGF-2 for periodontal intra-bony defects: A systematic review and meta-analysis. J. Clin. Periodontol. 2015, 42, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Yu, F.; Xu, X.; Li, C.; Huang, D.; Zhou, X.; Ye, L.; Zheng, L. Evaluation of Recombinant Human FGF-2 and PDGF-BB in Periodontal Regeneration: A Systematic Review and Meta-Analysis. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Gronowicz, G.; Jacobs, E.; Peng, T.; Zhu, L.; Hurley, M.; Kuhn, L.T. Calvarial Bone Regeneration Is Enhanced by Sequential Delivery of FGF-2 and BMP-2 from Layer-by-Layer Coatings with a Biomimetic Calcium Phosphate Barrier Layer. Tissue Eng. Part. A 2017, 23, 1490–1501. [Google Scholar] [CrossRef]
- Ou, G.; Charles, L.; Matton, S.; Rodner, C.; Hurley, M.; Kuhn, L.; Gronowicz, G. Fibroblast Growth Factor-2 Stimulates the Proliferation of Mesenchyme-Derived Progenitor Cells From Aging Mouse and Human Bone. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2010, 65, 1051–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukarawan, W.; Nowwarote, N.; Kerdpon, P.; Pavasant, P.; Osathanon, T. Effect of basic fibroblast growth factor on pluripotent marker expression and colony forming unit capacity of stem cells isolated from human exfoliated deciduous teeth. Odontology 2013, 102, 160–166. [Google Scholar] [CrossRef]
- Ludwig, T.E.; Levenstein, M.E.; Jones, J.M.; Berggren, W.T.; Mitchen, E.R.; Frane, J.L.; Crandall, L.J.; Daigh, C.A.; Conard, K.R.; Piekarczyk, M.S.; et al. Derivation of human embryonic stem cells in defined conditions. Nat. Biotechnol. 2006, 24, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Varkey, M.; Kucharski, C.; Haque, T.; Sebald, W.; Uludağ, H. In Vitro Osteogenic Response of Rat Bone Marrow Cells to bFGF and BMP-2 Treatments. Clin. Orthop. Relat. Res. 2006, 443, 113–123. [Google Scholar] [CrossRef] [Green Version]
- Dunstan, C.R.; Boyce, R.; Boyce, B.F.; Garrett, I.R.; Izbicka, E.; Burgess, W.H.; Mundy, G.R. Systemic Administration of Acidic Fibroblast Growth Factor (FGF-1) Prevents Bone Loss and Increases New Bone Formation in Ovariectomized Rats. J. Bone Miner. Res. 1999, 14, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Kawaguchi, H.; Hanada, K.; Aoyama, I.; Hiyama, Y.; Nakamura, T.; Kuzutani, K.; Tamura, M.; Kurokawa, T.; Nakamura, K. Single local injection of recombinant fibroblast growth factor-2 stimulates healing of segmental bone defects in rabbits. J. Orthop. Res. 1998, 16, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, H.; Kurokawa, T.; Nakamura, K.; Matsushita, T.; Mamada, K.; Kawaguchi, H. Stimulation of bone formation by recombinant fibroblast growth factor-2 in callotasis bone lengthening of rabbits. Calcif. Tissue Int. 1999, 64, 542–546. [Google Scholar] [CrossRef]
- Nakamura, T.; Hara, Y.; Tagawa, M.; Tamura, M.; Yuge, T.; Fukuda, H.; Nigi, H. Recombinant Human Basic Fibroblast Growth Factor Accelerates Fracture Healing by Enhancing Callus Remodeling in Experimental Dog Tibial Fracture. J. Bone Miner. Res. 1998, 13, 942–949. [Google Scholar] [CrossRef]
- Kawaguchi, H.; Nakamura, K.; Tabata, Y.; Ikada, Y.; Aoyama, I.; Anzai, J.; Nakamura, T.; Hiyama, Y.; Tamura, M. Acceleration of fracture healing in nonhuman primates by fibroblast growth factor-2. J. Clin. Endocrinol. Metab. 2001, 86, 875–880. [Google Scholar] [CrossRef]
- Radomsky, M.L.; Aufdemorte, T.B.; Swain, L.D.; Fox, W.C.; Spiro, R.C.; Poser, J.W. Novel formulation of fibroblast growth factor-2 in a hyaluronan gel accelerates fracture healing in nonhuman primates. J. Orthop. Res. 1999, 17, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Kamo, K.; Miyakoshi, N.; Kasukawa, Y.; Sasaki, H.; Shimada, Y. Effects of single and cyclical local injections of basic fibroblast growth factor on cancellous bone defects in rabbits. J. Orthop. Sci. 2009, 14, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Hanada, K.; Tamura, M.; Shibanushi, T.; Nigi, H.; Tagawa, M.; Fukumoto, S.; Matsumoto, T. Stimulation of endosteal bone formation by systemic injections of recombinant basic fibroblast growth factor in rats. Endocrinology 1995, 136, 1276–1284. [Google Scholar] [CrossRef]
- Nagai, H.; Tsukuda, R.; Mayahara, H. Effects of basic fibroblast growth factor (bFGF) on bone formation in growing rats. Bone 1995, 16, 367–373. [Google Scholar] [CrossRef]
- Mayahara, H.; Ito, T.; Nagai, H.; Miyajima, H.; Tsukuda, R.; Taketomi, S.; Mizoguchi, J.; Kato, K. In VivoStimulation of Endosteal Bone Formation by Basic Fibroblast Growth Factor in Rats. Growth Factors 1993, 9, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Lane, N.E.; Yao, W.; Kumer, J.; Breuning, T.; Wronski, T.; Modin, G.; Kinney, J.H. Basic fibroblast growth factor partially restores trabecular bone architecture in osteopenic ovariectomized rats. Osteoporos. Int. 2003, 4, 374–382. [Google Scholar]
- Abbaspour, A.; Takata, S.; Sairyo, K.; Katoh, S.; Yukata, K.; Yasui, N. Continuous local infusion of fibroblast growth factor-2 enhances consolidation of the bone segment lengthened by distraction osteogenesis in rabbit experiment. Bone 2008, 42, 98–106. [Google Scholar] [CrossRef]
- Benington, L.; Rajan, G.; Locher, C.; Lim, L.Y. Fibroblast Growth Factor 2—A Review of Stabilisation Approaches for Clinical Applications. Pharmaceutics 2020, 12, 508. [Google Scholar] [CrossRef]
- Kuroda, Y.; Kawai, T.; Goto, K.; Matsuda, S. Clinical application of injectable growth factor for bone regeneration: A systematic review. Inflamm. Regen. 2019, 39, 20. [Google Scholar] [CrossRef]
- Arakawa, T.; Prestrelski, S.J.; Kenney, W.C.; Carpenter, J.F. Factors affecting short-term and long-term stabilities of proteins. Adv. Drug Deliv. Rev. 2001, 46, 307–326. [Google Scholar] [CrossRef]
- Tabata, Y.; Hijikata, S.; Ikada, Y. Enhanced vascularization and tissue granulation by basic fibroblast growth factor impregnated in gelatin hydrogels. J. Control. Release 1994, 31, 189–199. [Google Scholar] [CrossRef]
- Kawaguchi, H.; Jingushi, S.; Izumi, T.; Fukunaga, M.; Matsushita, T.; Nakamura, T.; Mizuno, K.; Nakamura, T.; Nakamura, K. Local application of recombinant human fibroblast growth factor-2 on bone repair: A dose–escalation prospective trial on patients with osteotomy. J. Orthop. Res. 2007, 25, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, H.; Oka, H.; Jingushi, S.; Izumi, T.; Fukunaga, M.; Sato, K.; Matsushita, T.; Nakamura, K. For the TESK Group A local application of recombinant human fibroblast growth factor 2 for tibial shaft fractures: A randomized, placebo-controlled trial. J. Bone Miner. Res. 2010, 25, 2735–2743. [Google Scholar] [CrossRef]
- Nakamura, S.; Kanatani, Y.; Kishimoto, S.; Nakamura, S.I.; Ohno, C.; Horio, T.; Masanori, F.; Hattori, H.; Tanaka, Y.; Kiyosawa, T.; et al. Controlled release of FGF-2 using fragmin/protamine microparticles and effect on neovascularization. J. Biomed. Mater. Res. A 2009, 91, 814–823. [Google Scholar] [CrossRef]
- Mammadov, R.; Mammadov, B.; Guler, M.O.; Tekinay, A.B. Growth Factor Binding on Heparin Mimetic Peptide Nanofibers. Biomacromolecules 2012, 13, 3311–3319. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kiick, K.L. Heparin-mimetic sulfated peptides with modulated affinities for heparin-binding peptides and growth factors. Peptides 2007, 28, 2125–2136. [Google Scholar] [CrossRef] [Green Version]
- Tardieu, M.; Gamby, C.; Avramoglou, T.; Jozefonvicz, J.; Barritault, D. Derivatized dextrans mimic heparin as stabilizers, potentiators, and protectors of acidic or basic FGF. J. Cell. Physiol. 1992, 150, 194–203. [Google Scholar] [CrossRef]
- Liekens, S.; Leali, D.; Neyts, J.; Esnouf, R.; Rusnati, M.; Dell’Era, P.; Maudgal, P.C.; De Clercq, E.; Presta, M. Modulation of Fibroblast Growth Factor-2 Receptor Binding, Signaling, and Mitogenic Activity by Heparin-Mimicking Polysulfonated Compounds. Mol. Pharmacol. 1999, 56, 204–213. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.H.; Paluck, S.J.; McGahran, A.J.; Maynard, H.D. Poly(vinyl sulfonate) Facilitates bFGF-Induced Cell Proliferation. Biomacromolecules 2015, 16, 2684–2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arunkumar, P.; Dougherty, J.A.; Weist, J.; Kumar, N.; Angelos, M.G.; Powell, H.M.; Khan, M. Sustained Release of Basic Fibroblast Growth Factor (bFGF) Encapsulated Polycaprolactone (PCL) Microspheres Promote Angiogenesis In Vivo. Nanomaterials 2019, 9, 1037. [Google Scholar] [CrossRef] [Green Version]
- Ali, Z.; Islam, A.; Sherrell, P.; Le-Moine, M.; Lolas, G.; Syrigos, K.; Rafat, M.; Jensen, L.D. Adjustable delivery of pro-angiogenic FGF-2 by alginate:collagen microspheres. Biol. Open 2018, 7, bio027060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murahashi, Y.; Yano, F.; Nakamoto, H.; Maenohara, Y.; Iba, K.; Yamashita, T.; Tanaka, S.; Ishihara, K.; Okamura, Y.; Moro, T.; et al. Multi-layered PLLA-nanosheets loaded with FGF-2 induce robust bone regeneration with controlled release in critical-sized mouse femoral defects. Acta Biomater. 2019, 85, 172–179. [Google Scholar] [CrossRef]
- Gromolak, S.; Krawczenko, A.; Antończyk, A.; Buczak, K.; Kiełbowicz, Z.; Klimczak, A. Biological Characteristics and Osteogenic Differentiation of Ovine Bone Marrow Derived Mesenchymal Stem Cells Stimulated with FGF-2 and BMP-2. Int. J. Mol. Sci. 2020, 21, 9726. [Google Scholar] [CrossRef]
- Akita, S.; Fukui, M.; Nakagawa, H.; Fujii, T.; Akino, K. Cranial bone defect healing is accelerated by mesenchymal stem cells induced by coadministration of bone morphogenetic protein-2 and basic fibroblast growth factor. Wound Repair Regen. 2004, 12, 252–259. [Google Scholar] [CrossRef]
- Hanada, K.; Dennis, J.E.; Caplan, A.I. Stimulatory Effects of Basic Fibroblast Growth Factor and Bone Morphogenetic Protein-2 on Osteogenic Differentiation of Rat Bone Marrow-Derived Mesenchymal Stem Cells. J. Bone Miner. Res. 1997, 12, 1606–1614. [Google Scholar] [CrossRef]
- Fujimura, K.; Bessho, K.; Okubo, Y.; Kusumoto, K.; Segami, N.; Iizuka, T. The effect of fibroblast growth factor-2 on the osteoinductive activity of recombinant human bone morphogenetic protein-2 in rat muscle. Arch. Oral Biol. 2002, 47, 577–584. [Google Scholar] [CrossRef]
- Takita, H.; Tsuruga, E.; Ono, I.; Kuboki, Y. Enhancement by bFGF of osteogenesis induced by rhBMP-2 in rats. Eur. J. Oral Sci. 1997, 105, 588–592. [Google Scholar] [CrossRef]
- Nakamura, Y.; Tensho, K.; Nakaya, H.; Nawata, M.; Okabe, T.; Wakitani, S. Low dose fibroblast growth factor-2 (FGF-2) enhances bone morphogenetic protein-2 (BMP-2)-induced ectopic bone formation in mice. Bone 2005, 36, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, A.D.; Olsen, B.R. Bone development. Bone 2015, 80, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Breeland, G.; Menezes, R.G. Embryology, Bone Ossification; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2019. [Google Scholar]
- Deckers, M.M.; Van Bezooijen, R.L.; Van Der Horst, G.; Hoogendam, J.; van Der Bent, C.; Papapoulos, S.E.; Löwik, C.W. Bone morphogenetic proteins stimulate angiogenesis through osteoblast-derived vascular endothelial growth factor A. Endocrinology 2002, 143, 1545–1553. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Cao, X. BMP signaling in skeletal development. Biochem. Biophys. Res. Commun. 2005, 328, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Usas, A.; Olshanski, A.; Ho, A.M.; Gearhart, B.; Cooper, G.M.; Huard, J. VEGF Improves, Whereas sFlt1 Inhibits, BMP2-Induced Bone Formation and Bone Healing Through Modulation of Angiogenesis. J. Bone Miner. Res. 2005, 20, 2017–2027. [Google Scholar] [CrossRef]
- Valdimarsdottir, G.; Goumans, M.-J.; Rosendahl, A.; Brugman, M.; Itoh, S.; Lebrin, F.; Sideras, P.; Dijke, P.T. Stimulation of Id1 Expression by Bone Morphogenetic Protein Is Sufficient and Necessary for Bone Morphogenetic Protein–Induced Activation of Endothelial Cells. Circulation 2002, 106, 2263–2270. [Google Scholar] [CrossRef] [PubMed]
- Salazar, V.S.; Gamer, L.W.; Rosen, V. BMP signalling in skeletal development, disease and repair. Nat. Rev. Endocrinol. 2016, 12, 203–221. [Google Scholar] [CrossRef]
- Carano, R.A.; Filvaroff, E.H. Angiogenesis and bone repair. Drug Discov. Today 2003, 8, 980–989. [Google Scholar] [CrossRef]
- Chen, G.; Deng, C.; Li, Y.P. TGF-beta and BMP signaling in osteoblast differentiation and bone formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef] [Green Version]
- Yang, E.Y.; Moses, H.L. Transforming growth factor beta 1-induced changes in cell migration, proliferation, and angiogenesis in the chicken chorioallantoic membrane. J. Cell Biol. 1990, 111, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Sabbieti, M.G.; Marchetti, L.; Gabrielli, M.G.; Menghi, M.; Materazzi, S.; Menghi, G.; Raisz, L.G.; Hurley, M.M. Prostaglandins differently regulate FGF-2 and FGF receptor expression and induce nuclear translocation in osteoblasts via MAPK kinase. Cell Tissue Res. 2004, 319, 267–278. [Google Scholar] [CrossRef]
- Sobue, T.; Gravely, T.; Hand, A.; Min, Y.K.; Pilbeam, C.; Raisz, L.G.; Zhang, X.; Larocca, D.; Florkiewicz, R.; Hurley, M.M. Regulation of fibroblast growth factor 2 and fibroblast growth factor receptors by transforming growth factor beta in human osteoblastic MG-63 cells. J. Bone Miner. Res. 2002, 17, 502–512. [Google Scholar] [CrossRef]
- Fiedler, J.; Röderer, G.; Günther, K.-P.; Brenner, R.E. BMP-2, BMP-4, and PDGF-bb stimulate chemotactic migration of primary human mesenchymal progenitor cells. J. Cell. Biochem. 2002, 87, 305–312. [Google Scholar] [CrossRef]
- Edwards, J.R.; Nyman, J.S.; Lwin, S.T.; Moore, M.M.; Esparza, J.; O’Quinn, E.C.; Hart, A.J.; Biswas, S.; Patil, C.A.; Lonning, S.; et al. Inhibition of TGF-beta signaling by 1D11 antibody treatment increases bone mass and quality in vivo. J. Bone Miner. Res. 2010, 25, 2419–2426. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollinger, J.O.; Hart, C.E.; Hirsch, S.N.; Lynch, S.; Friedlaender, G.E. Recombinant Human Platelet-Derived Growth Factor: Biology and Clinical Applications. J. Bone Jt. Surg. Am. Vol. 2008, 90, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.H.; Shipley, J.; Bergers, G.; Berger, J.E.; Helms, J.A.; Hanahan, D.; Shapiro, S.D.; Senior, R.M.; Werb, Z. MMP-9/Gelatinase B Is a Key Regulator of Growth Plate Angiogenesis and Apoptosis of Hypertrophic Chondrocytes. Cell 1998, 93, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Stickens, D.; Behonick, D.J.; Ortega, N.; Heyer, B.; Hartenstein, B.; Yu, Y.; Fosang, A.J.; Schorpp-Kistner, M.; Angel, P.; Werb, Z. Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development 2004, 131, 5883–5895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramasamy, S.K.; Kusumbe, A.P.; Wang, L.; Adams, R.H. Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature 2014, 507, 376–380. [Google Scholar] [CrossRef] [Green Version]




| Study | Application | Dosage and Timing | Administration and/or Scaffolding | Results |
|---|---|---|---|---|
| in vitro studies | ||||
| Gromolak et al. 2020 | Ovine bone marrow MSCs | FGF-2 (20 ng/mL) alone or in combination with BMP-2 (100 ng/mL) | In culture medium | Osteogenic differentiation induced by BMP-2 is amplified with FGF-2 supplementation. FGF-2 alone boosted proliferation of smaller cells, but without osteoblast-like structures in culture and decreased expression of osteogenic genes. |
| Li et al. 2014 | Murine calvarial osteoblasts | FGF-2 (1, 10, 20, 60 ng/mL) | In culture medium | Doses ≤10 ng/mL yielded higher cell proliferation Doses >10 ng/mL decreased proliferation Increased mineralization at all doses |
| Sukarawan et al. 2014 | Stem cells from human exfoliated deciduous teeth (SHEDs) | FGF-2 (10 ng/mL) | In culture medium | FGF-2 maintains cell stemness |
| Ou et al. 2010 | Murine calvarial and femur osteoprogenitor cells Human cancellous bone osteoprogenitor cells from young and old patients | rhFGF-2 (0.0016, 0.016, 0.16, or 1.6 ng/mL) 4, 24, 48, and 72 h | In culture medium | Accelerated proliferation at all doses FGF-2 induced proliferation diminished with age |
| Varkey et al. 2006 | Rat bone marrow cells | FGF-2 (2, 10, 50 ng/mL) and BMP-2 (50, 150, 500 ng/mL) over 3 weeks | In culture medium | Accelerated mineralization at 10 ng/mL but reduced at 50 ng/mL of FGF-2 Synergistic role of FGF-2 and BMP-2 in old rat cells |
| Animal models | ||||
| Novais et al. 2019 | Critical calvarial bone defects in nude mice | FGF (10 ng/mL) over 72 h | SHEDs in dense collagen matrices in osteogenic culture medium | Enhanced bone formation in calvarial critical size defect |
| Wang et al. 2019 | Mandibular defects in non-human primates | FGF-2 (0.25 μg/μL) | Calcium phosphate cement for BMP-2 carrier PGA gel for FGF-2 carrier | Promotion of periodontal regeneration |
| Anzai et al. 2016 | 2-wall periodontal defects in Beagle dogs | FGF-2 (3 mg/mL) vs. vehicle | Cellulose solution Direct injection in the defect | FGF-2 promoted regeneration in alveolar bone, cementum and periodontal ligament. |
| Charles et al. 2015 | Calvarial bone defects in old mice | FGF-2 (5 ng) and BMP-2 (2 μg) | Collagen hydroxyapatite discs | Enhanced bone filling in the central bone defect area when BMP-2 was supplemented with FGF-2 |
| Akita et al. 2004 | Calvarial defects in nude mice | FGF-2 (2.5 ng/mL) and BMP-2 (50 ng/mL) vs. FGF-2 alone, BMP-2 alone or vehicle | Transfected human MSCs in gelatin sponge carrier | Combination of FGF-2 and BMP-2 showed the most advanced bone formation within the defects |
| Clinical studies | ||||
| Cochran et al. 2016 | Patients with periodontal intrabony defects | rhFGF-2 (0.1%, 0.3%, 0.4%) or no application | β-TCP | Increased clinical attachment gain and bone fill at concentrations of 0.4% and 0.3% |
| De Santana et al. 2015 | Patients with periodontal intrabony defects | rhFGF-2 (4 mg/mL) vs. no application | Sodium hyaluronate gel | Enhanced clinical parameters of wound healing compared to negative control |
| Kitamura et al. 2001 | Patients with periodontal intrabony defects | rhFGF-2 (0.2%, 0.3%, 0.4%) or placebo | 3% hydroxypropyl cellulose gel | At 36 weeks, all defects showed bone fill except placebo 0.3% dose had the best radiographic outcomes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novais, A.; Chatzopoulou, E.; Chaussain, C.; Gorin, C. The Potential of FGF-2 in Craniofacial Bone Tissue Engineering: A Review. Cells 2021, 10, 932. https://doi.org/10.3390/cells10040932
Novais A, Chatzopoulou E, Chaussain C, Gorin C. The Potential of FGF-2 in Craniofacial Bone Tissue Engineering: A Review. Cells. 2021; 10(4):932. https://doi.org/10.3390/cells10040932
Chicago/Turabian StyleNovais, Anita, Eirini Chatzopoulou, Catherine Chaussain, and Caroline Gorin. 2021. "The Potential of FGF-2 in Craniofacial Bone Tissue Engineering: A Review" Cells 10, no. 4: 932. https://doi.org/10.3390/cells10040932