
Hypoxia, Metabolic Reprogramming, and Drug Resistance in Liver Cancer

Abstract
:1. Introduction
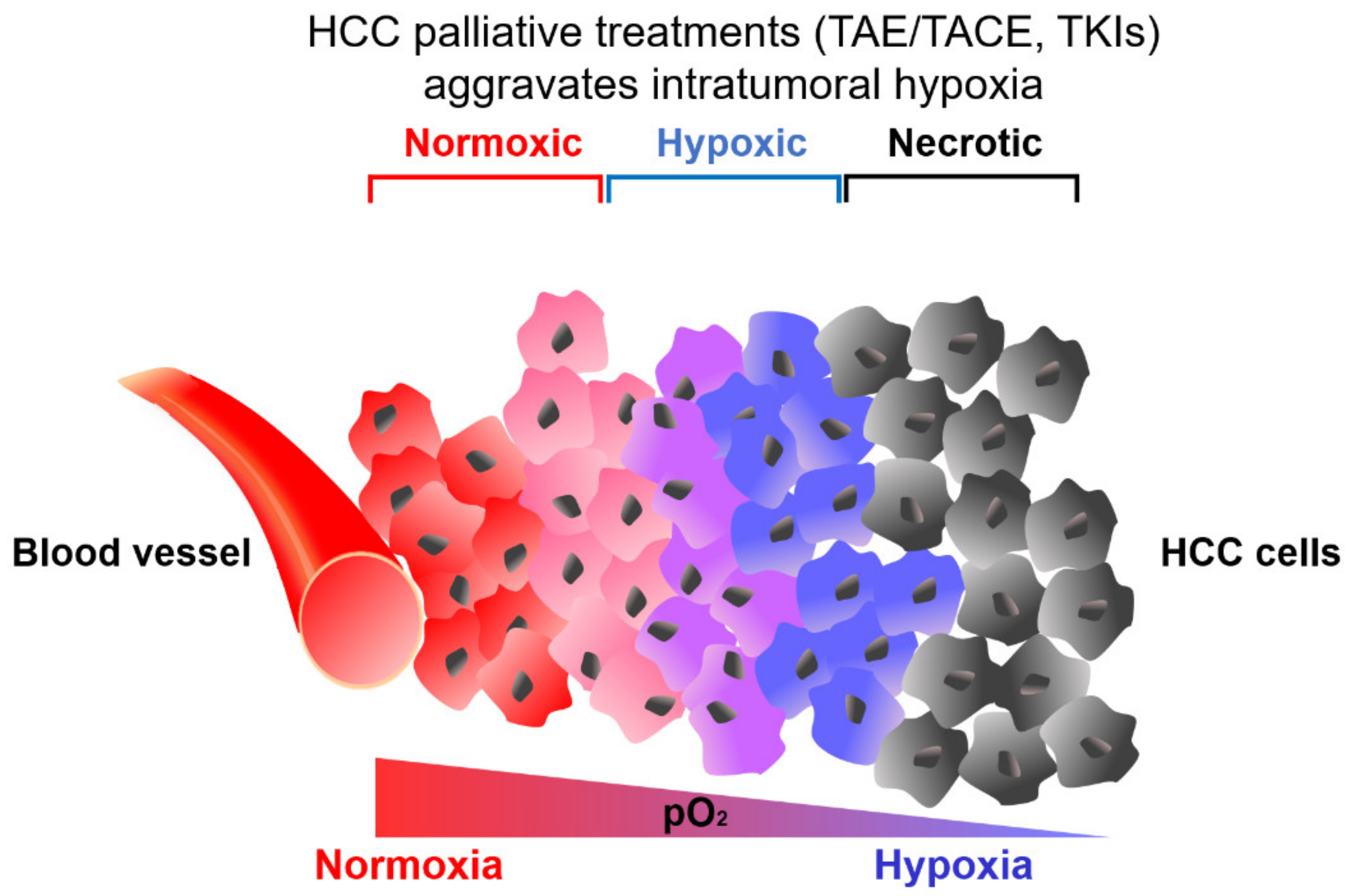
2. Hypoxic Tumor Microenvironment in HCC
3. HIF-Induced Metabolic Reprogramming under Hypoxia and Drug Resistance in HCC
3.1. HIF-Mediated Induction of Glucose Metabolism under Hypoxia
3.1.1. HIF-Mediated Induction of Glucose Metabolism under Hypoxia and TKI Resistance
3.1.2. HIF-Mediated Induction of Glucose Metabolism under Hypoxia and ICI Resistance
3.2. HIF-Mediated Induction of Lactate Metabolism under Hypoxia
HIF-Mediated Induction of Lactate Metabolism and ICI Resistance
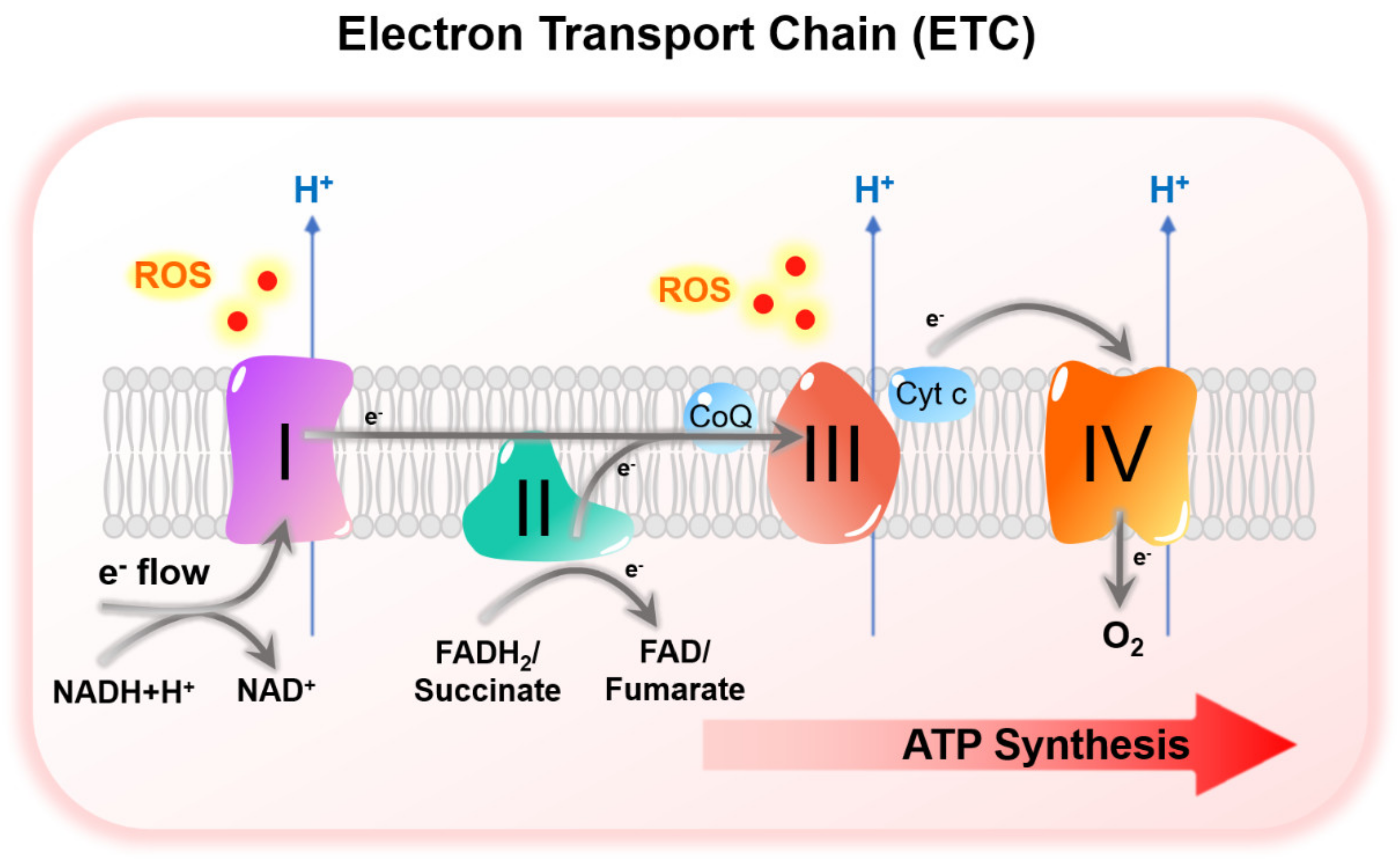
3.3. HIF-Mediated Suppression of Mitochondrial Metabolism under Hypoxia
HIF-Mediated Suppression of Mitochondrial Metabolism under Hypoxia and TKI Resistance
3.4. HIF-Mediated Induction of Serine Metabolism under Hypoxia
HIF-Mediated Induction of Serine Metabolism under Hypoxia and TKI Resistance
3.5. HIF-Mediated Induction of Adenosinergic Metabolism under Hypoxia
HIF-Mediated Induction of Adenosinergic Metabolism under Hypoxia and ICI Resistance
4. Targeting Hypoxic HCC to Overcome Drug Resistance
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Lou, T. Hypoxia inducible factors in hepatocellular carcinoma. Oncotarget 2017, 8, 46691–46703. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-Q.; Wu, X.; Gan, L.; Yang, X.-L.; Miao, Z.-H. Hypoxia induces universal but differential drug resistance and impairs anticancer mechanisms of 5-fluorouracil in hepatoma cells. Acta Pharmacol. Sin. 2017, 38, 1642–1654. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Kang, Y.-K.; Chen, Z.; Tsao, C.-J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.-S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Copur, M.S. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 2498, author reply 2498–2499. [Google Scholar]
- Ikeda, M.; Okusaka, T.; Mitsunaga, S.; Ueno, H.; Tamai, T.; Suzuki, T.; Hayato, S.; Kadowaki, T.; Okita, K.; Kumada, H. Safety and Pharmacokinetics of Lenvatinib in Patients with Advanced Hepatocellular Carcinoma. Clin. Cancer Res. 2015, 22, 1385–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.C.C.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.-K.; Yen, C.-J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Shi, F.; Shi, M.; Zeng, Z.; Qi, R.-Z.; Liu, Z.-W.; Zhang, J.-Y.; Yang, Y.-P.; Tien, P.; Wang, F.-S. PD-1 and PD-L1 upregulation promotes CD8+ T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int. J. Cancer 2010, 128, 887–896. [Google Scholar] [CrossRef]
- Pesce, S.; Greppi, M.; Grossi, F.; Del Zotto, G.; Moretta, L.; Sivori, S.; Genova, C.; Marcenaro, E. PD/1-PD-Ls Checkpoint: Insight on the Potential Role of NK Cells. Front. Immunol. 2019, 10, 1242. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours—implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P.; Höckel, M.; Mayer, A. Detection and Characterization of Tumor Hypoxia Using pO2 Histography. Antioxid. Redox Signal. 2007, 9, 1221–1236. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-B.; Cheng, Q.; Geng, W.; Ling, C.-C.; Liu, Y.; Ng, K.T.-P.; Yam, J.W.P.; Guan, X.-Y.; Lo, C.-M.; Man, K. Enhancement of cisplatin-based TACE by a hemoglobin-based oxygen carrier in an orthotopic rat HCC model. Artif. Cells Nanomed. Biotechnol. 2013, 42, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Riedl, C.C.; Brader, P.; Zanzonico, P.B.; Chun, Y.S.; Woo, Y.; Singh, P.; Carlin, S.; Wen, B.; Ling, C.C.; Hricak, H.; et al. Imaging Hypoxia in Orthotopic Rat Liver Tumors with Iodine 124–labeled Iodoazomycin Galactopyranoside PET. Radiology 2008, 248, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Wada, H.; Nagano, H.; Yamamoto, H.; Yang, Y.; Kondo, M.; Ota, H.; Nakamura, M.; Yoshioka, S.; Kato, H.; Damdinsuren, B.; et al. Expression pattern of angiogenic factors and prognosis after hepatic resection in hepatocellular carcinoma: Importance of angiopoietin-2 and hypoxia-induced factor-1a. Liver Int. 2006, 26, 414–423. [Google Scholar] [CrossRef]
- Amann, T.; Maegdefrau, U.; Hartmann, A.; Agaimy, A.; Marienhagen, J.; Weiss, T.; Stoeltzing, O.; Warnecke, C.; Schölmerich, J.; Oefner, P.J.; et al. GLUT1 Expression Is Increased in Hepatocellular Carcinoma and Promotes Tumorigenesis. Am. J. Pathol. 2009, 174, 1544–1552. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Li, X.; Sun, X.; Wang, J.; Yang, X.; Zhou, X.; Liu, X.; Liu, W.; Yuan, J.; Yao, L.; et al. Combined Aberrant Expression of NDRG2 and LDHA Predicts Hepatocellular Carcinoma Prognosis and Mediates the Anti-tumor Effect of Gemcitabine. Int. J. Biol. Sci. 2019, 15, 1771–1786. [Google Scholar] [CrossRef]
- Huang, W.-J.; Jeng, Y.-M.; Lai, H.-S.; Fong, I.-U.; Sheu, F.-Y.B.; Lai, P.-L.; Yuan, R.-H. Expression of Hypoxic Marker Carbonic Anhydrase IX Predicts Poor Prognosis in Resectable Hepatocellular Carcinoma. PLoS ONE 2015, 10, e0119181. [Google Scholar] [CrossRef] [PubMed]
- Helmlinger, G.; Yuan, F.; Dellian, M.; Jain, R.K. Interstitial pH and pO2 gradients in solid tumors in vivo: High-resolution measurements reveal a lack of correlation. Nat. Med. 1997, 3, 177–182. [Google Scholar] [CrossRef]
- Sun, X.; Jiang, H.; Jiang, X.; Tan, H.; Meng, Q.; Sun, B.; Xu, R.; Krissansen, G.W. Antisense Hypoxia-Inducible Factor-1α Augments Transcatheter Arterial Embolization in the Treatment of Hepatocellular Carcinomas in Rats. Hum. Gene Ther. 2009, 20, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Korotchkina, L.G.; Patel, M.S. Site Specificity of Four Pyruvate Dehydrogenase Kinase Isoenzymes toward the Three Phosphorylation Sites of Human Pyruvate Dehydrogenase. J. Biol. Chem. 2001, 276, 37223–37229. [Google Scholar] [CrossRef] [Green Version]
- Korotchkina, L.G.; Patel, M.S. Mutagenesis Studies of the Phosphorylation Sites of Recombinant Human Pyruvate Dehydrogenase. SITE-SPECIFIC REGULATION. J. Biol. Chem. 1995, 270, 14297–14304. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Li, G.; Zhu, H.; Dong, X.; Zhao, D.; Jiang, X.; Li, J.; Qiao, H.; Ni, S.; Sun, X. 2-Methoxyestradiol synergizes with sorafenib to suppress hepatocellular carcinoma by simultaneously dysregulating hypoxia-inducible factor-1 and -2. Cancer Lett. 2014, 355, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, D.; Park, Y.; Andrabi, S.A.; Shelton, L.M.; Gilkes, D.M.; Semenza, G.L. PHGDH Expression Is Required for Mitochondrial Redox Homeostasis, Breast Cancer Stem Cell Maintenance, and Lung Metastasis. Cancer Res. 2016, 76, 4430–4442. [Google Scholar] [CrossRef] [Green Version]
- Coriat, R.; Nicco, C.; Chéreau, C.; Mir, O.; Alexandre, J.; Ropert, S.; Weill, B.; Chaussade, S.; Goldwasser, F.; Batteux, F. Sorafenib-Induced Hepatocellular Carcinoma Cell Death Depends on Reactive Oxygen Species Production In Vitro and In Vivo. Mol. Cancer Ther. 2012, 11, 2284–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1alpha. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavo, A.C.; Brown, R.S.; Wahl, R.L.; Clavo, A.C.; Brown, R.S.; Wahl, R.L. Fluorodeoxyglucose uptake in human cancer cell lines is increased by hypoxia. J. Nucl. Med. 1995, 36, 1625–1632. [Google Scholar]
- Xia, H.; Chen, J.; Gao, H.; Kong, S.N.; Deivasigamani, A.; Shi, M.; Xie, T.; Hui, K.M. Hypoxia-induced modulation of glucose transporter expression impacts 18F-fluorodeoxyglucose PET-CT imaging in hepatocellular carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2019, 47, 787–797. [Google Scholar] [CrossRef]
- Pedersen, P.L.; Mathupala, S.; Rempel, A.; Geschwind, J.; Ko, Y.H. Mitochondrial bound type II hexokinase: A key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim. et Biophys. Acta (BBA) Bioenerg. 2002, 1555, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Gwak, G.-Y.; Yoon, J.-H.; Kim, K.M.; Lee, H.-S.; Chung, J.W.; Gores, G.J. Hypoxia stimulates proliferation of human hepatoma cells through the induction of hexokinase II expression. J. Hepatol. 2005, 42, 358–364. [Google Scholar] [CrossRef]
- Guo, W.; Qiu, Z.; Wang, Z.; Wang, Q.; Tan, N.; Chen, T.; Chen, Z.; Huang, S.; Gu, J.; Li, J.; et al. MiR-199a-5p is negatively associated with malignancies and regulates glycolysis and lactate production by targeting hexokinase 2 in liver cancer. Hepatology 2015, 62, 1132–1144. [Google Scholar] [CrossRef]
- Gao, H.; Hao, Y.; Zhou, X.; Li, H.; Liu, F.; Zhu, H.; Song, X.; Niu, Z.; Ni, Q.; Chen, M.; et al. Prognostic value of glucose transporter 3 expression in hepatocellular carcinoma. Oncol. Lett. 2019, 19, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Jiang, F.; Ge, Z.; Chen, B.; Yu, J.; Xin, M.; Wang, J.; An, L.; Wei, J.; Wu, L. Fructose-Bisphosphate Aldolase A Regulates Hypoxic Adaptation in Hepatocellular Carcinoma and Involved with Tumor Malignancy. Dig. Dis. Sci. 2019, 64, 3215–3227. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Kunjithapatham, R.; Geschwind, J.-F. Glyceraldehyde-3-Phosphate Dehydrogenase: A Promising Target for Molecular Therapy in Hepatocellular Carcinoma. Oncotarget 2012, 3, 940–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.-F.; Feng, X.-S.; Chen, H.; Duan, Z.-J.; Wang, L.-X.; Yang, D.; Liu, P.-X.; Zhang, Q.-P.; Jin, Y.-L.; Sun, Z.-G.; et al. Prognostic significance of synergistic hexokinase-2 and beta2-adrenergic receptor expression in human hepatocelluar carcinoma after curative resection. BMC Gastroenterol. 2016, 16, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.-J.; Yu, S.J.; Na, J.; Kim, K.; Cho, Y.Y.; Lee, Y.B.; Cho, E.J.; Lee, J.-H.; Kim, Y.J.; Youn, H.; et al. Hexokinase-II Inhibition Synergistically Augments the Anti-tumor Efficacy of Sorafenib in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.G.; Han, Z.T.; He, S.H.; Wu, X.D.; Chen, T.R.; Shao, C.H.; Chen, D.L.; Su, N.; Chen, Y.M.; Wang, T.; et al. HIF1/2alpha mediates hypoxia-induced LDHA expression in human pancreatic cancer cells. Oncotarget 2017, 8, 24840–24852. [Google Scholar] [CrossRef]
- Chen, H.-L.; Ouyang, H.-Y.; Le, Y.; Jiang, P.; Tang, H.; Yu, Z.-S.; He, M.-K.; Tang, Y.-Q.; Shi, M. Aberrant MCT4 and GLUT1 expression is correlated with early recurrence and poor prognosis of hepatocellular carcinoma after hepatectomy. Cancer Med. 2018, 7, 5339–5350. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Lai, R.K.-H.; Xu, I.M.-J.; Chiu, D.K.-C.; Tse, A.P.-W.; Wei, L.L.; Law, D.C.-T.; Lee, D.; Wong, C.-M.; Wong, M.P.; Ming-Jing, X.I.; et al. NDUFA4L2 Fine-tunes Oxidative Stress in Hepatocellular Carcinoma. Clin. Cancer Res. 2016, 22, 3105–3117. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Chiu, D.K.-C.; Tse, A.P.-W.; Law, C.-T.; Xu, I.M.-J.; Lee, D.; Chen, M.; Lai, R.K.-H.; Yuen, V.W.-H.; Cheu, J.W.-S.; Ho, D.W.-H.; et al. Hypoxia regulates the mitochondrial activity of hepatocellular carcinoma cells through HIF/HEY1/PINK1 pathway. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Wu, X.; Wang, J.; Liu, F.; Ao, P.; Li, X.; Zheng, H.; Wu, D.; Zhang, N.; She, J.; Yuan, J. Correlation of PDK1 expression with clinicopathologic features and prognosis of hepatocellular carcinoma. OncoTargets Ther. 2016, 9, 5597–5602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, J.; Rong, Y.; Luo, C.L.; Li, S.; Jiang, X.; Weng, H.; Chen, H.; Zhang, W.W.; Xie, W.; Wang, F.B. Up-Regulation of hsa-miR-210 Promotes Venous Metastasis and Predicts Poor Prognosis in Hepatocellular Carcinoma. Front. Oncol. 2018, 8, 569. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Sun, Y.; Jiang, M.; Li, Y.; Tian, Y.; Xue, W.; Ding, N.; Cheng, C.; Li, J.; Miao, X.; et al. Glyceraldehyde-3-phosphate dehydrogenase promotes liver tumorigenesis by modulating phosphoglycerate dehydrogenase. Hepatology 2017, 66, 631–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Song, L.; Wan, Q.; Wu, G.; Li, X.; Wang, Y.; Wang, J.; Liu, Z.; Zhong, X.; He, X.; et al. cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res. 2015, 25, 429–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Huang, Y.; Jiang, C.; Ou, H.; Guo, B.; Liao, H.; Li, X.; Yang, D. Methylenetetrahydrofolate dehydrogenase 2 overexpression is associated with tumor aggressiveness and poor prognosis in hepatocellular carcinoma. Dig. Liver Dis. 2016, 48, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Xu, I.M.-J.; Chiu, D.K.-C.; Lai, R.K.-H.; Tse, A.P.-W.; Li, L.L.; Law, C.-T.; Tsang, F.H.-C.; Wei, L.L.; Chan, C.Y.-K.; et al. Folate cycle enzyme MTHFD1L confers metabolic advantages in hepatocellular carcinoma. J. Clin. Investig. 2017, 127, 1856–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wang, L.; Chen, X.; Li, L.; Li, Y.; Ping, Y.; Huang, L.; Yue, D.; Zhang, Z.; Wang, F.; et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-beta-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 2017, 6, e1320011. [Google Scholar] [CrossRef] [Green Version]
- Chiu, D.K.-C.; Tse, A.P.-W.; Xu, I.M.-J.; Di Cui, J.; Lai, R.K.-H.; Li, L.L.; Koh, H.-Y.; Tsang, F.H.-C.; Wei, L.L.; Wong, C.-M.; et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat. Commun. 2017, 8, 517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.L.; Shen, M.N.; Hu, B.; Wang, B.L.; Yang, W.J.; Lv, L.H.; Wang, H.; Zhou, Y.; Jin, A.L.; Sun, Y.F.; et al. CD73 promotes hepatocellular carcinoma progression and metastasis via activating PI3K/AKT signaling by inducing Rap1-mediated membrane localization of P110beta and predicts poor prognosis. J. Hematol. Oncol. 2019, 12, 37. [Google Scholar] [CrossRef]
- Cao, X.; Fang, L.; Gibbs, S.; Huang, Y.; Dai, Z.; Wen, P.; Zheng, X.; Sadee, W.; Sun, D. Glucose uptake inhibitor sensitizes cancer cells to daunorubicin and overcomes drug resistance in hypoxia. Cancer Chemother. Pharmacol. 2006, 59, 495–505. [Google Scholar] [CrossRef]
- Shen, Y.-C.; Ou, D.-L.; Hsu, C.; Lin, K.-L.; Chang, C.-Y.; Lin, C.-Y.; Liu, S.-H.; Cheng, A.-L. Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma. Br. J. Cancer 2012, 108, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Zheng, T.; Song, R.; Wang, J.; Yin, D.; Wang, L.; Liu, H.; Tian, L.; Fang, X.; Meng, X.; et al. Hypoxia-mediated sorafenib resistance can be overcome by EF24 through Von Hippel-Lindau tumor suppressor-dependent HIF-1α inhibition in hepatocellular carcinoma. Hepatology 2013, 57, 1847–1857. [Google Scholar] [CrossRef]
- Tesori, V.; Piscaglia, A.C.; Samengo, D.; Barba, M.; Bernardini, C.; Scatena, R.; Pontoglio, A.; Castellini, L.; Spelbrink, J.; Maulucci, G.; et al. The multikinase inhibitor Sorafenib enhances glycolysis and synergizes with glycolysis blockade for cancer cell killing. Sci. Rep. 2015, 5, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, R.; Wani, N.; Ghoshal, K.; Jacob, S.T.; Motiwala, T. Sorafenib and 2-Deoxyglucose Synergistically Inhibit Proliferation of Both Sorafenib-Sensitive and -Resistant HCC Cells by Inhibiting ATP Production. Gene Expr. 2017, 17, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Lee, D.; Law, C.-T.; Zhang, M.S.; Shen, J.; Chin, D.W.-C.; Zhang, A.; Tsang, F.H.-C.; Wong, C.L.-S.; Ng, I.O.-L.; et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Urasaki, Y.; Heath, L.; Xu, C.W. Coupling of Glucose Deprivation with Impaired Histone H2B Monoubiquitination in Tumors. PLoS ONE 2012, 7, e36775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Rathmell, J.C.; MacIntyre, A.N. Metabolic Reprogramming towards Aerobic Glycolysis Correlates with Greater Proliferative Ability and Resistance to Metabolic Inhibition in CD8 versus CD4 T Cells. PLoS ONE 2014, 9, e104104. [Google Scholar] [CrossRef] [Green Version]
- Keating, S.E.; Zaiatz-Bittencourt, V.; Loftus, R.M.; Keane, C.; Brennan, K.; Finlay, D.K.; Gardiner, C.M. Metabolic Reprogramming Supports IFN-gamma Production by CD56bright NK Cells. J. Immunol. 2016, 196, 2552–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Guan, D.; Wang, S.; Chai, L.Y.A.; Xu, S.; Lam, K.-P. Glycolysis and Oxidative Phosphorylation Play Critical Roles in Natural Killer Cell Receptor-Mediated Natural Killer Cell Functions. Front. Immunol. 2020, 11, 202. [Google Scholar] [CrossRef] [Green Version]
- Escobar, G.; Mangani, D.; Anderson, A.C. T cell factor 1: A master regulator of the T cell response in disease. Sci. Immunol. 2020, 5, eabb9726. [Google Scholar] [CrossRef] [PubMed]
- Bannoud, N.; Dalotto-Moreno, T.; Kindgard, L.; García, P.A.; Blidner, A.G.; Mariño, K.V.; Rabinovich, G.A.; Croci, D.O. Hypoxia Supports Differentiation of Terminally Exhausted CD8 T Cells. Front. Immunol. 2021, 12, 660944. [Google Scholar] [CrossRef]
- Mah-Som, A.; Rashidi, A.; Keppel, M.P.; Saucier, N.; Moore, E.; Alinger, J.B.; Tripathy, S.K.; Agarwal, S.K.; Jeng, E.K.; Wong, H.C.; et al. Glycolytic requirement for NK cell cytotoxicity and cytomegalovirus control. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- DE LA Cruz, K.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [Green Version]
- Sheng, S.L.; Liu, J.J.; Dai, Y.H.; Sun, X.G.; Xiong, X.P.; Huang, G. Knockdown of lactate dehydrogenase A suppresses tumor growth and metastasis of human hepatocellular carcinoma. FEBS J. 2012, 279, 3898–3910. [Google Scholar] [CrossRef]
- Lim, K.S.; Lim, K.J.; Price, A.C.; A Orr, B.; Eberhart, C.G.; Bar, E.E. Inhibition of monocarboxylate transporter-4 depletes stem-like glioblastoma cells and inhibits HIF transcriptional response in a lactate-independent manner. Oncogene 2013, 33, 4433–4441. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Yang, H.; Zhang, Y.; Wei, H.; Zhu, Z.; Zhu, B.; Yang, M.; Cao, W.; Wang, L.; Wu, Z. Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene 2017, 36, 5829–5839. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Völkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Harmon, C.; Robinson, M.W.; Hand, F.; AlMuaili, D.; Mentor, K.; Houlihan, D.D.; Hoti, E.; Lynch, L.; Geoghegan, J.; O’Farrelly, C. Lactate-Mediated Acidification of Tumor Microenvironment Induces Apoptosis of Liver-Resident NK Cells in Colorectal Liver Metastasis. Cancer Immunol. Res. 2018, 7, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zuo, H.; Xiong, H.; Kolar, M.; Chu, Q.; Saghatelian, A.; Siegwart, D.J.; Wan, Y. Gpr132 sensing of lactate mediates tumor–macrophage interplay to promote breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, 580–585. [Google Scholar] [CrossRef] [Green Version]
- Comito, G.; Iscaro, A.; Bacci, M.; Morandi, A.; Ippolito, L.; Parri, M.; Montagnani, I.; Raspollini, M.R.; Serni, S.; Simeoni, L.; et al. Lactate modulates CD4+ T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene 2019, 38, 3681–3695. [Google Scholar] [CrossRef] [PubMed]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-Derived Lactate Modifies Antitumor Immune Response: Effect on Myeloid-Derived Suppressor Cells and NK Cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.J.; Vignali, P.D.A.; Mullett, S.J.; Overacre-Delgoffe, A.E.; Peralta, R.M.; Grebinoski, S.; Menk, A.V.; Rittenhouse, N.L.; DePeaux, K.; Whetstone, R.D.; et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nat. Cell Biol. 2021, 591, 645–651. [Google Scholar] [CrossRef]
- Renner, K.; Bruss, C.; Schnell, A.; Koehl, G.; Becker, H.M.; Fante, M.; Menevse, A.-N.; Kauer, N.; Blazquez, R.; Hacker, L.; et al. Restricting Glycolysis Preserves T Cell Effector Functions and Augments Checkpoint Therapy. Cell Rep. 2019, 29, 135–150.e9. [Google Scholar] [CrossRef] [Green Version]
- Daneshmandi, S.; Wegiel, B.; Seth, P. Blockade of Lactate Dehydrogenase-A (LDH-A) Improves Efficacy of Anti-Programmed Cell Death-1 (PD-1) Therapy in Melanoma. Cancers 2019, 11, 450. [Google Scholar] [CrossRef]
- Dupuy, F.; Tabariès, S.; Andrzejewski, S.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; Gao, D.; Leung, S.; Amir, E.; et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab. 2015, 22, 577–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFate, T.; Mohyeldin, A.; Lu, H.; Thakar, J.; Henriques, J.; Halim, N.D.; Wu, H.; Schell, M.J.; Tsang, T.M.; Teahan, O.; et al. Pyruvate Dehydrogenase Complex Activity Controls Metabolic and Malignant Phenotype in Cancer Cells. J. Biol. Chem. 2008, 283, 22700–22708. [Google Scholar] [CrossRef] [Green Version]
- Tello, D.; Balsa, E.; Acosta-Iborra, B.; Fuertes-Yebra, E.; Elorza, A.; Ordóñez, Á.; Corral-Escariz, M.; Soro, I.; Lopez, J.A.; Perales-Clemente, E.; et al. Induction of the Mitochondrial NDUFA4L2 Protein by HIF-1α Decreases Oxygen Consumption by Inhibiting Complex I Activity. Cell Metab. 2011, 14, 768–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, P.-H.; Huang, S.-C.; Lim, S.-N.; Chou, C.-H.; Yeh, T.-S.; Chen, T.-C.; Yen, T.-H.; Su, M.-Y.; Chiu, C.-T.; Yeh, C.-T.; et al. Complex IV subunit 1 defect predicts postoperative survival in hepatocellular carcinoma. Oncol. Lett. 2014, 7, 1430–1438. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Kasim, V.; Yan, X.; Li, L.; Meliala, I.T.S.; Huang, C.; Li, Z.; Lei, K.; Song, G.; Zheng, X.; et al. Yin Yang 1 facilitates hepatocellular carcinoma cell lipid metabolism and tumor progression by inhibiting PGC-1beta-induced fatty acid oxidation. Theranostics 2019, 9, 7599–7615. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.L.; Lorenz, N.I.; Klann, K.; Münch, C.; Depner, C.; Steinbach, J.P.; Ronellenfitsch, M.W.; Luger, A.-L. Serine-dependent redox homeostasis regulates glioblastoma cell survival. Br. J. Cancer 2020, 122, 1391–1398. [Google Scholar] [CrossRef]
- Ye, J.; Fan, J.; Venneti, S.; Wan, Y.-W.; Pawel, B.R.; Zhang, J.; Finley, L.W.; Lu, C.; Lindsten, T.; Cross, J.; et al. Serine Catabolism Regulates Mitochondrial Redox Control during Hypoxia. Cancer Discov. 2014, 4, 1406–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- To, W.L.; Kumar, P.; Marshall, J. Hypoxia is an effective stimulus for vesicular release of ATP from human umbilical vein endothelial cells. Placenta 2015, 36, 759–766. [Google Scholar] [CrossRef] [Green Version]
- Di Virgilio, F.; Adinolfi, E. Extracellular purines, purinergic receptors and tumor growth. Oncogene 2017, 36, 293–303. [Google Scholar] [CrossRef]
- Vaupel, P.; Mayer, A. Hypoxia-Driven Adenosine Accumulation: A Crucial Microenvironmental Factor Promoting Tumor Progression. In Advances in Experimental Medicine and Biology; Elwell, C.E., Leung, T.S., Harrison, D.K., Eds.; Springer Science and Business Media LLC: Berlin, Germany, 2016; Volume 876, pp. 177–183. [Google Scholar]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [Green Version]
- Beavis, P.A.; Divisekera, U.; Paget, C.; Chow, M.T.; John, L.B.; Devaud, C.; Dwyer, K.; Stagg, J.; Smyth, M.J.; Darcy, P.K. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 14711–14716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagg, J.; Divisekera, U.; Duret, H.; Sparwasser, T.; Teng, M.; Darcy, P.; Smyth, M. CD73-Deficient Mice Have Increased Antitumor Immunity and Are Resistant to Experimental Metastasis. Cancer Res. 2011, 71, 2892–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayan, D.; Barkauskas, D.S.; Stannard, K.; Sult, E.; Buonpane, R.; Takeda, K.; Teng, M.W.L.; Sachsenmeier, K.; Hay, C.; Smyth, M.J. Selective activation of anti-CD73 mechanisms in control of primary tumors and metastases. Oncoimmunology 2017, 6, e1312044. [Google Scholar] [CrossRef]
- Stagg, J.; Beavis, P.; Divisekera, U.; Liu, M.C.; Möller, A.; Darcy, P.; Smyth, M.; Diaconu, I.; Cerullo, V.; Hirvinen, M.L.; et al. CD73-Deficient Mice Are Resistant to Carcinogenesis. Cancer Res. 2012, 72, 2190–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, C.; Miele, L.; Porta, A.; Pinto, A.; Morello, S. Myeloid-derived suppressor cells contribute to A2B adenosine receptor-induced VEGF production and angiogenesis in a mouse melanoma model. Oncotarget 2015, 6, 27478–27489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csoka, B.; Selmeczy, Z.; Koscso, B.; Nemeth, Z.H.; Pacher, P.; Murray, P.J.; Kepka-Lenhart, D.; Morris, S.M.; Gause, W.C.; Leibovich, S.J.; et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. 2012, 26, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Perrot, I.; Michaud, H.-A.; Giraudon-Paoli, M.; Augier, S.; Docquier, A.; Gros, L.; Courtois, R.; Déjou, C.; Jecko, D.; Becquart, O.; et al. Blocking antibodies targeting the CD39/CD73 immunosuppressive pathway unleash immune responses in combination cancer therapies. Cell Rep. 2019, 27, 2411–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, D.K.-C.; Xu, I.M.-J.; Lai, R.K.-H.; Tse, A.P.-W.; Wei, L.L.; Koh, H.-Y.; Li, L.L.; Lee, D.; Lo, R.C.-L.; Wong, C.-M.; et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology 2016, 64, 797–813. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Qian, D.Z.; Tan, Y.S.; Lee, K.; Gao, P.; Ren, Y.R.; Rey, S.; Hammers, H.; Chang, D.; Pili, R.; et al. Digoxin and other cardiac glycosides inhibit HIF-1 synthesis and block tumor growth. Proc. Natl. Acad. Sci. USA 2008, 105, 19579–19586. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.C.L.; Zhang, H.; Gilkes, D.M.; Chen, J.; Wei, H.; Chaturvedi, P.; Hubbi, M.E.; Semenza, G.L. Inhibitors of hypoxia-inducible factor 1 block breast cancer metastatic niche formation and lung metastasis. J. Mol. Med. 2012, 90, 803–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheuermann, T.H.; Tomchick, D.; Machius, M.; Guo, Y.; Bruick, R.K.; Gardner, K.H. Artificial ligand binding within the HIF2 PAS-B domain of the HIF2 transcription factor. Proc. Natl. Acad. Sci. USA 2009, 106, 450–455. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zheng, L.; Chen, J.; Sun, Y.; Lin, H.; Jin, R.-A.; Tang, M.; Liang, X.; Cai, X. Increasing AR by HIF-2α inhibitor (PT-2385) overcomes the side-effects of sorafenib by suppressing hepatocellular carcinoma invasion via alteration of pSTAT3, pAKT and pERK signals. Cell Death Dis. 2017, 8, e3095. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, S.; Bruick, R.K.; Josey, J.A.; et al. On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature 2016, 539, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.-L.; Liu, L.-P.; Niu, L.; Sun, Y.-F.; Yang, X.-R.; Fan, J.; Ren, J.-W.; Chen, G.G.; Lai, P.B. Downregulation and pro-apoptotic effect of hypoxia-inducible factor 2 alpha in hepatocellular carcinoma. Oncotarget 2016, 7, 34571–34581. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.-X.; Xu, Y.; Yang, X.-R.; Wang, W.-M.; Bai, H.; Shi, R.-Y.; Nayar, S.K.; Devbhandari, R.P.; He, Y.-Z.; Zhu, Q.; et al. Hypoxia inducible factor 2 alpha inhibits hepatocellular carcinoma growth through the transcription factor dimerization partner 3/ E2F transcription factor 1-dependent apoptotic pathway. Hepatology 2012, 57, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Blanco, C.; Fernández-Palanca, P.; Fondevila, F.; González-Gallego, J.; Mauriz, J.L. Prognostic and clinicopathological significance of hypoxia-inducible factors 1α and 2α in hepatocellular carcinoma: A systematic review with meta-analysis. Ther. Adv. Med. Oncol. 2021, 13, 1758835920987071. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dong, J.; Jia, L.; Zhao, T.; Lang, M.; Li, Z.; Lan, C.; Li, X.; Hao, J.; Wang, H.; et al. HIF-2-dependent expression of stem cell factor promotes metastasis in hepatocellular carcinoma. Cancer Lett. 2017, 393, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Bangoura, G.; Liu, Z.S.; Qian, Q.; Jiang, C.Q.; Yang, G.F.; Jing, S. Prognostic significance of HIF-2alpha/EPAS1 expression in hepatocellular carcinoma. World J. Gastroenterol. 2007, 13, 3176–3182. [Google Scholar] [CrossRef]
- Bao, M.H.-R.; Yang, C.; Tse, A.P.-W.; Wei, L.; Lee, D.; Zhang, M.S.; Goh, C.C.; Chiu, D.K.-C.; Yuen, V.W.-H.; Law, C.-T.; et al. Genome-wide CRISPR-Cas9 knockout library screening identified PTPMT1 in cardiolipin synthesis is crucial to survival in hypoxia in liver cancer. Cell Rep. 2021, 34, 108676. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic Pathways | Genes | HIF-Inducible | Expression in Human HCC |
|---|---|---|---|
| Glucose Metabolism (Activated) | GLUT1, GLUT3, HK2, ALDA and GAPDH | Yes [30] | Overexpressed [19,32,36,37,38,39,40] |
| PFKL, ALDC, TPI, PGK1, ENO1 and PKM | Yes [30] | Undetermined | |
| Lactate Metabolism (Activated) | LDHA and MCT4 | Yes [26,41,42] | Overexpressed [20,43] |
| Mitochondrial Metabolism (Suppressed) | PDK1, NDUFA4L2, COX4-2, miR-210, HEY1 | Yes [44,45,46,47,48,49] | Overexpressed [46,50,51] |
| MXI-1 | Yes [52] | Undetermined | |
| Serine Synthesis Pathway and Folate Cycle (Activated) | PHGDH, PSPH, SHMT2, MTHFD2 and MTHFD1L | Yes [28] | Overexpressed [53,54,55,56] |
| PSAT | Yes [28] | Undetermined | |
| Adenosinergic Metabolism (Activated) | CD39/CD39L1 and CD73 | Yes [57] | Overexpressed [58,59] |
| Inhibitors | Targets | Combination Treatment | Synergistic Effects on Hypoxic HCC |
|---|---|---|---|
| 2-DG | Glycolysis | Sorafenib (TKI) | Reduced cell viability, induced oxidative stress and apoptosis [63,64] |
| 3-BP | HK2 (glycolysis) | Sorafenib (TKI) | Reduced cell viability in vitro and suppressed HCC progression in vivo [40] |
| DCA | PDK1 (mitochondrial activity) | Sorafenib (TKI) | Promoted apoptosis and induced oxidative stress in vitro and suppress HCC progression in vivo [61] |
| NCT-503 | PHGDH (SSP) | Sorafenib, Regorafenib and Lenvatinib (TKIs) | Promoted apoptosis and induced oxidative stress in vitro and suppressed HCC progression in vivo [65] |
| POM-1 | CD39L1 (adenosinergic metabolism) | Anti-PD-1 and anti-CTLA-4 antibodies (ICIs) | Promoted lymphocyte infiltration and suppressed HCC progression in vivo [58] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bao, M.H.-R.; Wong, C.C.-L. Hypoxia, Metabolic Reprogramming, and Drug Resistance in Liver Cancer. Cells 2021, 10, 1715. https://doi.org/10.3390/cells10071715
Bao MH-R, Wong CC-L. Hypoxia, Metabolic Reprogramming, and Drug Resistance in Liver Cancer. Cells. 2021; 10(7):1715. https://doi.org/10.3390/cells10071715
Chicago/Turabian StyleBao, Macus Hao-Ran, and Carmen Chak-Lui Wong. 2021. "Hypoxia, Metabolic Reprogramming, and Drug Resistance in Liver Cancer" Cells 10, no. 7: 1715. https://doi.org/10.3390/cells10071715