
Hypoxia-Driven Effects in Cancer: Characterization, Mechanisms, and Therapeutic Implications


Abstract
:1. Introduction
2. Effects of Hypoxia on Cancer Molecular and Cellular Characteristics
2.1. Mutagenesis and Impaired DNA Repair
2.2. Metastasis
2.3. The CSC Phenotype
2.4. Resistance to Radio- and Chemo-Therapy
3. Key Drivers of Hypoxia-Mediated Resistance and Metastasis
3.1. Hypoxia-Inducible Factors
3.2. Oxoglutarate-Dependent Dioxygenases
3.3. The Unfolded Protein Response Pathway
3.4. Other Emerging Pathways, from Exosomes to Noncoding RNAs
4. From Mechanisms to Therapeutics
4.1. Drugs That Target the Hypoxic Tumor Microenvironment
4.2. Therapies Targeting Hypoxia-Mediated Pathways
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hammarlund, E.U.; Flashman, E.; Mohlin, S.; Licausi, F. Oxygen-sensing mechanisms across eukaryotic kingdoms and their roles in complex multicellularity. Science 2020, 370, 6515. [Google Scholar] [CrossRef]
- Zhang, Q.; Yan, Q.; Yang, H.; Wei, W. Oxygen sensing and adaptability won the 2019 Nobel Prize in Physiology or medicine. Genes Dis. 2019, 6, 328–332. [Google Scholar] [CrossRef]
- Henley, S.J.; Ward, E.M.; Scott, S.; Ma, J.; Anderson, R.N.; Firth, A.U.; Thomas, C.C.; Islami, F.; Weir, H.K.; Lewis, D.R. Annual Report to the Nation on the Status of Cancer, part I: National cancer statistics. Cancer 2020, 126, 2225–2249. [Google Scholar] [CrossRef]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaupel, P.; Höckel, M.; Mayer, A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid. Redox Signal. 2007, 9, 1221–1236. [Google Scholar] [CrossRef]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Butturini, E.; Carcereri de Prati, A.; Boriero, D.; Mariotto, S. Tumor dormancy and interplay with hypoxic tumor microenvironment. Int. J. Mol. Sci. 2019, 20, 4305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Schödel, J.; Ratcliffe, P.J. Mechanisms of hypoxia signalling: New implications for nephrology. Nat. Rev. Nephrol. 2019, 15, 641–659. [Google Scholar] [CrossRef]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef]
- Cockman, M.E.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.W.; Clifford, S.C.; Maher, E.R.; Pugh, C.W.; Ratcliffe, P.J.; Maxwell, P.H. Hypoxia inducible factor-α binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2000, 275, 25733–25741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losman, J.A.; Kaelin, W.G., Jr. What a difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013, 27, 836–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losman, J.-A.; Koivunen, P.; Kaelin, W.G. 2-Oxoglutarate-dependent dioxygenases in cancer. Nat. Rev. Cancer 2020, 20, 710–726. [Google Scholar] [CrossRef]
- Bayer, C.; Vaupel, P. Acute versus chronic hypoxia in tumors. Strahlenther. Onkol. 2012, 188, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E. Radiation-modifying effect of oxygen in synchronized cells pre-treated with acute or prolonged hypoxia. Int. J. Radiat. Biol. 1996, 70, 319–326. [Google Scholar] [CrossRef]
- Holmquist-Mengelbier, L.; Fredlund, E.; Löfstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, Å.; Gradin, K. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell 2006, 10, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Godet, I.; Shin, Y.J.; Ju, J.A.; Ye, I.C.; Wang, G.; Gilkes, D.M. Fate-mapping post-hypoxic tumor cells reveals a ROS-resistant phenotype that promotes metastasis. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Alqawi, O.; Wang, H.P.; Espiritu, M.; Singh, G. Chronic hypoxia promotes an aggressive phenotype in rat prostate cancer cells. Free Radic. Res. 2007, 41, 788–797. [Google Scholar] [CrossRef]
- Pires, I.M.; Bencokova, Z.; Milani, M.; Folkes, L.K.; Li, J.-L.; Stratford, M.R.; Harris, A.L.; Hammond, E.M. Effects of acute versus chronic hypoxia on DNA damage responses and genomic instability. Cancer Res. 2010, 70, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Tafani, M.; Pucci, B.; Russo, A.; Schito, L.; Pellegrini, L.; Perrone, G.A.; Villanova, L.; Salvatori, L.; Ravenna, L.; Petrangeli, E. Modulators of HIF1α and NFkB in cancer treatment: Is it a rational approach for controlling malignant progression? Front. Pharmacol. 2013, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, T.Y.; Rockwell, S.; Glazer, P.M. Genetic instability induced by the tumor microenvironment. Cancer Res. 1996, 56, 5754–5757. [Google Scholar] [PubMed]
- Papp-Szabó, E.; Josephy, P.D.; Coomber, B.L. Microenvironmental influences on mutagenesis in mammary epithelial cells. Int. J. Cancer 2005, 116, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Narayanan, L.; Rockwell, S.; Glazer, P.M. Diminished DNA repair and elevated mutagenesis in mammalian cells exposed to hypoxia and low pH. Cancer Res. 2000, 60, 4372–4376. [Google Scholar]
- Coquelle, A.; Rozier, L.; Dutrillaux, B.; Debatisse, M. Induction of multiple double-strand breaks within an hsr by meganucleaseI-SceI expression or fragile site activation leads to formation of double minutes and other chromosomal rearrangements. Oncogene 2002, 21, 7671–7679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cam, H.; Easton, J.B.; High, A.; Houghton, P.J. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1α. Mol. Cell 2010, 40, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Freiberg, R.A.; Hammond, E.M.; Dorie, M.J.; Welford, S.M.; Giaccia, A.J. DNA damage during reoxygenation elicits a Chk2-dependent checkpoint response. Mol. Cell Biol. 2006, 26, 1598–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, D.A.; Tan, T.-T.; Rabson, A.B.; Anderson, D.; Degenhardt, K.; White, E. Hypoxia and defective apoptosis drive genomic instability and tumorigenesis. Genes Dev. 2004, 18, 2095–2107. [Google Scholar] [CrossRef] [Green Version]
- Aparicio, T.; Baer, R.; Gautier, J. DNA double-strand break repair pathway choice and cancer. DNA Repair 2014, 19, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Dynan, W.S.; Yoo, S. Interaction of Ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Res. 1998, 26, 1551–1559. [Google Scholar] [CrossRef] [Green Version]
- Mihaylova, V.T.; Bindra, R.S.; Yuan, J.; Campisi, D.; Narayanan, L.; Jensen, R.; Giordano, F.; Johnson, R.S.; Rockwell, S.; Glazer, P.M. Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol. Cell. Biol. 2003, 23, 3265–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindra, R.S.; Gibson, S.L.; Meng, A.; Westermark, U.; Jasin, M.; Pierce, A.J.; Bristow, R.G.; Classon, M.K.; Glazer, P.M. Hypoxia-induced down-regulation of BRCA1 expression by E2Fs. Cancer Res. 2005, 65, 11597–11604. [Google Scholar] [CrossRef] [Green Version]
- Bindra, R.S.; Glazer, P.M. Co-repression of mismatch repair gene expression by hypoxia in cancer cells: Role of the Myc/Max network. Cancer Lett. 2007, 252, 93–103. [Google Scholar] [CrossRef]
- Bindra, R.; Glazer, P. Repression of RAD51 gene expression by E2F4/p130 complexes in hypoxia. Oncogene 2007, 26, 2048–2057. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Zhang, Y.; Chen, S.; Weng, X.; Rao, Y.; Fang, H. Mechanism and current progress of Poly ADP-ribose polymerase (PARP) inhibitors in the treatment of ovarian cancer. Biomed. Pharmacother. 2020, 123, 109661. [Google Scholar] [CrossRef]
- Kaplan, A.R.; Glazer, P.M. Impact of hypoxia on DNA repair and genome integrity. Mutagenesis 2020, 35, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Young, S.; Marshall, R.; Hill, R. Hypoxia induces DNA overreplication and enhances metastatic potential of murine tumor cells. Proc. Natl. Acad. Sci. USA 1988, 85, 9533–9537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Z.-F.; Zhao, T.-T.; Wang, Z.-N.; Xu, Y.-Y.; Mao, X.-Y.; Wu, J.-H.; Liu, X.-Y.; Xu, H.; You, Y.; Xu, H.-M. Influence of different hypoxia models on metastatic potential of SGC-7901 gastric cancer cells. Tumor Biol. 2014, 35, 6801–6808. [Google Scholar] [CrossRef]
- Cairns, R.A.; Hill, R.P. Acute hypoxia enhances spontaneous lymph node metastasis in an orthotopic murine model of human cervical carcinoma. Cancer Res. 2004, 64, 2054–2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rofstad, E.K.; Gaustad, J.V.; Egeland, T.A.; Mathiesen, B.; Galappathi, K. Tumors exposed to acute cyclic hypoxic stress show enhanced angiogenesis, perfusion and metastatic dissemination. Int. J. Cancer 2010, 127, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.C.; Lebedev, A.; Aten, E.; Madsen, K.; Marciano, L.; Kolb, H.C. The clinical importance of assessing tumor hypoxia: Relationship of tumor hypoxia to prognosis and therapeutic opportunities. Antioxid. Redox Signal. 2014, 21, 1516–1554. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.A.; Giaccia, A.J. Hypoxia, gene expression, and metastasis. Cancer Metastasis Rev. 2007, 26, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Giaccia, A.J. Lysyl oxidase mediates hypoxic control of metastasis. Cancer Res. 2006, 66, 10238–10241. [Google Scholar] [CrossRef] [Green Version]
- Muz, B.; De La Puente, P.; Azab, F.; Ghobrial, I.M.; Azab, A.K. Hypoxia promotes dissemination and colonization in new bone marrow niches in Waldenström macroglobulinemia. Mol. Cancer Res. 2015, 13, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Fluegen, G.; Avivar-Valderas, A.; Wang, Y.; Padgen, M.R.; Williams, J.K.; Nobre, A.R.; Calvo, V.; Cheung, J.F.; Bravo-Cordero, J.J.; Entenberg, D. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat. Cell Biol. 2017, 19, 120–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.; Sceneay, J.; Gödde, N.; Kinwel, T.; Ham, S.; Thompson, E.W.; Humbert, P.O.; Möller, A. Intermittent hypoxia induces a metastatic phenotype in breast cancer. Oncogene 2018, 37, 4214–4225. [Google Scholar] [CrossRef]
- Muinao, T.; Boruah, H.P.D.; Pal, M. Diagnostic and Prognostic Biomarkers in ovarian cancer and the potential roles of cancer stem cells–An updated review. Exp. Cell Res. 2018, 362, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Giraud, J.; Chambonnier, L.; Dubus, P.; Wittkop, L.; Belleannée, G.; Collet, D.; Soubeyran, I.; Evrard, S.; Rousseau, B. Characterization of biomarkers of tumorigenic and chemoresistant cancer stem cells in human gastric carcinoma. Clin. Cancer Res. 2017, 23, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Corro, C.; Moch, H. Biomarker discovery for renal cancer stem cells. J. Pathol. Clin. Res. 2018, 4, 3–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Xu, J.; Tang, L.; Guan, X. Breast cancer stem cell: The roles and therapeutic implications. Cell. Mol. Life Sci. 2017, 74, 951–966. [Google Scholar] [CrossRef] [PubMed]
- Smit, J.K.; Faber, H.; Niemantsverdriet, M.; Baanstra, M.; Bussink, J.; Hollema, H.; van Os, R.P.; Plukker, J.T.M.; Coppes, R.P. Prediction of response to radiotherapy in the treatment of esophageal cancer using stem cell markers. Radiother. Oncol. 2013, 107, 434–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- Zomer, A.; Ellenbroek, S.I.J.; Ritsma, L.; Beerling, E.; Vrisekoop, N.; Van Rheenen, J. Brief report: Intravital imaging of cancer stem cell plasticity in mammary tumors. Stem Cells 2013, 31, 602–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jögi, A.; Øra, I.; Nilsson, H.; Lindeheim, Å.; Makino, Y.; Poellinger, L.; Axelson, H.; Påhlman, S. Hypoxia alters gene expression in human neuroblastoma cells toward an immature and neural crest-like phenotype. Proc. Natl. Acad. Sci. USA 2002, 99, 7021–7026. [Google Scholar] [CrossRef] [Green Version]
- Helczynska, K.; Kronblad, Å.; Jögi, A.; Nilsson, E.; Beckman, S.; Landberg, G.; Påhlman, S. Hypoxia promotes a dedifferentiated phenotype in ductal breast carcinoma in situ. Cancer Res. 2003, 63, 1441–1444. [Google Scholar]
- Chang, S.; Park, B.; Choi, K.; Moon, Y.; Lee, H.Y.; Park, H. Hypoxic reprograming of H3K27me3 and H3K4me3 at the INK 4A locus. FEBS Lett. 2016, 590, 3407–3415. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, J.; Zhang, Z.; Zhou, W.; Wang, A.J.; Heddleston, J.M.; Pinna, C.M.; Hubaud, A.; Stadler, B.; Choi, M.; Bar, M. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011, 71, 4640–4652. [Google Scholar] [CrossRef] [Green Version]
- Eastham, A.M.; Spencer, H.; Soncin, F.; Ritson, S.; Merry, C.L.; Stern, P.L.; Ward, C.M. Epithelial-mesenchymal transition events during human embryonic stem cell differentiation. Cancer Res. 2007, 67, 11254–11262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, S.D.; Morand, G.B.; Alobaid, F.A.; Hier, M.P.; Mlynarek, A.M.; Alaoui-Jamali, M.A.; Kowalski, L.P. Epithelial-mesenchymal transition (EMT) markers have prognostic impact in multiple primary oral squamous cell carcinoma. Clin. Exp. Metastasis 2015, 32, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Azab, A.K.; Hu, J.; Quang, P.; Azab, F.; Pitsillides, C.; Awwad, R.; Thompson, B.; Maiso, P.; Sun, J.D.; Hart, C.P. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood 2012, 119, 5782–5794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Renaud, S.; Guenot, D.; Falcoz, P.-E.; Massard, G.; Beau-Faller, M. Role of hypoxia in epithelial-to-mesenchymal transition (EMT) in non-small cell lung cancer (NSCLC). Eur. Respir. J. 2014, 44, 814. [Google Scholar]
- Cheng, Z.-X.; Sun, B.; Wang, S.-J.; Gao, Y.; Zhang, Y.-M.; Zhou, H.-X.; Jia, G.; Wang, Y.-W.; Kong, R.; Pan, S.-H. Nuclear factor-κb–dependent epithelial to mesenchymal transition induced by HIF-1α activation in pancreatic cancer cells under hypoxic conditions. PLoS ONE 2011, 6, e23752. [Google Scholar] [CrossRef]
- Cheng, Z.-X.; Wang, D.-W.; Liu, T.; Liu, W.-X.; Xia, W.-B.; Xu, J.; Zhang, Y.-H.; Qu, Y.-K.; Guo, L.-Q.; Ding, L. Effects of the HIF-1α and NF-κB loop on epithelial-mesenchymal transition and chemoresistance induced by hypoxia in pancreatic cancer cells. Oncol. Rep. 2014, 31, 1891–1898. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, Y.; Li, L.; Kong, R.; Pan, S.; Ji, L.; Liu, H.; Chen, H.; Sun, B. Hyperoside induces apoptosis and inhibits growth in pancreatic cancer via Bcl-2 family and NF-κB signaling pathway both in vitro and in vivo. Tumor Biol. 2016, 37, 7345–7355. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.N.; Du, W.; Brekken, R.A. Behind the Wheel of Epithelial Plasticity in KRAS-Driven Cancers. Front. Oncol. 2019, 9, 1049. [Google Scholar] [CrossRef] [Green Version]
- Buck, E.; Eyzaguirre, A.; Barr, S.; Thompson, S.; Sennello, R.; Young, D.; Iwata, K.K.; Gibson, N.W.; Cagnoni, P.; Haley, J.D. Loss of homotypic cell adhesion by epithelial-mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition. Mol. Cancer Ther. 2007, 6, 532–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrader, M.; Pino, M.S.; Brown, G.; Black, P.; Adam, L.; Bar-Eli, M.; Dinney, C.P.; McConkey, D.J. Molecular correlates of gefitinib responsiveness in human bladder cancer cells. Mol. Cancer Ther. 2007, 6, 277–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frederiksen, L.J.; Siemens, D.R.; Heaton, J.P.; Maxwell, L.R.; Adams, M.A.; Graham, C.H. Hypoxia induced resistance to doxorubicin in prostate cancer cells is inhibited by low concentrations of glyceryl trinitrate. J. Urol. 2003, 170, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Witta, S.E.; Gemmill, R.M.; Hirsch, F.R.; Coldren, C.D.; Hedman, K.; Ravdel, L.; Helfrich, B.; Dziadziuszko, R.; Chan, D.C.; Sugita, M. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006, 66, 944–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurrey, N.K.; Jalgaonkar, S.P.; Joglekar, A.V.; Ghanate, A.D.; Chaskar, P.D.; Doiphode, R.Y.; Bapat, S.A. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 2009, 27, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Hsu, W.-H.; Han, J.; Xia, Y.; DePinho, R.A. Cancer Stemness Meets Immunity: From Mechanism to Therapy. Cell Rep. 2021, 34, 108597. [Google Scholar] [CrossRef]
- Miranda, A.; Hamilton, P.T.; Zhang, A.W.; Pattnaik, S.; Becht, E.; Mezheyeuski, A.; Bruun, J.; Micke, P.; de Reynies, A.; Nelson, B.H. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9020–9029. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.-C.; Chao, Y.-J.; Hsieh, M.-H.; Tung, H.-L.; Wang, H.-C.; Shan, Y.-S. Low CD8+ T cell infiltration and high PD-L1 expression are associated with level of CD44+/CD133+ cancer stem cells and predict an unfavorable prognosis in pancreatic cancer. Cancers 2019, 11, 541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thews, O.; Riemann, A.; Nowak, M.; Gekle, M. Impact of hypoxia-related tumor acidosis on cytotoxicity of different chemotherapeutic drugs in vitro and in vivo. In Oxygen Transport to Tissue XXXVI; Springer: Berlin/Heidelberg, Germany, 2014; pp. 51–58. [Google Scholar]
- Graham, K.; Unger, E. Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int. J. Nanomed. 2018, 13, 6049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roizin-towle, L.; Hall, E.J. The effect of bleomycin on aerated and hypoxic cells in vitro, in combination with irradiation. Int. J. Radiat. Oncol. Biol. Phys. 1979, 5, 1491–1494. [Google Scholar]
- Conley, S.J.; Gheordunescu, E.; Kakarala, P.; Newman, B.; Korkaya, H.; Heath, A.N.; Clouthier, S.G.; Wicha, M.S. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl. Acad. Sci. USA 2012, 109, 2784–2789. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Zhou, L.; Huang, J.; Xiao, X. Effect of multidrug resistance 1/P-glycoprotein on the hypoxia-induced multidrug resistance of human laryngeal cancer cells. Oncol. Lett. 2016, 12, 1569–1574. [Google Scholar] [CrossRef] [Green Version]
- Flamant, L.; Roegiers, E.; Pierre, M.; Hayez, A.; Sterpin, C.; De Backer, O.; Arnould, T.; Poumay, Y.; Michiels, C. TMEM45A is essential for hypoxia-induced chemoresistance in breast and liver cancer cells. BMC Cancer 2012, 12, 391. [Google Scholar] [CrossRef] [Green Version]
- Mottram, J. A factor of importance in the radio sensitivity of tumours. Br. J. Radiol. 1936, 9, 606–614. [Google Scholar] [CrossRef]
- Evans, S.M.; Koch, C.J. Prognostic significance of tumor oxygenation in humans. Cancer Lett. 2003, 195, 1–16. [Google Scholar] [CrossRef]
- Chaplin, D.; Durand, R.; Olive, P. Acute hypoxia in tumors: Implications for modifiers of radiation effects. Int. J. Radiat. Oncol. Biol. Phys. 1986, 12, 1279–1282. [Google Scholar] [CrossRef]
- Hsieh, C.-H.; Lee, C.-H.; Liang, J.-A.; Yu, C.-Y.; Shyu, W.-C. Cycling hypoxia increases U87 glioma cell radioresistance via ROS induced higher and long-term HIF-1 signal transduction activity. Oncol. Rep. 2010, 24, 1629–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, Y.; Yashiro, M.; Fuyuhiro, Y.; Kashiwagi, S.; Matsuoka, J.; Hirakawa, T.; Noda, S.; Aomatsu, N.; Hasegawa, T.; Matsuzaki, T. Effects of acute and chronic hypoxia on the radiosensitivity of gastric and esophageal cancer cells. Anticancer. Res. 2011, 31, 3369–3375. [Google Scholar] [PubMed]
- Zheng, Y.; Liu, L.; Chen, C.; Ming, P.; Huang, Q.; Li, C.; Cao, D.; Xu, X.; Ge, W. The extracellular vesicles secreted by lung cancer cells in radiation therapy promote endothelial cell angiogenesis by transferring miR-23a. PeerJ 2017, 5, e3627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haider, S.; McIntyre, A.; van Stiphout, R.G.; Winchester, L.M.; Wigfield, S.; Harris, A.L.; Buffa, F.M. Genomic alterations underlie a pan-cancer metabolic shift associated with tumour hypoxia. Genome Biol. 2016, 17, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntyre, A.; Harris, A.L. Metabolic and hypoxic adaptation to anti-angiogenic therapy: A target for induced essentiality. EMBO Mol. Med. 2015, 7, 368–379. [Google Scholar] [CrossRef]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.; Yim, S.; Park, H. The cancer driver genes IDH1/2, JARID1C/KDM5C, and UTX/KDM6A: Crosstalk between histone demethylation and hypoxic reprogramming in cancer metabolism. Exp. Mol. Med. 2019, 51, 66. [Google Scholar] [CrossRef] [Green Version]
- Pilon-Thomas, S.; Kodumudi, K.N.; El-Kenawi, A.E.; Russell, S.; Weber, A.M.; Luddy, K.; Damaghi, M.; Wojtkowiak, J.W.; Mulé, J.J.; Ibrahim-Hashim, A. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. 2016, 76, 1381–1390. [Google Scholar] [CrossRef] [Green Version]
- Hatfield, S.M.; Sitkovsky, M. A2A adenosine receptor antagonists to weaken the hypoxia-HIF-1α driven immunosuppression and improve immunotherapies of cancer. Curr. Opin. Pharmacol. 2016, 29, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Kathagen, A.; Schulte, A.; Balcke, G.; Phillips, H.S.; Martens, T.; Matschke, J.; Günther, H.S.; Soriano, R.; Modrusan, Z.; Sandmann, T. Hypoxia and oxygenation induce a metabolic switch between pentose phosphate pathway and glycolysis in glioma stem-like cells. Acta Neuropathol. 2013, 126, 763–780. [Google Scholar] [CrossRef]
- Kathagen-Buhmann, A.; Schulte, A.; Weller, J.; Holz, M.; Herold-Mende, C.; Glass, R.; Lamszus, K. Glycolysis and the pentose phosphate pathway are differentially associated with the dichotomous regulation of glioblastoma cell migration versus proliferation. Neuro Oncol. 2016, 18, 1219–1229. [Google Scholar] [CrossRef] [Green Version]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative phosphorylation as an emerging target in cancer therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Demere, Z.; Nair, K.; Ali, A.; Ferraro, G.B.; Natoli, T.; Deik, A.; Petronio, L.; Tang, A.A.; Zhu, C. A metastasis map of human cancer cell lines. Nature 2020, 588, 331–336. [Google Scholar] [CrossRef]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A review in the theme: Cellular responses to hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef] [Green Version]
- Briggs, K.J.; Koivunen, P.; Cao, S.; Backus, K.M.; Olenchock, B.A.; Patel, H.; Zhang, Q.; Signoretti, S.; Gerfen, G.J.; Richardson, A.L. Paracrine induction of HIF by glutamate in breast cancer: EglN1 senses cysteine. Cell 2016, 166, 126–139. [Google Scholar] [CrossRef] [Green Version]
- Jiao, M.; Nan, K.-J. Activation of PI3 kinase/Akt/HIF-1α pathway contributes to hypoxia-induced epithelial-mesenchymal transition and chemoresistance in hepatocellular carcinoma. Int. J. Oncol. 2012, 40, 461–468. [Google Scholar]
- Jögi, A.; Ehinger, A.; Hartman, L.; Alkner, S. Expression of HIF-1α is related to a poor prognosis and tamoxifen resistance in contralateral breast cancer. PLoS ONE 2019, 14, e0226150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaus, A.; Fathi, O.; Tatjana, T.-W.; Bruno, N.; Oskar, K. Expression of hypoxia-associated protein HIF-1α in follicular thyroid cancer is associated with distant metastasis. Pathol. Oncol. Res. 2018, 24, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Li, H.; Liu, L.; Cheng, J. Expression levels of PTEN, HIF-1alpha, and VEGF as prognostic factors in ovarian cancer. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2596–2603. [Google Scholar] [PubMed]
- Lin, C.-S.; Liu, T.-C.; Lee, M.-T.; Yang, S.-F.; Tsao, T.C.-Y. Independent prognostic value of hypoxia-inducible factor 1-alpha expression in small cell lung cancer. Int. J. Med. Sci. 2017, 14, 785. [Google Scholar] [CrossRef] [Green Version]
- Bottini, A.; Harris, A.L.; Fox, S.B. Hypoxia-Inducible Factor-1AExpression Predicts a Poor Response to Primary ChemoendocrineTherapy and Disease-Free Survival in Primary Human Breast Cancer. Clin. Cancer Res. 2006, 2006, 15. [Google Scholar]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 47–71. [Google Scholar] [CrossRef]
- Schödel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood J. Am. Soc. Hematol. 2011, 117, e207–e217. [Google Scholar] [CrossRef] [Green Version]
- Kelly, B.D.; Hackett, S.F.; Hirota, K.; Oshima, Y.; Cai, Z.; Berg-Dixon, S.; Rowan, A.; Yan, Z.; Campochiaro, P.A.; Semenza, G.L. Cell type–specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ. Res. 2003, 93, 1074–1081. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.C.-L.; Gilkes, D.M.; Zhang, H.; Chen, J.; Wei, H.; Chaturvedi, P.; Fraley, S.I.; Wong, C.-M.; Khoo, U.-S.; Ng, I.O.-L. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc. Natl. Acad. Sci. USA 2011, 108, 16369–16374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V.; Kim, J.-W.; Gao, P.; Yustein, J. The interplay between MYC and HIF in cancer. Nat. Rev. Cancer 2008, 8, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.E.; Simon, M.C. From stem cells to cancer stem cells: HIF takes the stage. Curr. Opin. Cell Biol. 2012, 24, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.-H.; Wu, M.-Z.; Chiou, S.-H.; Chen, P.-M.; Chang, S.-Y.; Liu, C.-J.; Teng, S.-C.; Wu, K.-J. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Chafe, S.C.; McDonald, P.C.; Saberi, S.; Nemirovsky, O.; Venkateswaran, G.; Burugu, S.; Gao, D.; Delaidelli, A.; Kyle, A.H.; Baker, J.H. Targeting hypoxia-induced carbonic anhydrase IX enhances immune-checkpoint blockade locally and systemically. Cancer Immunol. Res. 2019, 7, 1064–1078. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Bai, X.; Chen, W.; Ma, T.; Hu, Q.; Liang, C.; Xie, S.; Chen, C.; Hu, L.; Xu, S. Wnt/β-catenin signaling enhances hypoxia-induced epithelial–mesenchymal transition in hepatocellular carcinoma via crosstalk with hif-1α signaling. Carcinogenesis 2013, 34, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhöfer, N.; Kong, C.; Le, Q.-T.; Chi, J.-T.A.; Jeffrey, S.S.; Giaccia, A.J. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Saatci, O.; Kaymak, A.; Raza, U.; Ersan, P.G.; Akbulut, O.; Banister, C.E.; Sikirzhytski, V.; Tokat, U.M.; Aykut, G.; Ansari, S.A. Targeting lysyl oxidase (LOX) overcomes chemotherapy resistance in triple negative breast cancer. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Moreno Roig, E.; Groot, A.J.; Yaromina, A.; Hendrickx, T.C.; Barbeau, L.M.; Giuranno, L.; Dams, G.; Ient, J.; Olivo Pimentel, V.; van Gisbergen, M.W. HIF-1α and HIF-2α Differently Regulate the Radiation Sensitivity of NSCLC Cells. Cells 2019, 8, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.Y.; Perera, S.; Zhou, B.; Carretero, J.; Yeh, J.J.; Heathcote, S.A.; Jackson, A.L.; Nikolinakos, P.; Ospina, B.; Naumov, G. HIF2α cooperates with RAS to promote lung tumorigenesis in mice. J. Clin. Investig. 2009, 119, 2160–2170. [Google Scholar] [CrossRef] [Green Version]
- Bishop, T.; Ratcliffe, P.J. HIF hydroxylase pathways in cardiovascular physiology and medicine. Circ. Res. 2015, 117, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Masson, N.; Ratcliffe, P.J. Hypoxia signaling pathways in cancer metabolism: The importance of co-selecting interconnected physiological pathways. Cancer Metab. 2014, 2, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Kapitsinou, P.P.; Liu, Q.; Unger, T.L.; Rha, J.; Davidoff, O.; Keith, B.; Epstein, J.A.; Moores, S.L.; Erickson-Miller, C.L.; Haase, V.H. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood J. Am. Soc. Hematol. 2010, 116, 3039–3048. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.R.; Xue, X.; Shah, Y.M. Intestinal hypoxia-inducible factor-2α (HIF-2α) is critical for efficient erythropoiesis. J. Biol. Chem. 2011, 286, 19533–19540. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Zhang, H.; Dinavahi, R.; Li, F.; Xiang, Y.; Raman, V.; Bhujwalla, Z.M.; Felsher, D.W.; Cheng, L.; Pevsner, J. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell 2007, 12, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Agani, F.; Jiang, B.-H. Oxygen-independent regulation of HIF-1: Novel involvement of PI3K/AKT/mTOR pathway in cancer. Curr. Cancer Drug Targets 2013, 13, 245–251. [Google Scholar] [CrossRef]
- Lee, K.; Qian, D.Z.; Rey, S.; Wei, H.; Liu, J.O.; Semenza, G.L. Anthracycline chemotherapy inhibits HIF-1 transcriptional activity and tumor-induced mobilization of circulating angiogenic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2353–2358. [Google Scholar] [CrossRef] [Green Version]
- Erler, J.T.; Bennewith, K.L.; Cox, T.R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.-T.; Giaccia, A.J. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Qiao, B.; Liu, Q.; Zhang, W. Upregulation of extracellular matrix metalloproteinase inducer promotes hypoxia-induced epithelial-mesenchymal transition in esophageal cancer. Mol. Med. Rep. 2015, 12, 7419–7424. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wong, C.; Wei, H.; Gilkes, D.; Korangath, P.; Chaturvedi, P.; Schito, L.; Chen, J.; Krishnamachary, B.; Winnard, P.T. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene 2012, 31, 1757–1770. [Google Scholar] [CrossRef]
- Hanna, S.C.; Krishnan, B.; Bailey, S.T.; Moschos, S.J.; Kuan, P.-F.; Shimamura, T.; Osborne, L.D.; Siegel, M.B.; Duncan, L.M.; O’Brien, E.T. HIF1α and HIF2α independently activate SRC to promote melanoma metastases. J. Clin. Investig. 2013, 123, 2078–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schito, L.; Rey, S.; Tafani, M.; Zhang, H.; Wong, C.C.-L.; Russo, A.; Russo, M.A.; Semenza, G.L. Hypoxia-inducible factor 1-dependent expression of platelet-derived growth factor B promotes lymphatic metastasis of hypoxic breast cancer cells. Proc. Natl. Acad. Sci. USA 2012, 109, E2707–E2716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.; Zong, X. Aberrant cancer metabolism in epithelial–mesenchymal transition and cancer metastasis: Mechanisms in cancer progression. Crit. Rev. Oncol. Hematol. 2017, 115, 13–22. [Google Scholar] [CrossRef]
- Dupuy, F.; Tabariès, S.; Andrzejewski, S.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; Gao, D.; Leung, S.; Amir, E. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 2015, 22, 577–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF Transcription Factors, Inflammation, and Immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wang, L.; Chen, X.; Li, L.; Li, Y.; Ping, Y.; Huang, L.; Yue, D.; Zhang, Z.; Wang, F. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 2017, 6, e1320011. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keith, B.; Simon, M.C. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007, 129, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, M.; Kimura, S.; Kuroda, J.; Ashihara, E.; Kawatani, M.; Osada, H.; Umezawa, K.; Yasui, E.; Imoto, M.; Tsuruo, T. Glyoxalase-I is a novel target against Bcr-Abl+ leukemic cells acquiring stem-like characteristics in a hypoxic environment. Cell Death Differ. 2010, 17, 1211–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, S.; Charbonneau, M.; Grandmont, S.; Richard, D.E.; Dubois, C.M. Transforming growth factor β1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J. Biol. Chem. 2006, 281, 24171–24181. [Google Scholar] [CrossRef] [Green Version]
- Deschoemaeker, S.; Di Conza, G.; Lilla, S.; Martín-Pérez, R.; Mennerich, D.; Boon, L.; Hendrikx, S.; Maddocks, O.D.; Marx, C.; Radhakrishnan, P. PHD 1 regulates p53-mediated colorectal cancer chemoresistance. EMBO Mol. Med. 2015, 7, 1350–1365. [Google Scholar] [CrossRef]
- Hinohara, K.; Wu, H.-J.; Vigneau, S.; McDonald, T.O.; Igarashi, K.J.; Yamamoto, K.N.; Madsen, T.; Fassl, A.; Egri, S.B.; Papanastasiou, M. KDM5 histone demethylase activity links cellular transcriptomic heterogeneity to therapeutic resistance. Cancer Cell 2018, 34, 939–953.e939. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, S.-M.; McGeary, M.K.; Krykbaeva, I.; Lai, L.; Jansen, D.J.; Kales, S.C.; Simeonov, A.; Hall, M.D.; Kelly, D.P. KDM5B promotes drug resistance by regulating melanoma-propagating cell subpopulations. Mol. Cancer Ther. 2019, 18, 706–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, A.A.; Laukka, T.; Myllykoski, M.; Ringel, A.E.; Booker, M.A.; Tolstorukov, M.Y.; Meng, Y.J.; Meier, S.R.; Jennings, R.B.; Creech, A.L.; et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science 2019, 363, 1217–1222. [Google Scholar] [CrossRef] [Green Version]
- Hakimi, A.A.; Chen, Y.-B.; Wren, J.; Gonen, M.; Abdel-Wahab, O.; Heguy, A.; Liu, H.; Takeda, S.; Tickoo, S.K.; Reuter, V.E. Clinical and pathologic impact of select chromatin-modulating tumor suppressors in clear cell renal cell carcinoma. Eur. Urol. 2013, 63, 848–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gossage, L.; Murtaza, M.; Slatter, A.F.; Lichtenstein, C.P.; Warren, A.; Haynes, B.; Marass, F.; Roberts, I.; Shanahan, S.J.; Claas, A. Clinical and pathological impact of VHL, PBRM1, BAP1, SETD2, KDM6A, and JARID1c in clear cell renal cell carcinoma. Genes Chromos. Cancer 2014, 53, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shilatifard, A. UTX mutations in human cancer. Cancer Cell 2019, 35, 168–176. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.J.; Park, J.H.; Park, M.; Won, H.Y.; Joo, H.s.; Lee, C.H.; Lee, J.Y.; Kong, G. UTX inhibits EMT-induced breast CSC properties by epigenetic repression of EMT genes in cooperation with LSD 1 and HDAC 1. EMBO Rep. 2015, 16, 1288–1298. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Samanta, D.; Lu, H.; Bullen, J.W.; Zhang, H.; Chen, I.; He, X.; Semenza, G.L. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. USA 2016, 113, E2047–E2056. [Google Scholar] [CrossRef] [Green Version]
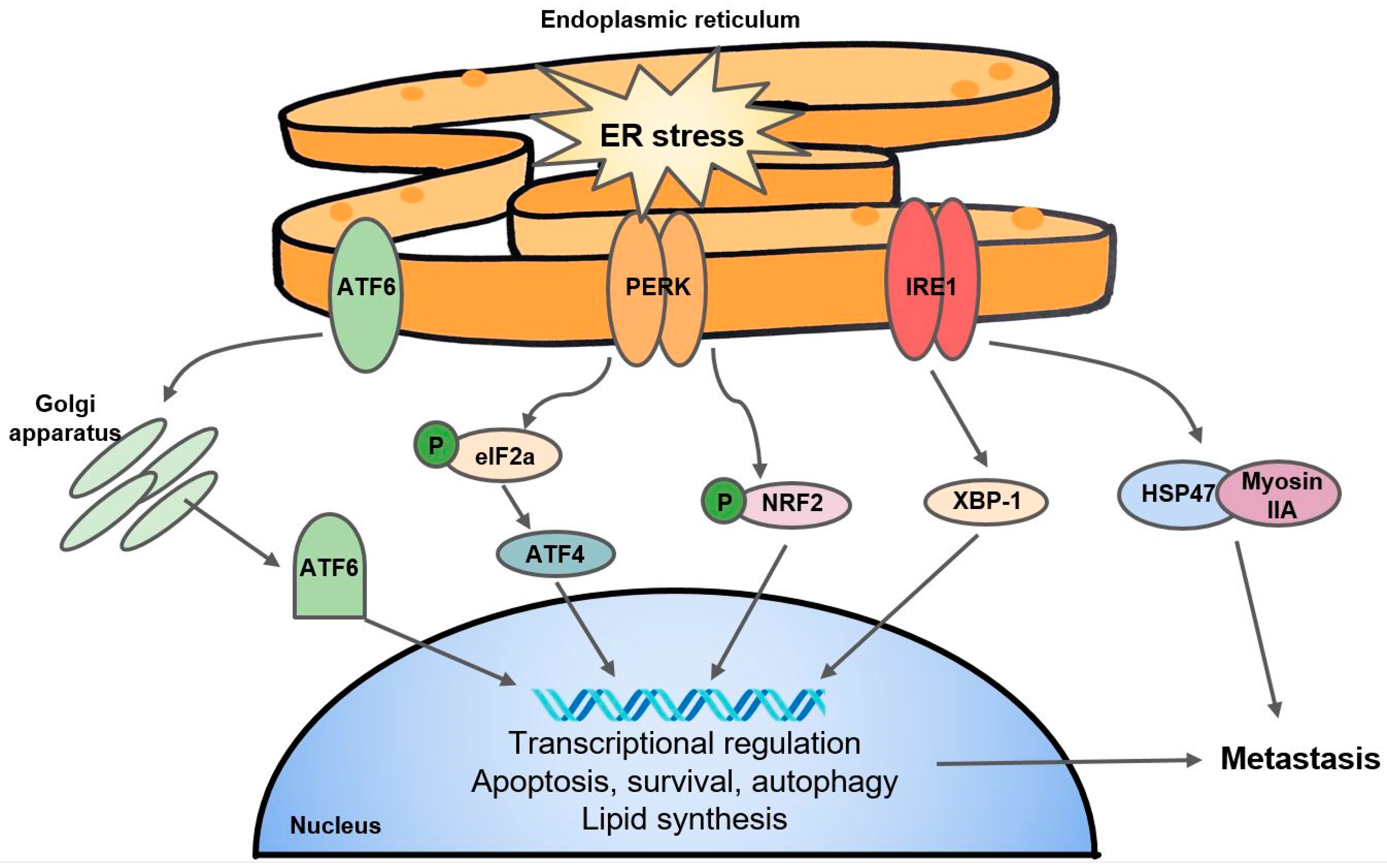
- Bartoszewska, S.; Collawn, J.F. Unfolded protein response (UPR) integrated signaling networks determine cell fate during hypoxia. Cell. Mol. Biol. Lett. 2020, 25, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Wouters, B.G.; Koritzinsky, M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat. Rev. Cancer 2008, 8, 851–864. [Google Scholar] [CrossRef]
- Feng, Y.-X.; Jin, D.X.; Sokol, E.S.; Reinhardt, F.; Miller, D.H.; Gupta, P.B. Cancer-specific PERK signaling drives invasion and metastasis through CREB3L1. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Rouschop, K.M.; Dubois, L.J.; Keulers, T.G.; van den Beucken, T.; Lambin, P.; Bussink, J.; van der Kogel, A.J.; Koritzinsky, M.; Wouters, B.G. PERK/eIF2α signaling protects therapy resistant hypoxic cells through induction of glutathione synthesis and protection against ROS. Proc. Natl. Acad. Sci. USA 2013, 110, 4622–4627. [Google Scholar] [CrossRef] [Green Version]
- Yoneda, A.; Minomi, K.; Tamura, Y. HSP47 promotes metastasis of breast cancer by interacting with myosin IIA via the unfolded protein response transducer IRE1α. Oncogene 2020, 39, 4519–4537. [Google Scholar] [CrossRef]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef]
- Dong, H.; Adams, N.M.; Xu, Y.; Cao, J.; Allan, D.S.; Carlyle, J.R.; Chen, X.; Sun, J.C.; Glimcher, L.H. The IRE1 endoplasmic reticulum stress sensor activates natural killer cell immunity in part by regulating c-Myc. Nat. Immunol. 2019, 20, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-R.; Chang, S.-Y.; Hong, E.-H.; Kwon, B.-E.; Kim, H.M.; Kim, Y.-J.; Lee, J.; Cho, H.-J.; Cheon, J.-H.; Ko, H.-J. Elevated endoplasmic reticulum stress reinforced immunosuppression in the tumor microenvironment via myeloid-derived suppressor cells. Oncotarget 2014, 5, 12331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sareddy, G.; Viswanadhapalli, S.; Surapaneni, P.; Suzuki, T.; Brenner, A.; Vadlamudi, R. Novel KDM1A inhibitors induce differentiation and apoptosis of glioma stem cells via unfolded protein response pathway. Oncogene 2017, 36, 2423–2434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köditz, J.; Nesper, J.; Wottawa, M.; Stiehl, D.P.; Camenisch, G.; Franke, C.; Myllyharju, J.; Wenger, R.H.; Katschinski, D.M. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood J. Am. Soc. Hematol. 2007, 110, 3610–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucharzewska, P.; Christianson, H.C.; Welch, J.E.; Svensson, K.J.; Fredlund, E.; Ringnér, M.; Mörgelin, M.; Bourseau-Guilmain, E.; Bengzon, J.; Belting, M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 7312–7317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, H.W.; Michael, M.Z.; Gleadle, J.M. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer 2012, 12, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Panigrahi, G.K.; Praharaj, P.P.; Peak, T.C.; Long, J.; Singh, R.; Rhim, J.S.; Abd Elmageed, Z.Y.; Deep, G. Hypoxia-induced exosome secretion promotes survival of African-American and Caucasian prostate cancer cells. Sci. Rep. 2018, 8, 1–13. [Google Scholar]
- Dorayappan, K.D.P.; Wanner, R.; Wallbillich, J.J.; Saini, U.; Zingarelli, R.; Suarez, A.A.; Cohn, D.E.; Selvendiran, K. Hypoxia-induced exosomes contribute to a more aggressive and chemoresistant ovarian cancer phenotype: A novel mechanism linking STAT3/Rab proteins. Oncogene 2018, 37, 3806–3821. [Google Scholar] [CrossRef]
- Umezu, T.; Tadokoro, H.; Azuma, K.; Yoshizawa, S.; Ohyashiki, K.; Ohyashiki, J.H. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood J. Am. Soc. Hematol. 2014, 124, 3748–3757. [Google Scholar] [CrossRef]
- Ramteke, A.; Ting, H.; Agarwal, C.; Mateen, S.; Somasagara, R.; Hussain, A.; Graner, M.; Frederick, B.; Agarwal, R.; Deep, G. Exosomes secreted under hypoxia enhance invasiveness and stemness of prostate cancer cells by targeting adherens junction molecules. Mol. Carcinog. 2015, 54, 554–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlaepfer, I.R.; Nambiar, D.K.; Ramteke, A.; Kumar, R.; Dhar, D.; Agarwal, C.; Bergman, B.; Graner, M.; Maroni, P.; Singh, R.P. Hypoxia induces triglycerides accumulation in prostate cancer cells and extracellular vesicles supporting growth and invasiveness following reoxygenation. Oncotarget 2015, 6, 22836. [Google Scholar] [CrossRef]
- Hwang, W.-L.; Lan, H.-Y.; Cheng, W.-C.; Huang, S.-C.; Yang, M.-H. Tumor stem-like cell-derived exosomal RNAs prime neutrophils for facilitating tumorigenesis of colon cancer. J. Hematol. Oncol. 2019, 12, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Gilkes, D.M.; Takano, N.; Xiang, L.; Luo, W.; Bishop, C.J.; Chaturvedi, P.; Green, J.J.; Semenza, G.L. Hypoxia-inducible factors and RAB22A mediate formation of microvesicles that stimulate breast cancer invasion and metastasis. Proc. Natl. Acad. Sci. USA 2014, 111, E3234–E3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Zhou, X.; Yao, Q.; Liu, Y.; Zhang, H.; Dong, Z. HIF-1-mediated production of exosomes during hypoxia is protective in renal tubular cells. Am. J. Physiol. Ren. Physiol. 2017, 313, F906–F913. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef]
- Han, X.-J.; Yang, Z.-J.; Jiang, L.-P.; Wei, Y.-F.; Liao, M.-F.; Qian, Y.; Li, Y.; Huang, X.; Wang, J.-B.; Xin, H.-B. Mitochondrial dynamics regulates hypoxia-induced migration and antineoplastic activity of cisplatin in breast cancer cells. Int. J. Oncol. 2015, 46, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Peiris-Pagès, M.; Bonuccelli, G.; Sotgia, F.; Lisanti, M.P. Mitochondrial fission as a driver of stemness in tumor cells: mDIVI1 inhibits mitochondrial function, cell migration and cancer stem cell (CSC) signalling. Oncotarget 2018, 9, 13254. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, C.; Chen, X.; Takada, M.; Fan, C.; Zheng, X.; Wen, H.; Liu, Y.; Wang, C.; Pestell, R.G.; et al. EglN2 associates with the NRF1-PGC1alpha complex and controls mitochondrial function in breast cancer. EMBO J. 2015, 34, 2953–2970. [Google Scholar] [CrossRef]
- Bao, M.H.-R.; Yang, C.; Tse, A.P.-W.; Wei, L.; Lee, D.; Zhang, M.S.; Goh, C.C.; Chiu, D.K.-C.; Yuen, V.W.-H.; Law, C.-T. Genome-wide CRISPR-Cas9 knockout library screening identified PTPMT1 in cardiolipin synthesis is crucial to survival in hypoxia in liver cancer. Cell Rep. 2021, 34, 108676. [Google Scholar] [CrossRef] [PubMed]
- Scharping, N.E.; Rivadeneira, D.B.; Menk, A.V.; Vignali, P.D.; Ford, B.R.; Rittenhouse, N.L.; Peralta, R.; Wang, Y.; Wang, Y.; DePeaux, K. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 2021, 22, 205–215. [Google Scholar] [CrossRef]
- Tang, T.; Yang, Z.; Zhu, Q.; Wu, Y.; Sun, K.; Alahdal, M.; Zhang, Y.; Xing, Y.; Shen, Y.; Xia, T. Up-regulation of miR-210 induced by a hypoxic microenvironment promotes breast cancer stem cell metastasis, proliferation, and self-renewal by targeting E-cadherin. FASEB J. 2018, 32, 6965–6981. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, P.; Lovat, F.; Fassan, M.; Casadei, L.; Cascione, L.; Jacob, N.K.; Carasi, S.; Palmieri, D.; Costinean, S.; Shapiro, C.L. Protective role of miR-155 in breast cancer through RAD51 targeting impairs homologous recombination after irradiation. Proc. Natl. Acad. Sci. USA 2014, 111, 4536–4541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The different mechanisms of cancer drug resistance: A brief review. Adv. Pharm. Bull. 2017, 7, 339. [Google Scholar] [CrossRef]
- Lin, C.; Yang, L. Long noncoding RNA in cancer: Wiring signaling circuitry. Trends Cell Biol. 2018, 28, 287–301. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, S.; Cai, G.; Kong, L.; Zhang, T.; Ren, Y.; Wu, Y.; Mei, M.; Zhang, L.; Wang, X. Long non coding RNA MALAT1 promotes tumor growth and metastasis by inducing epithelial-mesenchymal transition in oral squamous cell carcinoma. Sci. Rep. 2015, 5, 15972. [Google Scholar] [CrossRef] [Green Version]
- Shih, J.-W.; Chiang, W.-F.; Wu, A.T.; Wu, M.-H.; Wang, L.-Y.; Yu, Y.-L.; Hung, Y.-W.; Wang, W.-C.; Chu, C.-Y.; Hung, C.-L. Long noncoding RNA LncHIFCAR/MIR31HG is a HIF-1α co-activator driving oral cancer progression. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Wang, S.; Chen, J.; Wang, Z.; Liang, X.; Wang, X.; Jiang, J.; Lang, J.; Li, L. Long noncoding RNA HAS2-AS1 mediates hypoxia-induced invasiveness of oral squamous cell carcinoma. Mol. Carcinog. 2017, 56, 2210–2222. [Google Scholar] [CrossRef]
- Niu, Y.; Bao, L.; Chen, Y.; Wang, C.; Luo, M.; Zhang, B.; Zhou, M.; Wang, J.E.; Fang, Y.V.; Kumar, A. HIF2-induced long noncoding RNA RAB11B-AS1 promotes hypoxia-mediated angiogenesis and breast cancer metastasis. Cancer Res. 2020, 80, 964–975. [Google Scholar] [CrossRef]
- Jin, Y.; Che, X.; Qu, X.; Li, X.; Lu, W.; Wu, J.; Wang, Y.; Hou, K.; Li, C.; Zhang, X. CircHIPK3 promotes metastasis of gastric cancer via miR-653-5p/miR-338-3p-NRP1 axis under a long-term hypoxic microenvironment. Front. Oncol. 2020, 10, 1612. [Google Scholar] [CrossRef] [PubMed]
- Buurman, R.; Gürlevik, E.; Schäffer, V.; Eilers, M.; Sandbothe, M.; Kreipe, H.; Wilkens, L.; Schlegelberger, B.; Kühnel, F.; Skawran, B. Histone deacetylases activate hepatocyte growth factor signaling by repressing microRNA-449 in hepatocellular carcinoma cells. Gastroenterology 2012, 143, 811–820.e815. [Google Scholar] [CrossRef]
- Hsieh, Y.-L.; Tu, H.-J.; Pan, S.-L.; Liou, J.-P.; Yang, C.-R. Anti-metastatic activity of MPT0G211, a novel HDAC6 inhibitor, in human breast cancer cells in vitro and in vivo. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 992–1003. [Google Scholar] [CrossRef]
- Dong, J.; Zheng, N.; Wang, X.; Tang, C.; Yan, P.; Zhou, H.-B.; Huang, J. A novel HDAC6 inhibitor exerts an anti-cancer effect by triggering cell cycle arrest and apoptosis in gastric cancer. Eur. J. Pharmacol. 2018, 828, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Dowling, C.M.; Hollinshead, K.E.; Di Grande, A.; Pritchard, J.; Zhang, H.; Dillon, E.T.; Haley, K.; Papadopoulos, E.; Mehta, A.K.; Bleach, R. Multiple screening approaches reveal HDAC6 as a novel regulator of glycolytic metabolism in triple-negative breast cancer. Sci. Adv. 2021, 7, eabc4897. [Google Scholar] [CrossRef]
- Daskalaki, I.; Gkikas, I.; Tavernarakis, N. Hypoxia and selective autophagy in cancer development and therapy. Front. Cell Dev. Biol. 2018, 6, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noman, M.Z.; Janji, B.; Kaminska, B.; Van Moer, K.; Pierson, S.; Przanowski, P.; Buart, S.; Berchem, G.; Romero, P.; Mami-Chouaib, F. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 2011, 71, 5976–5986. [Google Scholar] [CrossRef] [Green Version]
- Baginska, J.; Viry, E.; Berchem, G.; Poli, A.; Noman, M.Z.; van Moer, K.; Medves, S.; Zimmer, J.; Oudin, A.; Niclou, S.P. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. USA 2013, 110, 17450–17455. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Feng, L.; Liang, C.; Yang, K.; Liu, Z. Ultrasound triggered tumor oxygenation with oxygen-shuttle nanoperfluorocarbon to overcome hypoxia-associated resistance in cancer therapies. Nano Lett. 2016, 16, 6145–6153. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.J.; Cosby, L.A.; Shansky, C.W.; Sartorelli, A.C. Potential bioreductive alkylating agents. 1. Benzoquinone derivatives. J. Med. Chem. 1972, 15, 1247–1252. [Google Scholar] [CrossRef]
- Williams, K.J.; Cowen, R.L.; Stratford, I.J. Hypoxia and oxidative stress in breast cancer Tumour hypoxia–therapeutic considerations. Breast Cancer Res. 2001, 3, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef]
- Sun, J.D.; Liu, Q.; Wang, J.; Ahluwalia, D.; Ferraro, D.; Wang, Y.; Duan, J.-X.; Ammons, W.S.; Curd, J.G.; Matteucci, M.D. Selective tumor hypoxia targeting by hypoxia-activated prodrug TH-302 inhibits tumor growth in preclinical models of cancer. Clin. Cancer Res. 2012, 18, 758–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, I.N.; Thomas, M.; Calder, E.D.; Conway, S.J.; Hammond, E.M. Clinical advances of hypoxia-activated prodrugs in combination with radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2017, 98, 1183–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegelberg, L.; Houben, R.; Niemans, R.; de Ruysscher, D.; Yaromina, A.; Theys, J.; Guise, C.P.; Smaill, J.B.; Patterson, A.V.; Lambin, P. Hypoxia-activated prodrugs and (lack of) clinical progress: The need for hypoxia-based biomarker patient selection in phase III clinical trials. Clin. Transl. Radiat. Oncol. 2019, 15, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallah, J.; Rini, B.I. HIF inhibitors: Status of current clinical development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef]
- Xu, R.; Wang, K.; Rizzi, J.P.; Huang, H.; Grina, J.A.; Schlachter, S.T.; Wang, B.; Wehn, P.M.; Yang, H.; Dixon, D.D. 3-[(1 S, 2 S, 3 R)-2, 3-Difluoro-1-hydroxy-7-methylsulfonylindan-4-yl] oxy-5-fluorobenzonitrile (PT2977), a hypoxia-inducible factor 2α (HIF-2α) inhibitor for the treatment of clear cell Renal cell carcinoma. J. Med. Chem. 2019, 62, 6876–6893. [Google Scholar] [CrossRef] [Green Version]
- Courtney, K.D.; Ma, Y.; de Leon, A.D.; Christie, A.; Xie, Z.; Woolford, L.; Singla, N.; Joyce, A.; Hill, H.; Madhuranthakam, A.J. HIF-2 complex dissociation, target inhibition, and acquired resistance with PT2385, a first-in-class HIF-2 inhibitor, in patients with clear cell renal cell carcinoma. Clin. Cancer Res. 2020, 26, 793–803. [Google Scholar] [CrossRef]
- Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R.M.; Harsh, G.R.; Brown, J.M. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Investig. 2010, 120, 694–705. [Google Scholar] [CrossRef]
- Hu, H.; Miao, X.-K.; Li, J.-Y.; Zhang, X.-W.; Xu, J.-J.; Zhang, J.-Y.; Zhou, T.-X.; Hu, M.-N.; Yang, W.-L.; Mou, L.-Y. YC-1 potentiates the antitumor activity of gefitinib by inhibiting HIF-1α and promoting the endocytic trafficking and degradation of EGFR in gefitinib-resistant non-small-cell lung cancer cells. Eur. J. Pharmacol. 2020, 874, 172961. [Google Scholar] [CrossRef]
- Wu, S.-L.; Li, Y.-J.; Liao, K.; Shi, L.; Zhang, N.; Liu, S.; Hu, Y.-Y.; Li, S.-L.; Wang, Y. 2-Methoxyestradiol inhibits the proliferation and migration and reduces the radioresistance of nasopharyngeal carcinoma CNE-2 stem cells via NF-κB/HIF-1 signaling pathway inactivation and EMT reversal. Oncol. Rep. 2017, 37, 793–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Y.; Yang, C.; Schovanek, J.; Wang, H.; Bullova, P.; Caisova, V.; Gupta, G.; Wolf, K.I.; Semenza, G.L.; Zhuang, Z. Anthracyclines suppress pheochromocytoma cell characteristics, including metastasis, through inhibition of the hypoxia signaling pathway. Oncotarget 2017, 8, 22313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, K.; Hu, L.; Liu, X.; Simon, J.M.; Ptacek, T.S.; Zheng, X.; Liao, C.; Baldwin, A.S.; Zhang, Q. USP37 promotes deubiquitination of HIF2α in kidney cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 13023–13032. [Google Scholar] [CrossRef] [PubMed]
- Lachenmayer, A.; Toffanin, S.; Cabellos, L.; Alsinet, C.; Hoshida, Y.; Villanueva, A.; Minguez, B.; Tsai, H.-W.; Ward, S.C.; Thung, S. Combination therapy for hepatocellular carcinoma: Additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. J. Hepatol. 2012, 56, 1343–1350. [Google Scholar] [CrossRef] [Green Version]
- Jin, N.; Lubner, S.J.; Mulkerin, D.L.; Rajguru, S.; Carmichael, L.; Chenv, H.; Holen, K.D.; LoConte, N.K. A phase II trial of a histone deacetylase inhibitor panobinostat in patients with low-grade neuroendocrine tumors. Oncologist 2016, 21, 785. [Google Scholar] [CrossRef] [Green Version]
- Liao, C.; Zhang, Y.; Fan, C.; Herring, L.E.; Liu, J.; Locasale, J.W.; Takada, M.; Zhou, J.; Zurlo, G.; Hu, L. Identification of BBOX1 as a therapeutic target in triple-negative breast cancer. Cancer Discov. 2020, 10, 1706–1721. [Google Scholar] [CrossRef] [PubMed]
- Holdstock, L.; Meadowcroft, A.M.; Maier, R.; Johnson, B.M.; Jones, D.; Rastogi, A.; Zeig, S.; Lepore, J.J.; Cobitz, A.R. Four-week studies of oral hypoxia-inducible factor–prolyl hydroxylase inhibitor GSK1278863 for treatment of anemia. J. Am. Soc. Nephrol. 2016, 27, 1234–1244. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Bratton, D.L.; Colgan, S.P. Targeting hypoxia signalling for the treatment of ischaemic and inflammatory diseases. Nat. Rev. Drug Discov. 2014, 13, 852–869. [Google Scholar] [CrossRef] [Green Version]
- Si, Y.; Liu, J.; Shen, H.; Zhang, C.; Wu, Y.; Huang, Y.; Gong, Z.; Xue, J.; Liu, T. Fisetin decreases TET 1 activity and CCNY/CDK 16 promoter 5hmC levels to inhibit the proliferation and invasion of renal cancer stem cell. J. Cell. Mol. Med. 2019, 23, 1095–1105. [Google Scholar] [CrossRef]
- Miyake, Y.; Itoh, Y.; Hatanaka, A.; Suzuma, Y.; Suzuki, M.; Kodama, H.; Arai, Y.; Suzuki, T. Identification of novel lysine demethylase 5-selective inhibitors by inhibitor-based fragment merging strategy. Bioorg. Med. Chem. 2019, 27, 1119–1129. [Google Scholar] [CrossRef]
- Ye, Q.; Holowatyj, A.; Wu, J.; Liu, H.; Zhang, L.; Suzuki, T.; Yang, Z.-Q. Genetic alterations of KDM4 subfamily and therapeutic effect of novel demethylase inhibitor in breast cancer. Am. J. Cancer Res. 2015, 5, 1519. [Google Scholar]
- Hoyle, R.G.; Wang, H.; Cen, Y.; Zhang, Y.; Li, J. IOX1 Suppresses Wnt Target Gene Transcription and Colorectal Cancer Tumorigenesis through Inhibition of KDM3 Histone Demethylases. Mol. Cancer Ther. 2021, 20, 191–202. [Google Scholar] [CrossRef]
- Hu, Q.; Chen, J.; Zhang, J.; Xu, C.; Yang, S.; Jiang, H. IOX1, a JMJD2A inhibitor, suppresses the proliferation and migration of vascular smooth muscle cells induced by angiotensin II by regulating the expression of cell cycle-related proteins. Int. J. Mol. Med. 2016, 37, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, H.P.; Smitheman, K.N.; Kamat, C.D.; Soong, D.; Federowicz, K.E.; Van Aller, G.S.; Schneck, J.L.; Carson, J.D.; Liu, Y.; Butticello, M. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell 2015, 28, 57–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Su, R.; Sheng, Y.; Dong, L.; Dong, Z.; Xu, H.; Ni, T.; Zhang, Z.S.; Zhang, T.; Li, C. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell 2019, 35, 677–691.e610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, M.; Shimizu, T.; Mabuchi, M.; Horiike, K.; Kitae, K.; Hase, H.; Ueda, Y.; Tsujikawa, K.; Tanaka, A. Novel metabolically stable PCA-1/ALKBH3 inhibitor has potent antiproliferative effects on DU145 cells in vivo. Anticancer. Res. 2018, 38, 211–218. [Google Scholar]
- Nigam, R.; Babu, K.R.; Ghosh, T.; Kumari, B.; Akula, D.; Rath, S.N.; Das, P.; Anindya, R.; Khan, F.A. Indenone derivatives as inhibitor of human DNA dealkylation repair enzyme AlkBH3. Bioorg. Med. Chem. 2018, 26, 4100–4112. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Xie, H.; Liu, X.; Potjewyd, F.; James, L.I.; Wilkerson, E.M.; Herring, L.E.; Xie, L.; Chen, X.; Cabrera, J.C. TBK1 is a synthetic lethal target in cancer with VHL loss. Cancer Discov. 2020, 10, 460–475. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Aref, A.R.; Lizotte, P.H.; Ivanova, E.; Stinson, S.; Zhou, C.W.; Bowden, M.; Deng, J.; Liu, H.; Miao, D. Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer Discov. 2018, 8, 196–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, N.; Pires, I.M.; Bencokova, Z.; Coackley, C.; Luoto, K.R.; Bhogal, N.; Lakshman, M.; Gottipati, P.; Oliver, F.J.; Helleday, T. Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Res. 2010, 70, 8045–8054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaprakash, P.; Ai, M.; Liu, A.; Budhani, P.; Bartkowiak, T.; Sheng, J.; Ager, C.; Nicholas, C.; Jaiswal, A.R.; Sun, Y. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Investig. 2018, 128, 5137–5149. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Liang, X.; Wang, P.; Hu, Y.; Qi, Y.; Jin, Y.; Du, Y.; Fang, C.; Tian, J. A Hepatocellular Carcinoma Targeting Nanostrategy with Hypoxia-Ameliorating and Photothermal Abilities that, Combined with Immunotherapy, Inhibits Metastasis and Recurrence. ACS Nano 2020, 14, 12679–12696. [Google Scholar] [CrossRef]
- Fong, L.; Hotson, A.; Powderly, J.D.; Sznol, M.; Heist, R.S.; Choueiri, T.K.; George, S.; Hughes, B.G.; Hellmann, M.D.; Shepard, D.R. Adenosine 2A receptor blockade as an immunotherapy for treatment-refractory renal cell cancer. Cancer Discov. 2020, 10, 40–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halpin-Veszeleiova, K.; Hatfield, S.M. Oxygenation and A2AR blockade to eliminate hypoxia/HIF-1α-adenosinergic immunosuppressive axis and improve cancer immunotherapy. Curr. Opin. Pharmacol. 2020, 53, 84–90. [Google Scholar] [CrossRef]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Uribesalgo, I.; Hoffmann, D.; Zhang, Y.; Kavirayani, A.; Lazovic, J.; Berta, J.; Novatchkova, M.; Pai, T.P.; Wimmer, R.A.; László, V. Apelin inhibition prevents resistance and metastasis associated with anti-angiogenic therapy. EMBO Mol. Med. 2019, 11, e9266. [Google Scholar] [CrossRef]
- Maione, F.; Capano, S.; Regano, D.; Zentilin, L.; Giacca, M.; Casanovas, O.; Bussolino, F.; Serini, G.; Giraudo, E. Semaphorin 3A overcomes cancer hypoxia and metastatic dissemination induced by antiangiogenic treatment in mice. J. Clin. Investig. 2012, 122, 1832–1848. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Target Protein(s) | Disease Context | References |
|---|---|---|---|
| GSK1278863 | PHDs | Anemia of chronic kidney disease | [220] |
| FG-4592 | PHDs | Anemia of chronic kidney disease | [221] |
| Fisetin | TET1 | Renal cancer stem cells | [222] |
| Compound 10 and 13 | KDM5 | Lung cancer cell line A549 | [223] |
| NCDM-32B | KDM4 | Basal-like breast cancer | [224] |
| IOX1 | KDM3, KDM4 | Colorectal cancer and vascular smooth muscle cells in atherosclerosis | [225,226] |
| GSK2879552 | KDM1A | Small cell lung carcinoma | [227] |
| FB23 and FB23-2 | FTO | Acute myeloid leukemia | [228] |
| Compound 7l | ALKBH3 | Prostate cancer cell line DU145 | [229] |
| Indenone derivative Compound 5c | ALKBH3 | Lung cancer cell line A549 | [230] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, R.; Liao, C.; Zhang, Q. Hypoxia-Driven Effects in Cancer: Characterization, Mechanisms, and Therapeutic Implications. Cells 2021, 10, 678. https://doi.org/10.3390/cells10030678
Shi R, Liao C, Zhang Q. Hypoxia-Driven Effects in Cancer: Characterization, Mechanisms, and Therapeutic Implications. Cells. 2021; 10(3):678. https://doi.org/10.3390/cells10030678
Chicago/Turabian StyleShi, Rachel, Chengheng Liao, and Qing Zhang. 2021. "Hypoxia-Driven Effects in Cancer: Characterization, Mechanisms, and Therapeutic Implications" Cells 10, no. 3: 678. https://doi.org/10.3390/cells10030678