
p66α Suppresses Breast Cancer Cell Growth and Migration by Acting as Co-Activator of p53

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Cell Culture, Transfections, and Viral Infection
2.3. Western Blot, Immunofluorescence, Co-IP and GST Pull Down
2.4. Reverse Transcription PCR (RT-PCR) and Quantitative Real-Time PCR (qRT-PCR)
2.5. Chromatin Immunoprecipitation (ChIP)
2.6. Luciferase Reporter Assay
2.7. Cell Counting Assay
2.8. Cell Migration Assay
2.9. Immunoprecipitation Mass Spectrometry
2.10. Subcutaneous Xenograft Assay
2.11. Lung Metastasis Assay and Hematoxylin and Eosin (H&E) Staining
2.12. Survival Analysis and Correlation Analysis
2.13. Statistical Analysis
3. Results
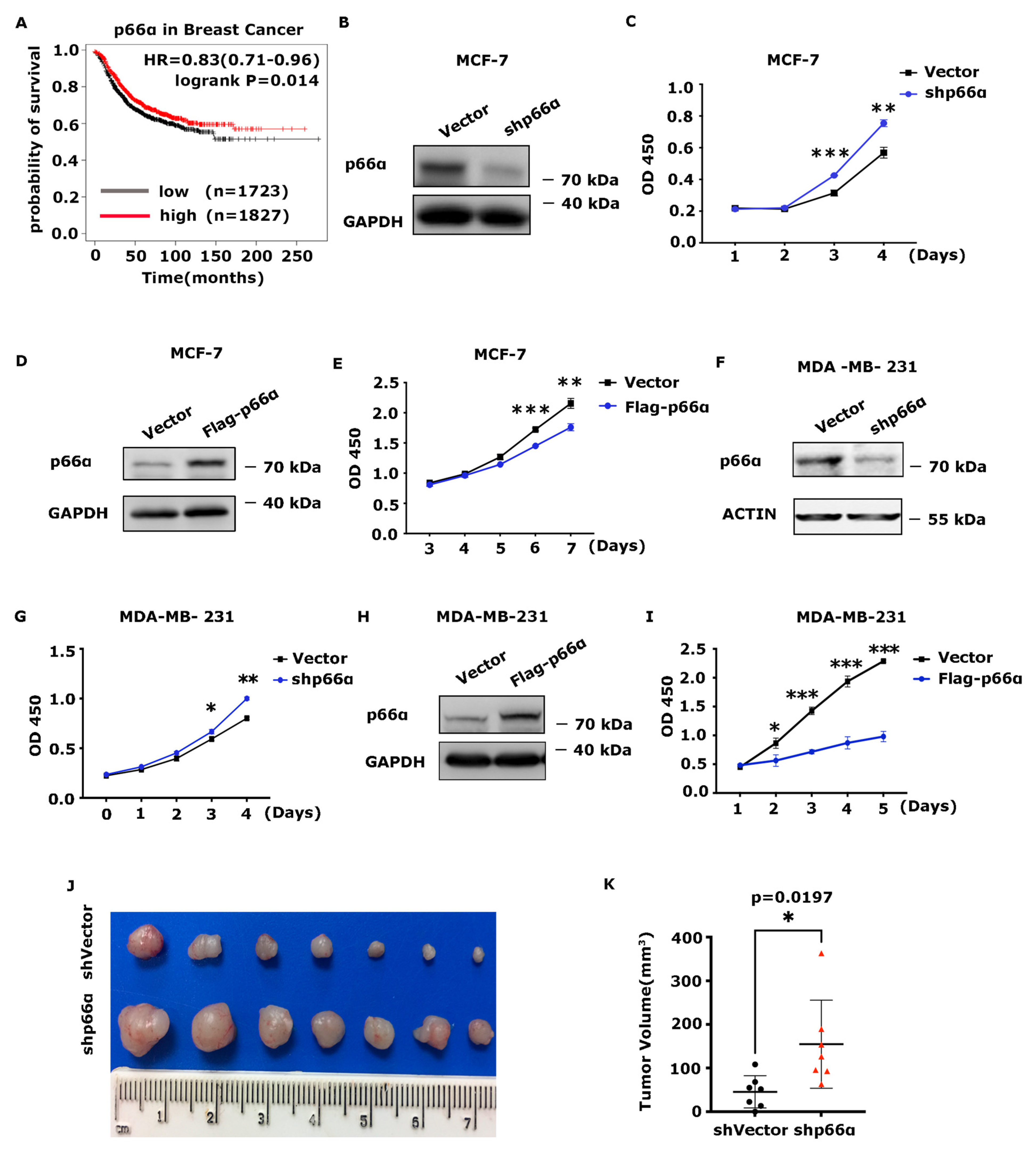
3.1. p66α Inhibits Breast Cancer Cell Growth
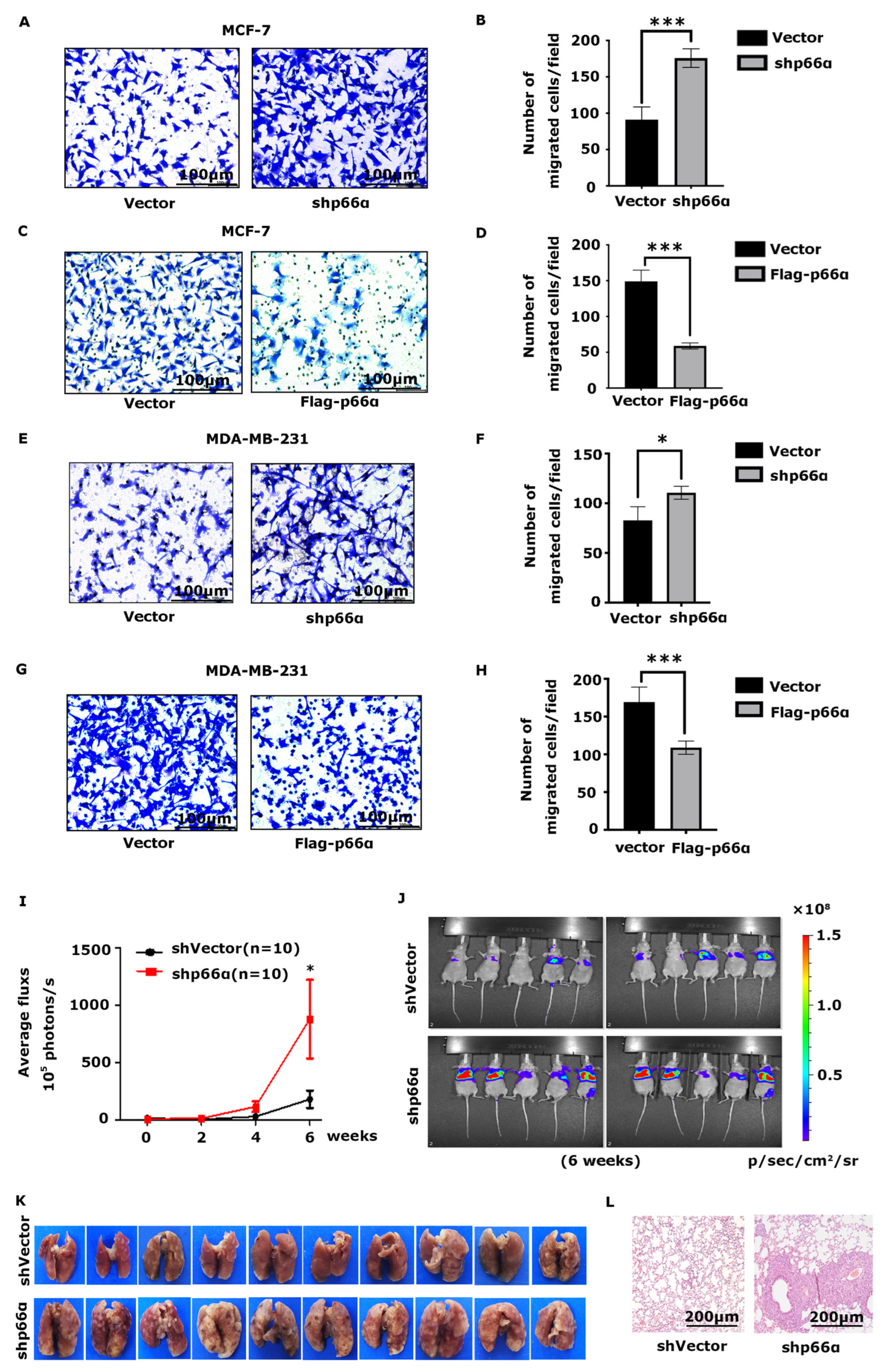
3.2. p66α Suppresses Breast Cancer Cell Migration and Metastasis
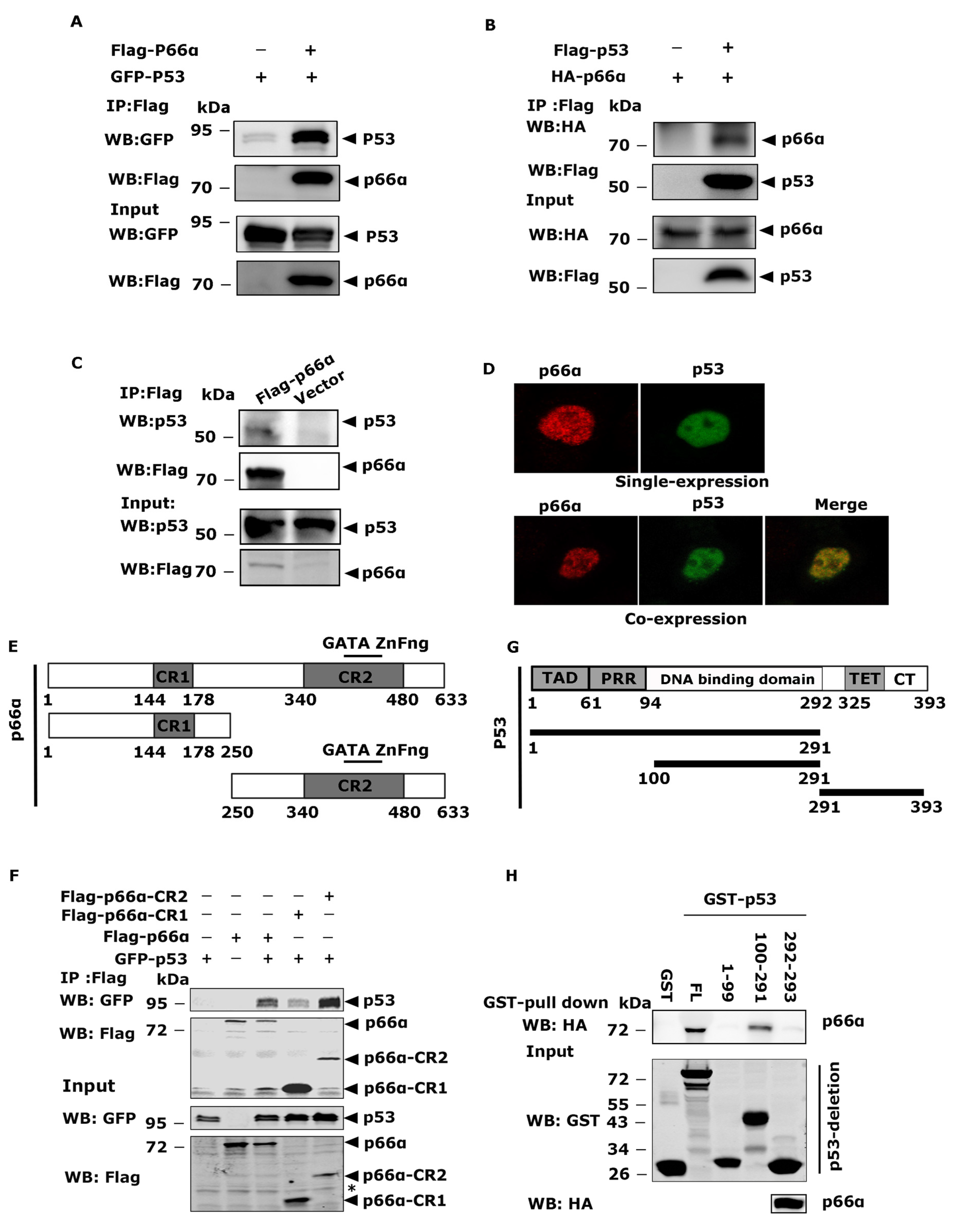
3.3. p66α Interacts with p53
3.4. The CR2 Domain of p66α and the DNA-Binding Domain of p53 Are Essential for the Interaction
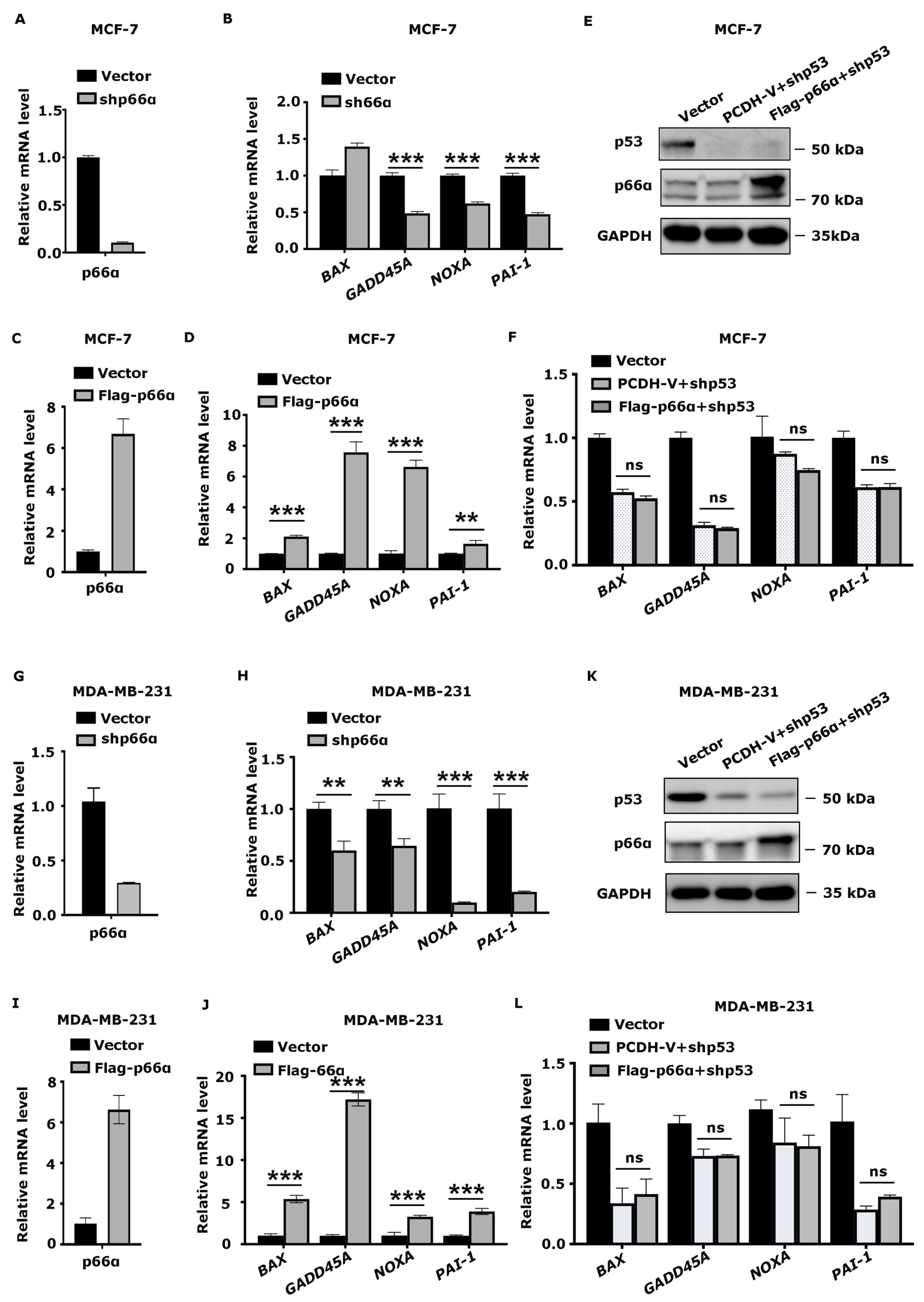
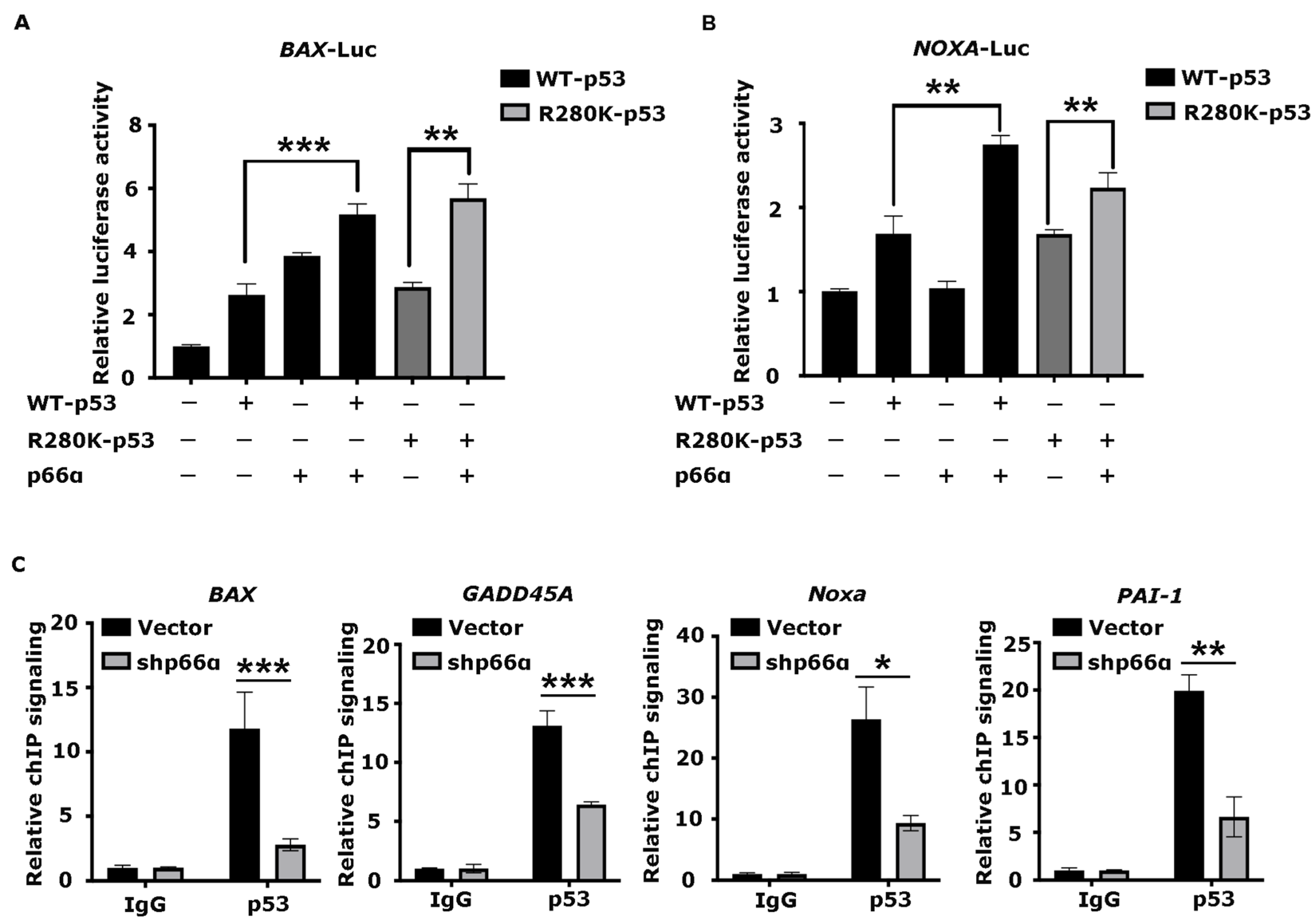
3.5. p66α Promotes the Expression of p53 Target Genes
3.6. p66α Promotes the Transactivity of p53 by Enhancing p53 Binding at Target Promoters
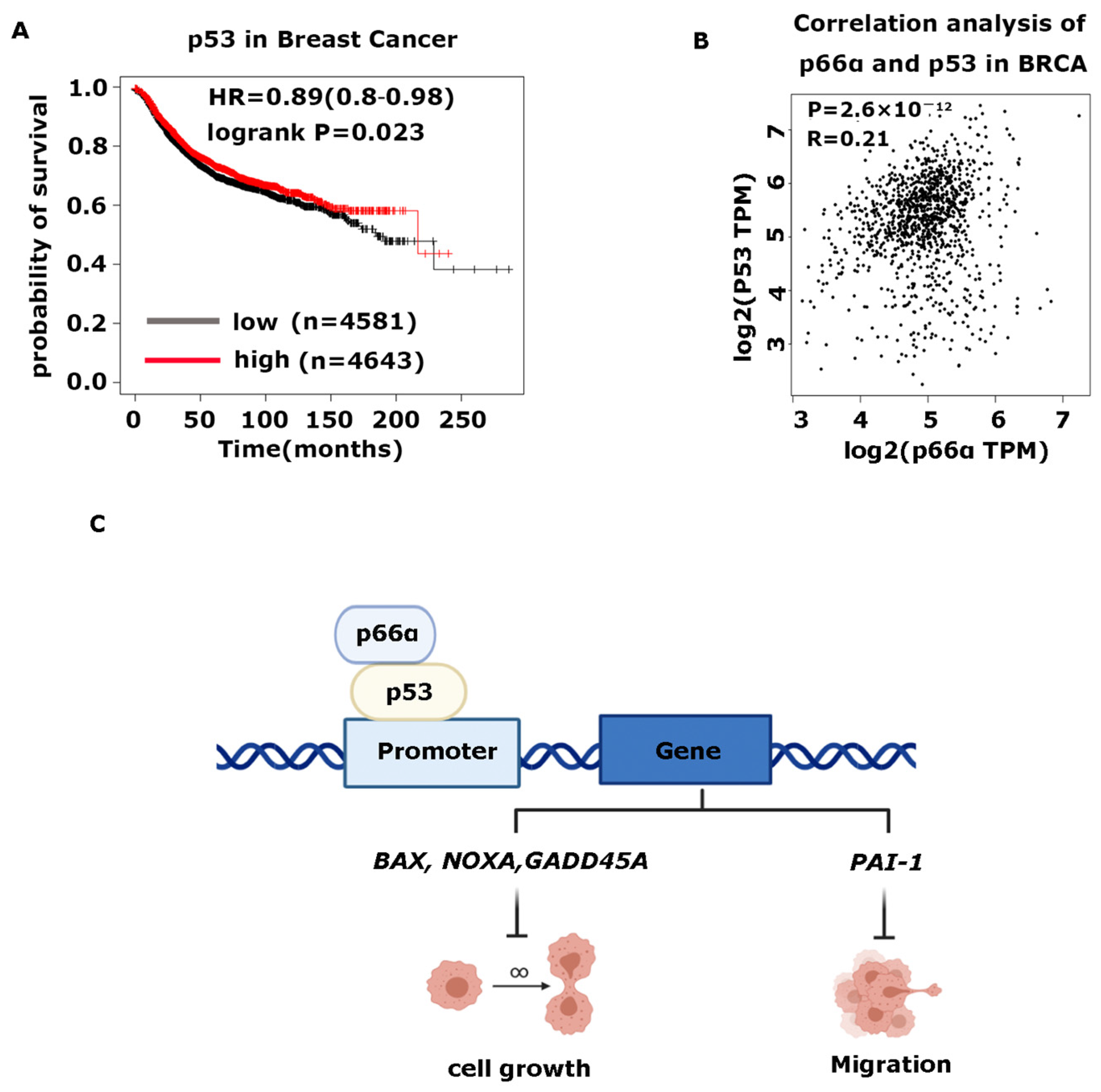
3.7. High Expression of p66α and p53 Is Positively Correlated and Predicts Good Prognosis in Breast Cancer
4. Discussion
4.1. p66α Functions as a Tumor Suppressor of Breast Cancer Cells
4.2. p66α Functions as a Potent Co-Activator of p53 by Enhancing Its DNA-Binding Activity
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brackertz, M.; Boeke, J.; Zhang, R.; Renkawitz, R. Two highly related p66 proteins comprise a new family of potent transcriptional repressors interacting with MBD2 and MBD3. J. Biol. Chem. 2002, 277, 40958–40966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eom, K.S.; Cheong, J.S.; Lee, S.J. Structural Analyses of Zinc Finger Domains for Specific Interactions with DNA. J. Microbiol. Biotechnol. 2016, 26, 2019–2029. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.; McLachlan, A.D.; Klug, A. Repetitive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes. EMBO J. 1985, 4, 1609–1614. [Google Scholar] [CrossRef]
- Schulz, R.A. GATA transcription factors in heart and blood cell development and disease. Semin. Cell Dev. Biol. 2005, 16, 69. [Google Scholar] [CrossRef]
- Patient, R.K.; McGhee, J.D. The GATA family (vertebrates and invertebrates). Curr. Opin. Genet. Dev. 2002, 12, 416–422. [Google Scholar] [CrossRef]
- Omichinski, J.G.; Clore, G.M.; Schaad, O.; Felsenfeld, G.; Trainor, C.; Appella, E.; Stahl, S.J.; Gronenborn, A.M. NMR structure of a specific DNA complex of Zn-containing DNA binding domain of GATA-1. Science 1993, 261, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Merika, M.; Orkin, S.H. DNA-binding specificity of GATA family transcription factors. Mol. Cell. Biol. 1993, 13, 3999–4010. [Google Scholar] [CrossRef]
- Kowalski, K.; Liew, C.K.; Matthews, J.M.; Gell, D.A.; Crossley, M.; Mackay, J.P. Characterization of the conserved interaction between GATA and FOG family proteins. J. Biol. Chem. 2002, 277, 35720–35729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.Y.; Evans, T. Distinct roles for the two cGATA-1 finger domains. Mol. Cell Biol. 1992, 12, 4562–4570. [Google Scholar] [CrossRef] [PubMed]
- Brackertz, M.; Gong, Z.; Leers, J.; Renkawitz, R. p66alpha and p66beta of the Mi-2/NuRD complex mediate MBD2 and histone interaction. Nucleic Acids Res. 2006, 34, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Torchy, M.P.; Hamiche, A.; Klaholz, B.P. Structure and function insights into the NuRD chromatin remodeling complex. Cell Mol. Life Sci. 2015, 72, 2491–2507. [Google Scholar] [CrossRef]
- Ding, X.; Liu, S.; Tian, M.; Zhang, W.; Zhu, T.; Li, D.; Wu, J.; Deng, H.; Jia, Y.; Xie, W.; et al. Activity-induced histone modifications govern Neurexin-1 mRNA splicing and memory preservation. Nat. Neurosci. 2017, 20, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Kim, J.S.; Son, S.H.; Lim, C.S.; Eum, H.Y.; Ha, D.H.; Park, M.A.; Baek, E.J.; Ryu, B.Y.; Kang, H.C.; et al. Mbd2-CP2c loop drives adult-type globin gene expression and definitive erythropoiesis. Nucleic Acids Res. 2018, 46, 4933–4949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mor, N.; Rais, Y.; Sheban, D.; Peles, S.; Aguilera-Castrejon, A.; Zviran, A.; Elinger, D.; Viukov, S.; Geula, S.; Krupalnik, V.; et al. Neutralizing Gatad2a-Chd4-Mbd3/NuRD Complex Facilitates Deterministic Induction of Naive Pluripotency. Cell Stem Cell 2018, 23, 412–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pergola, G.; Di Carlo, P.; D’Ambrosio, E.; Gelao, B.; Fazio, L.; Papalino, M.; Monda, A.; Scozia, G.; Pietrangelo, B.; Attrotto, M.; et al. DRD2 co-expression network and a related polygenic index predict imaging, behavioral and clinical phenotypes linked to schizophrenia. Transl. Psychiatry 2017, 7, e1006. [Google Scholar] [CrossRef] [PubMed]
- Torretta, S.; Rampino, A.; Basso, M.; Pergola, G.; Di Carlo, P.; Shin, J.H.; Kleinman, J.E.; Hyde, T.M.; Weinberger, D.R.; Masellis, R.; et al. NURR1 and ERR1 Modulate the Expression of Genes of a DRD2 Coexpression Network Enriched for Schizophrenia Risk. J. Neurosci. 2020, 40, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kang, J.; Deng, X.; Guo, B.; Wu, B.; Fan, Y. Knockdown of GATAD2A suppresses cell proliferation in thyroid cancer in vitro. Oncol. Rep. 2017, 37, 2147–2152. [Google Scholar] [CrossRef] [PubMed]
- Whitton, L.; Apostolova, G.; Rieder, D.; Dechant, G.; Rea, S.; Donohoe, G.; Morris, D.W. Genes regulated by SATB2 during neurodevelopment contribute to schizophrenia and educational attainment. PLoS Genet. 2018, 14, e1007515. [Google Scholar] [CrossRef]
- Yu, T.; Ding, Y.; Zhang, Y.; Liu, Y.; Li, Y.; Lei, J.; Zhou, J.; Song, S.; Hu, B. Circular RNA GATAD2A promotes H1N1 replication through inhibiting autophagy. Vet. Microbiol. 2019, 231, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Zviran, A.; Mor, N.; Rais, Y.; Gingold, H.; Peles, S.; Chomsky, E.; Viukov, S.; Buenrostro, J.D.; Scognamiglio, R.; Weinberger, L.; et al. Deterministic Somatic Cell Reprogramming Involves Continuous Transcriptional Changes Governed by Myc and Epigenetic-Driven Modules. Cell Stem Cell 2019, 24, 328–341.e329. [Google Scholar] [CrossRef] [Green Version]
- Gnanapragasam, M.N.; Scarsdale, J.N.; Amaya, M.L.; Webb, H.D.; Desai, M.A.; Walavalkar, N.M.; Wang, S.Z.; Zu Zhu, S.; Ginder, G.D.; Williams, D.C., Jr. p66Alpha-MBD2 coiled-coil interaction and recruitment of Mi-2 are critical for globin gene silencing by the MBD2-NuRD complex. Proc. Natl. Acad. Sci. USA 2011, 108, 7487–7492. [Google Scholar] [CrossRef] [Green Version]
- Spruijt, C.G.; Luijsterburg, M.S.; Menafra, R.; Lindeboom, R.G.; Jansen, P.W.; Edupuganti, R.R.; Baltissen, M.P.; Wiegant, W.W.; Voelker-Albert, M.C.; Matarese, F.; et al. ZMYND8 Co-localizes with NuRD on Target Genes and Regulates Poly(ADP-Ribose)-Dependent Recruitment of GATAD2A/NuRD to Sites of DNA Damage. Cell Rep. 2016, 17, 783–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Zhang, X.; Liu, Y.; Wang, L.; Ye, Y.; Tan, X.; Pu, J.; Zhang, Q.; Bao, X.; Wei, X.; et al. GATA zinc finger domain-containing protein 2A (GATAD2A) deficiency reactivates fetal haemoglobin in patients with beta-thalassaemia through impaired formation of methyl-binding domain protein 2 (MBD2)-containing nucleosome remodelling and deacetylation (NuRD) complex. Br. J. Haematol. 2021, 193, 1220–1227. [Google Scholar] [CrossRef]
- Gong, Z.; Brackertz, M.; Renkawitz, R. SUMO modification enhances p66-mediated transcriptional repression of the Mi-2/NuRD complex. Mol. Cell Biol. 2006, 26, 4519–4528. [Google Scholar] [CrossRef] [Green Version]
- Kantor, B.; Makedonski, K.; Shemer, R.; Razin, A. Expression and localization of components of the histone deacetylases multiprotein repressory complexes in the mouse preimplantation embryo. Gene Expr. Patterns 2003, 3, 697–702. [Google Scholar] [CrossRef]
- Marino, S.; Nusse, R. Mutants in the mouse NuRD/Mi2 component P66alpha are embryonic lethal. PLoS ONE 2007, 2, e519. [Google Scholar] [CrossRef] [Green Version]
- Xin, J.; Zhang, Z.; Su, X.; Wang, L.; Zhang, Y.; Yang, R. Epigenetic Component p66a Modulates Myeloid-Derived Suppressor Cells by Modifying STAT3. J. Immunol. 2017, 198, 2712–2720. [Google Scholar] [CrossRef] [Green Version]
- Kar, S.P.; Beesley, J.; Amin Al Olama, A.; Michailidou, K.; Tyrer, J.; Kote-Jarai, Z.; Lawrenson, K.; Lindstrom, S.; Ramus, S.J.; Thompson, D.J.; et al. Genome-Wide Meta-Analyses of Breast, Ovarian, and Prostate Cancer Association Studies Identify Multiple New Susceptibility Loci Shared by at Least Two Cancer Types. Cancer Discov. 2016, 6, 1052–1067. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Song, J.; Lu, Y.; Fall, K.; Chen, X.; Fang, F.; Landén, M.; Hultman, C.M.; Czene, K.; Sullivan, P.; et al. A shared genetic contribution to breast cancer and schizophrenia. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shu, K.X.; Li, B.; Wu, L.X. The p53 network: p53 and its downstream genes. Colloids Surf. B Biointerfaces 2007, 55, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Prives, C.; Hall, P.A. The p53 pathway. J. Pathol. 1999, 187, 112–126. [Google Scholar] [CrossRef]
- Agarwal, M.L.; Agarwal, A.; Taylor, W.R.; Stark, G.R. p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc. Natl. Acad. Sci. USA 1995, 92, 8493–8497. [Google Scholar] [CrossRef] [Green Version]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H.; Norman, J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell. Biol. 2011, 192, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.; Piwnica-Worms, D.; Piwnica-Worms, H. Contribution of p53 to metastasis. Cancer Discov. 2014, 4, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teodoro, J.G.; Evans, S.K.; Green, M.R. Inhibition of tumor angiogenesis by p53: A new role for the guardian of the genome. J. Mol. Med. 2007, 85, 1175–1186. [Google Scholar] [CrossRef]
- Walerych, D.; Napoli, M.; Collavin, L.; Del Sal, G. The rebel angel: Mutant p53 as the driving oncogene in breast cancer. Carcinogenesis 2012, 33, 2007–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef]
- Resnick, M.A.; Inga, A. Functional mutants of the sequence-specific transcription factor p53 and implications for master genes of diversity. Proc. Natl. Acad. Sci. USA 2003, 100, 9934–9939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, J.J.; Inga, A.; Conway, K.; Edmiston, S.; Carey, L.A.; Wu, L.; Resnick, M.A. Altered-Function p53 Missense Mutations Identified in Breast Cancers Can Have Subtle Effects on Transactivation. Mol. Cancer Res. 2010, 8, 701–716. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Han, S.Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424–8429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Deppert, W. Transcriptional activities of mutant p53: When mutations are more than a loss. J. Cell Biochem. 2004, 93, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Weisz, L.; Oren, M.; Rotter, V. Transcription regulation by mutant p53. Oncogene 2007, 26, 2202–2211. [Google Scholar] [CrossRef] [Green Version]
- Fontemaggi, G.; Dell’Orso, S.; Trisciuoglio, D.; Shay, T.; Melucci, E.; Fazi, F.; Terrenato, I.; Mottolese, M.; Muti, P.; Domany, E.; et al. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat. Struct. Mol. Biol. 2009, 16, U1086–U1111. [Google Scholar] [CrossRef]
- Girardini, J.E.; Napoli, M.; Piazza, S.; Rustighi, A.; Marotta, C.; Radaelli, E.; Capaci, V.; Jordan, L.; Quinlan, P.; Thompson, A.; et al. A Pin1/Mutant p53 Axis Promotes Aggressiveness in Breast Cancer. Cancer Cell 2011, 20, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Roger, L.; Jullien, L.; Gire, V.; Roux, P. Gain of oncogenic function of p53 mutants regulates E-cadherin expression uncoupled from cell invasion in colon cancer cells. J. Cell Sci. 2010, 123, 1295–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Z.; Peng, H.; Ayyanathan, K.; Yan, K.P.; Langer, E.M.; Longmore, G.D.; Rauscher, F.J., 3rd. The LIM protein AJUBA recruits protein arginine methyltransferase 5 to mediate SNAIL-dependent transcriptional repression. Mol. Cell Biol. 2008, 28, 3198–3207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Z.; Peng, H.; White, D.E.; Negorev, D.G.; Maul, G.G.; Feng, Y.; Longmore, G.D.; Waxman, S.; Zelent, A.; Rauscher, F.J., 3rd. LIM protein Ajuba functions as a nuclear receptor corepressor and negatively regulates retinoic acid signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 2938–2943. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.H.; Zou, X.Q.; Qian, W.L.; Weng, X.L.; Zhang, L.; Zhang, L.; Wang, S.; Cao, X.; Ma, L.; Wei, G.; et al. Enhanced PAPSS2/VCAN sulfation axis is essential for Snail-mediated breast cancer cell migration and metastasis. Cell Death Differ. 2019, 26, 565–579. [Google Scholar] [CrossRef] [Green Version]
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast cancer. Lancet 2021, 397, 1750–1769. [Google Scholar] [CrossRef]
- Barzaman, K.; Karami, J.; Zarei, Z.; Hosseinzadeh, A.; Kazemi, M.H.; Moradi-Kalbolandi, S.; Safari, E.; Farahmand, L. Breast cancer: Biology, biomarkers, and treatments. Int. Immunopharmacol. 2020, 84. [Google Scholar] [CrossRef] [PubMed]
- Avery-Kiejda, K.A.; Mathe, A.; Scott, R.J. Genome-wide miRNA, gene and methylation analysis of triple negative breast cancer to identify changes associated with lymph node metastases. Genom. Data 2017, 14, 1–4. [Google Scholar] [CrossRef]
- Sporikova, Z.; Koudelakova, V.; Trojanec, R.; Hajduch, M. Genetic Markers in Triple-Negative Breast Cancer. Clin. Breast Cancer 2018, 18, E841–E850. [Google Scholar] [CrossRef]
- Howard, F.M.; Olopade, O.I. Epidemiology of Triple-Negative Breast Cancer A Review. Cancer J. 2021, 27, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.D.; Noskov, S.Y.; Lim, C. Factors governing loss and rescue of DNA binding upon single and double mutations in the p53 core domain. Nucleic Acids Res. 2002, 30, 1563–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malcikova, J.; Tichy, B.; Damborsky, J.; Kabathova, J.; Trbusek, M.; Mayer, J.; Pospisilova, S. Analysis of the DNA-binding activity of p53 mutants using functional protein microarrays and its relationship to transcriptional activation. Biol. Chem. 2010, 391, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Boutell, J.M.; Hart, D.J.; Godber, B.L.; Kozlowski, R.Z.; Blackburn, J.M. Functional protein microarrays for parallel characterisation of p53 mutants. Proteomics 2004, 4, 1950–1958. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Zhang, Y.; Zhang, J.; Zhang, D.; Li, M.; Yan, H.; Zhang, H.; Song, L.; Wang, J.; Hou, Z.; et al. p66α Suppresses Breast Cancer Cell Growth and Migration by Acting as Co-Activator of p53. Cells 2021, 10, 3593. https://doi.org/10.3390/cells10123593
Zhang Q, Zhang Y, Zhang J, Zhang D, Li M, Yan H, Zhang H, Song L, Wang J, Hou Z, et al. p66α Suppresses Breast Cancer Cell Growth and Migration by Acting as Co-Activator of p53. Cells. 2021; 10(12):3593. https://doi.org/10.3390/cells10123593
Chicago/Turabian StyleZhang, Qun, Yihong Zhang, Jie Zhang, Dan Zhang, Mengying Li, Han Yan, Hui Zhang, Liwei Song, Jiamin Wang, Zhaoyuan Hou, and et al. 2021. "p66α Suppresses Breast Cancer Cell Growth and Migration by Acting as Co-Activator of p53" Cells 10, no. 12: 3593. https://doi.org/10.3390/cells10123593