
Cardiac Glycosides as Autophagy Modulators


Abstract
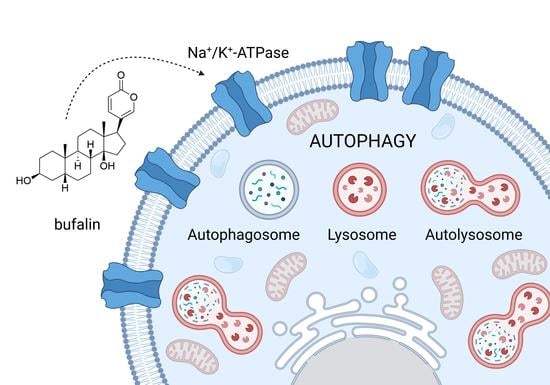
:
1. Introduction
2. Cardiac Glycosides and Sodium-Potassium ATPase
3. Autophagy
4. Cardiac Glycosides as Autophagy Modulators
4.1. Bufalin
4.2. Digoxin
4.3. Ouabain
4.4. Digitoxin
4.5. Lanatoside C, Strophantidin, Peruvoside, Convallatoxin
4.6. Oleandrin, Divaricoside, Proscillaridin A, Glucoevatromonoside, Neriifolin
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3-MA | 3-methyladenine |
| A2780 | Human cells from ovarian carcinoma |
| A549 | Human cells derived from non-small cell lung carcinoma |
| Akt | Protein kinase B |
| AMPK | Adenosine monophosphate-activated protein kinase |
| ATG | Autophagy-related protein |
| ATG5 | Autophagy-related gene 5 |
| ATG7 | Autophagy-related gene 7 |
| ATP | Adenosine triphosphate |
| Bcl2 | B-cell lymphoma 2 |
| BGC-823 | Human cells derived from gastric carcinoma |
| BiP | Binding immunoglobulin protein |
| BJ | Human primary fibroblasts from foreskin |
| BRAF | V-raf murine sarcoma viral oncogene homolog B1 |
| Caco-2 | Human cells from colorectal carcinoma |
| CGs | Cardiac glycosides |
| CHOP | CCAAT/enhancer-binding protein homologous protein |
| COVID-19 | Coronavirus disease 2019 |
| CT26 | N-nitroso-N-methylurethane-induced, undifferentiated colon carcinoma cell line |
| DAMPs | Damage(Danger)-associated molecular patterns |
| EGFR | The epidermal growth factor receptor |
| eIF2α | Eukaryotic translation initiation factor 2α |
| ER | Endoplasmic reticulum |
| ERK1/2 | Extracellular signal-regulated protein kinases 1 and 2 |
| GES-1 | Gastric mucous epithelium cells |
| Grb2 | Growth factor receptor-bound protein 2 |
| GRP78 | Binding immunoglobulin protein |
| H460 | Human cells derived from non-small cell lung carcinoma |
| HA22T | Human cells from hepatocellular carcinoma |
| HCCLM3 | Human cells from hepatocellular carcinoma |
| HCMV | Human cytomegalovirus |
| HCT 116 | Human cells from colorectal carcinoma |
| HeLa | Human cells derived from cervical carcinoma |
| Hep3B | Human cells from hepatocellular carcinoma |
| HepG2 | Human cells from hepatocellular carcinoma |
| HT-29 | Human cells from colorectal carcinoma |
| Huh7 | Human cells from hepatocellular carcinoma |
| IC50 | Half-maximal inhibitory concentrations |
| ICD | Immunogenic cell death |
| I/R | Ischemia/reperfusion |
| IGROV-1 | Human cells from ovarian carcinoma |
| IP3 | Inositol 1,4,5-trisphosphate |
| JNK | c-Jun N-terminal kinases |
| LC3-II | Microtubule-associated protein 1A/1B-light chain 3-II |
| LN-229 | Human cells derived from glioblastoma |
| MCF-7 | Human cells from breast carcinoma |
| Mcl-1 | Myeloid-cell leukemia 1 |
| MDA-MB-231 | Human cells from breast carcinoma |
| MEK | Mitogen-activated protein kinase |
| MEK 1/2 | Mitogen-activated protein kinase kinases 1 and 2 |
| MAPK | Mitogen-activated protein kinase |
| MHCC97H | Human cells derived from hepatocarcinoma |
| mTOR | Mammalian target of rapamycin |
| NHE | Na+/H+-exchanger |
| NF- κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NKA | Sodium-potassium ATPase |
| NMTC | Non-medullary thyroid cancer |
| OCCC | Ovarian clear cell carcinoma |
| PA | Palmitic acid |
| PANC-1 | Human cells from pancreatic carcinoma |
| PARP | Poly(adenosine diphosphate-ribose) polymerase |
| PAS | Phagophore assembly site |
| PERK | Protein kinase R-like endoplasmic reticulum kinase |
| PI3K | Phosphoinositide 3-kinase |
| PLC | Phospholipase C |
| PP2 | 1-tert-butyl-3-(4-chlorophenyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine) |
| Rab7 | Ras-related protein |
| Raf | Rapidly accelerated fibrosarcoma protein |
| Ras | Rat sarcoma virus protein |
| RAW 264.7 | Mouse macrophages |
| ROS | Reactive oxygen species |
| SCC2095 | Cells derived from oral squamous carcinoma |
| SGC 7901 | Human cells derived from gastric carcinoma |
| SH-SY5Y | Human cells from neuroblastoma |
| Shc | Src homology 2 domain-containing transforming protein |
| SK-OV-3 | Human cells from ovarian carcinoma |
| siRNA | Small interfering ribonucleic acid (siRNA |
| Sos | Son of sevenless protein |
| Src | Proto-oncogene tyrosine-protein kinase Src |
| Tau | Tubulin-associated unit |
| TFEB | Transcription factor EB |
| TOV-21G | Human cells from ovarian carcinoma |
| TRAIL | Tumor necrosis factor-related apoptosis-inducing ligand |
| U-2 OS | Human cells from osteosarcoma |
| U-87 MG | Human cells derived from glioblastoma |
| ULK1 | Unc-51 like autophagy activating kinase |
| Wnt | Wingless-related integration site protein |
References
- Rahib, L.; Wehner, M.R.; Matrisian, L.M.; Nead, K.T. Estimated projection of US cancer incidence and death to 2040. JAMA Network Open 2021, 4, e214708. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Bakowski, M.A.; Beutler, N.; Wolff, K.C.; Kirkpatrick, M.G.; Chen, E.; Nguyen, T.-T.H.; Riva, L.; Shaabani, N.; Parren, M.; Ricketts, J.; et al. Drug repurposing screens identify chemical entities for the development of COVID-19 interventions. Nat. Commun. 2021, 12, 3309. [Google Scholar] [CrossRef]
- Armando, R.G.; Mengual Gómez, D.L.; Gomez, D.E. New drugs are not enough-drug repositioning in oncology: An update. Int. J. Oncol. 2020, 56, 651–684. [Google Scholar] [CrossRef] [Green Version]
- Bessen, H.A. Therapeutic and toxic effects of digitalis: William Withering, 1785. J. Emerg. Med. 1986, 4, 243–248. [Google Scholar] [CrossRef]
- Kelly, R.A. Cardiac glycosides and congestive heart failure. Am. J. Cardiol. 1990, 65, E10–E16. [Google Scholar] [CrossRef]
- Shrestha, T.; Kopp, B.; Bisset, N.G. The Moraceae-based dart poisons of South America. Cardiac glycosides of Maquira and Naucleopsis species. J. Ethnopharmacol. 1992, 37, 129–143. [Google Scholar] [CrossRef]
- Fisch, C. William Withering: An account of the foxglove and some of its medical uses 1785-1985. J. Am. Coll. Cardiol. 1985, 5, 1A–2A. [Google Scholar] [CrossRef] [Green Version]
- Shiratori, O. Growth inhibitory effect of cardiac glycosides and aglycones on neoplastic cells: In vitro and in vivo studies. GANN Jpn. J. Cancer Res. 1967, 58, 521–528. [Google Scholar]
- Schönfeld, W.; Weiland, J.; Lindig, C.; Masnyk, M.; Kabat, M.M.; Kurek, A.; Wicha, J.; Repke, K.R. The lead structure in cardiac glycosides is 5 beta, 14 beta-androstane-3 beta 14-diol. Naunyn Schmiedebergs Arch. Pharmacol. 1985, 329, 414–426. [Google Scholar] [CrossRef]
- Melero, C.P.; Medarde, M.; San Feliciano, A. A short review on cardiotonic steroids and their aminoguanidine analogues. Molecules 2000, 5, 51–81. [Google Scholar] [CrossRef] [Green Version]
- Hollman, A. Plants and cardiac glycosides. Br. Heart J. 1985, 54, 258–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Luo, H.; Wang, H.; Hou, H. Preparative isolation of bufalin and cinobufagin from Chinese traditional medicine ChanSu. J. Chromatogr. Sci. 2008, 46, 81–85. [Google Scholar] [CrossRef] [Green Version]
- Skou, J.C. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim. Biophys. Acta 1957, 23, 394–401. [Google Scholar] [CrossRef]
- Bejček, J.; Spiwok, V.; Kmoníčková, E.; Rimpelová, S. Na+/K+-ATPase revisited: On its mechanism of action, role in cancer, and activity modulation. Molecules 2021, 26, 1905. [Google Scholar] [CrossRef]
- Geering, K. Functional roles of Na,K-ATPase subunits. Curr. Opin. Nephrol. Hypertens. 2008, 17, 526–532. [Google Scholar] [CrossRef]
- de Souza, W.F.; Barbosa, L.A.; Liu, L.; de Araujo, W.M.; de-Freitas-Junior, J.C.M.; Fortunato-Miranda, N.; Fontes, C.F.L.; Morgado-Díaz, J.A. Ouabain-induced alterations of the apical junctional complex involve α1 and β1 Na,K-ATPase downregulation and ERK1/2 activation independent of caveolae in colorectal cancer cells. J. Membr. Biol. 2014, 247, 23–33. [Google Scholar] [CrossRef]
- Geering, K. The functional role of beta subunits in oligomeric P-type ATPases. J. Bioenerg. Biomembr. 2001, 33, 425–438. [Google Scholar] [CrossRef]
- Clausen, M.V.; Hilbers, F.; Poulsen, H. The structure and function of the Na,K-ATPase isoforms in health and disease. Front. Physiol. 2017, 8, 371. [Google Scholar] [CrossRef]
- Béguin, P.; Wang, X.; Firsov, D.; Puoti, A.; Claeys, D.; Horisberger, J.D.; Geering, K. The gamma subunit is a specific component of the Na,K-ATPase and modulates its transport function. EMBO J. 1997, 16, 4250–4260. [Google Scholar] [CrossRef] [Green Version]
- Sweadner, K.J.; Rael, E. The FXYD gene family of small ion transport regulators or channels: cDNA sequence, protein signature sequence, and expression. Genomics 2000, 68, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, E.; Philipp, G.; Scholz, H. Cardiac glycoside receptor, (Na+ + K+)-ATPase activity and force of contraction in rat heart. Biochem. Pharmacol. 1980, 29, 3219–3229. [Google Scholar] [CrossRef]
- Grupp, I.; Im, W.B.; Lee, C.O.; Lee, S.W.; Pecker, M.S.; Schwartz, A. Relation of sodium pump inhibition to positive inotropy at low concentrations of ouabain in rat heart muscle. J. Physiol. 1985, 360, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, S.; Lindholm, P.; Gullbo, J.; Larsson, R.; Bohlin, L.; Claeson, P. Cytotoxicity of digitoxin and related cardiac glycosides in human tumor cells. Anti-cancer Drugs 2001, 12, 475–483. [Google Scholar] [CrossRef]
- Kaplan, J.G. Membrane cation transport and the control of proliferation of mammalian cells. Annu. Rev. Physiol. 1978, 40, 19–41. [Google Scholar] [CrossRef] [PubMed]
- Bortner, C.D.; Hughes, F.M., Jr.; Cidlowski, J.A. A primary role for K+ and Na+ efflux in the activation of apoptosis. J. Biol. Chem. 1997, 272, 32436–32442. [Google Scholar] [CrossRef] [Green Version]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef]
- Wingrave, J.M.; Schaecher, K.E.; Sribnick, E.A.; Wilford, G.G.; Ray, S.K.; Hazen-Martin, D.J.; Hogan, E.L.; Banik, N.L. Early induction of secondary injury factors causing activation of calpain and mitochondria-mediated neuronal apoptosis following spinal cord injury in rats. J. Neurosci. Res. 2003, 73, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Arisaka, H.; Ikeda, U.; Takayasu, T.; Takeda, K.; Natsume, T.; Hosoda, S. Ouabain inhibits intracellular pH recovery from acidosis in cultured mouse heart cells. J. Mol. Cell. Cardiol. 1988, 20, 1–3. [Google Scholar] [CrossRef]
- Rich, I.N.; Worthington-White, D.; Garden, O.A.; Musk, P. Apoptosis of leukemic cells accompanies reduction in intracellular pH after targeted inhibition of the Na+/H+ exchanger. Blood 2000, 95, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Tian, J.; Liu, L.; Pierre, S.; Liu, J.; Shapiro, J.; Xie, Z.J. Identification of a pool of non-pumping Na/K-ATPase. J. Biol. Chem. 2007, 282, 10585–10593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Cai, T. Na+-K+–ATPase-mediated signal transduction: From protein interaction to cellular function. Mol. Interv. 2003, 3, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Askari, A.; Xie, Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J. Biol. Chem. 2000, 275, 27832–27837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, M.; Wang, H.; Tian, J.; Xie, Z. Src-mediated inter-receptor cross-talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J. Biol. Chem. 2002, 277, 18694–18702. [Google Scholar] [CrossRef] [Green Version]
- Kometiani, P.; Liu, L.; Askari, A. Digitalis-induced signaling by Na+/K+-ATPase in human breast cancer cells. Mol. Pharmacol. 2005, 67, 929–936. [Google Scholar] [CrossRef] [Green Version]
- Liang, M.; Cai, T.; Tian, J.; Qu, W.; Xie, Z.J. Functional characterization of Src-interacting Na/K-ATPase using RNA interference assay. J. Biol. Chem. 2006, 281, 19709–19719. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Liu, J.; Garlid, K.D.; Shapiro, J.I.; Xie, Z. Involvement of mitogen-activated protein kinases and reactive oxygen species in the inotropic action of ouabain on cardiac myocytes. A potential role for mitochondrial K(ATP) channels. In Cardiac Cell Biology. Developments in Molecular and Cellular Biochemistry; Kardami, E., Hryshko, L., Mesaeli, N., Eds.; Springer: Boston, MA, USA, 2003; Volume 39, pp. 181–187. [Google Scholar] [CrossRef]
- Xie, Z.; Kometiani, P.; Liu, J.; Li, J.; Shapiro, J.I.; Askari, A. Intracellular reactive oxygen species mediate the linkage of Na+/K+-ATPase to hypertrophy and its marker genes in cardiac myocytes. J. Biol. Chem. 1999, 274, 19323–19328. [Google Scholar] [CrossRef] [Green Version]
- Dolmetsch, R.E.; Xu, K.; Lewis, R.S. Calcium oscillations increase the efficiency and specificity of gene expression. Nature 1998, 392, 933–936. [Google Scholar] [CrossRef]
- Miyakawa-Naito, A.; Uhlén, P.; Lal, M.; Aizman, O.; Mikoshiba, K.; Brismar, H.; Zelenin, S.; Aperia, A. Cell signaling microdomain with Na,K-ATPase and inositol 1,4,5-trisphosphate receptor generates calcium oscillations. J. Biol. Chem. 2003, 278, 50355–50361. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Cai, T.; Tian, J.; Ivanov, A.V.; Giovannucci, D.R.; Xie, Z. Na/K-ATPase tethers phospholipase C and IP3 receptor into a calcium-regulatory complex. Mol. Biol. Cell 2005, 16, 4034–4045. [Google Scholar] [CrossRef] [Green Version]
- Barwe, S.P.; Anilkumar, G.; Moon, S.Y.; Zheng, Y.; Whitelegge, J.P.; Rajasekaran, S.A.; Rajasekaran, A.K. Novel role for Na,K-ATPase in phosphatidylinositol 3-kinase signaling and suppression of cell motility. Mol. Biol. Cell 2005, 16, 1082–1094. [Google Scholar] [CrossRef]
- Navé, B.T.; Ouwens, M.; Withers, D.J.; Alessi, D.R.; Shepherd, P.R. Mammalian target of rapamycin is a direct target for protein kinase B: Identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem. J. 1999, 344, 427–431. [Google Scholar] [CrossRef]
- Wick, M.J.; Dong, L.Q.; Riojas, R.A.; Ramos, F.J.; Liu, F. Mechanism of phosphorylation of protein kinase B/Akt by a constitutively active 3-phosphoinositide-dependent protein kinase-1. J. Biol. Chem. 2000, 275, 40400–40406. [Google Scholar] [CrossRef] [Green Version]
- Yudowski, G.A.; Efendiev, R.; Pedemonte, C.H.; Katz, A.I.; Berggren, P.-O.; Bertorello, A.M. Phosphoinositide-3 kinase binds to a proline-rich motif in the Na+,K+-ATPase α subunit and regulates its trafficking. Proc. Natl. Acad. Sci. USA 2000, 97, 6556–6561. [Google Scholar] [CrossRef] [Green Version]
- Graef, M.; Friedman, J.R.; Graham, C.; Babu, M.; Nunnari, J. ER exit sites are physical and functional core autophagosome biogenesis components. Mol. Biol. Cell 2013, 24, 2918–2931. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Wijshake, T.; Zou, Z.; Chen, B.; Zhong, L.; Xiao, G.; Xie, Y.; Doench, J.G.; Bennett, L.; Levine, B. Tumor-suppressor function of Beclin 1 in breast cancer cells requires E-cadherin. Proc. Natl. Acad. Sci. USA 2021, 118, e2020478118. [Google Scholar] [CrossRef]
- Cho, D.H.; Jo, Y.K.; Kim, S.C.; Park, I.J.; Kim, J.C. Down-regulated expression of ATG5 in colorectal cancer. Anticancer Res. 2012, 32, 4091. [Google Scholar] [PubMed]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Cayo, A.; Segovia, R.; Venturini, W.; Moore-Carrasco, R.; Valenzuela, C.; Brown, N. mTOR activity and autophagy in senescent cells, a complex partnership. Int. J. Mol. Sci. 2021, 22, 8149. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Yang, L.; Zhang, X.; Ma, Y.; Li, Y.; Dong, L.; Zong, Z.; Hua, X.; Su, D.; Li, H.; et al. Autophagy promotes metastasis and glycolysis by upregulating MCT1 expression and Wnt/β-catenin signaling pathway activation in hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 2018, 37, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.; Davis, T.; Loos, B.; Sishi, B.; Huisamen, B.; Strijdom, H.; Engelbrecht, A.M. Autophagy is essential for the maintenance of amino acids and ATP levels during acute amino acid starvation in MDAMB231 cells. Cell Biochem. Funct. 2018, 36, 65–79. [Google Scholar] [CrossRef]
- Goldsmith, J.; Levine, B.; Debnath, J. Autophagy and cancer metabolism. Methods Enzymol. 2014, 542, 25–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple cell death modalities and their key features (Review). World Acad. Sci. J. 2020, 2, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M., Jr.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Zhang, K.; Hu, P. The role of autophagy in acute myocardial infarction. Front. Pharmacol. 2019, 10, 551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nah, J.; Zablocki, D.; Sadoshima, J. Autosis: A new target to prevent cell death. JACC: Basic Transl. Sci. 2020, 5, 857–869. [Google Scholar] [CrossRef]
- Miller, S.C.; Huang, R.; Sakamuru, S.; Shukla, S.J.; Attene-Ramos, M.S.; Shinn, P.; Van Leer, D.; Leister, W.; Austin, C.P.; Xia, M. Identification of known drugs that act as inhibitors of NF-kappaB signaling and their mechanism of action. Biochem. pharmacol. 2010, 79, 1272–1280. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Huang, W.; Jozwik, C.; Lin, Y.; Glasman, M.; Caohuy, H.; Srivastava, M.; Esposito, D.; Gillette, W.; Hartley, J.; et al. Cardiac glycosides inhibit TNF-α/NF-κB signaling by blocking recruitment of TNF receptor-associated death domain to the TNF receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 9631–9636. [Google Scholar] [CrossRef] [Green Version]
- Bereta, J.; Cohen, M.C.; Bereta, M. Stimulatory effect of ouabain on VCAM-1 and iNOS expression in murine endothelial cells: Involvement of NF-kappa B. FEBS Lett. 1995, 377, 21–25. [Google Scholar] [CrossRef] [Green Version]
- Farghaly, H.S.M.; Ashry, I.E.-S.M.; Hareedy, M.S. High doses of digoxin increase the myocardial nuclear factor-kB and CaV1.2 channels in healthy mice. A possible mechanism of digitalis toxicity. Biomed. Pharmacother. 2018, 105, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qiu, Q.; Shen, J.J.; Li, D.D.; Jiang, X.J.; Si, S.Y.; Shao, R.G.; Wang, Z. Cardiac glycosides induce autophagy in human non-small cell lung cancer cells through regulation of dual signaling pathways. Int. J. Biochem. Cell Biol. 2012, 44, 1813–1824. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-dependent ferroptosis: Machinery and regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar] [CrossRef]
- Goodall, M.L.; Cramer, S.D.; Thorburn, A. Autophagy complexes cell death by necroptosis. Oncotarget 2016, 7, 50818–50819. [Google Scholar] [CrossRef]
- Ahmed, A.; Tait, S.W.G. Targeting immunogenic cell death in cancer. Mol. Oncol. 2020, 14, 2994–3006. [Google Scholar] [CrossRef]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef]
- Škubník, J.; Pavlíčková, V.; Rimpelová, S. Cardiac glycosides as immune system modulators. Biomolecules 2021, 11, 659. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Hsieh, S.-Y.; Fan, Y.-T.; Wei, W.-C.; Hsiao, P.-W.; Tsai, D.-H.; Wu, T.-S.; Yang, N.-S. Necroptosis promotes autophagy-dependent upregulation of DAMP and results in immunosurveillance. Autophagy 2018, 14, 778–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeVorkin, L.; Choutka, C.; Gorski, S.M. Chapter 24—The interplay between autophagy and apoptosis. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Hayat, M.A., Ed.; Academic Press: Amsterdam, The Neatherlands, 2014; pp. 369–383. [Google Scholar] [CrossRef]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Ng, T.T.H.; Sham, K.W.Y.; Zhang, L.; Chan, M.T.V.; Wu, W.K.K.; Cheng, C.H.K. Bufalin, a traditional Chinese medicine compound, prevents tumor formation in two murine models of colorectal cancer. Cancer Prev. Res. (Phila.) 2019, 12, 653–666. [Google Scholar] [CrossRef] [Green Version]
- Meng, Z.; Yang, P.; Shen, Y.; Bei, W.; Zhang, Y.; Ge, Y.; Newman, R.A.; Cohen, L.; Liu, L.; Thornton, B.; et al. Pilot study of huachansu in patients with hepatocellular carcinoma, nonsmall-cell lung cancer, or pancreatic cancer. Cancer 2009, 115, 5309–5318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.M.; Chan, W.Y.; Yu, S.; Zhao, J.; Cheng, C.H. Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation. Free Radic. Biol. Med. 2011, 51, 1365–1375. [Google Scholar] [CrossRef]
- Li, D.D.; Wang, L.L.; Deng, R.; Tang, J.; Shen, Y.; Guo, J.F.; Wang, Y.; Xia, L.P.; Feng, G.K.; Liu, Q.Q.; et al. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene 2009, 28, 886–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.M.; Tsai, Y.; Wan, L.; Tsai, F.J. Bufalin induces G2/M phase arrest and triggers autophagy via the TNF, JNK, BECN-1 and ATG8 pathway in human hepatoma cells. Int. J. Oncol. 2013, 43, 338–348. [Google Scholar] [CrossRef]
- Miao, Q.; Bi, L.L.; Li, X.; Miao, S.; Zhang, J.; Zhang, S.; Yang, Q.; Xie, Y.H.; Zhang, J.; Wang, S.W. Anticancer effects of bufalin on human hepatocellular carcinoma HepG2 cells: Roles of apoptosis and autophagy. Int. J. Mol. Sci. 2013, 14, 1370–1382. [Google Scholar] [CrossRef]
- Shen, S.; Zhang, Y.; Wang, Z.; Zhang, R.; Gong, X. Bufalin induces the interplay between apoptosis and autophagy in glioma cells through endoplasmic reticulum stress. Int. J. Biol. Sci. 2014, 10, 212–224. [Google Scholar] [CrossRef]
- Zhao, H.; Li, Q.; Pang, J.; Jin, H.; Li, H.; Yang, X. Blocking autophagy enhances the pro-apoptotic effect of bufalin on human gastric cancer cells through endoplasmic reticulum stress. Biol. Open 2017, 6, 1416–1422. [Google Scholar] [CrossRef] [Green Version]
- Sheng, X.; Zhu, P.; Qin, J.; Li, Q. The biological role of autophagy in regulating and controlling the proliferation of liver cancer cells induced by bufalin. Oncol. Rep. 2018, 39, 2931–2941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollman, A. Drugs for atrial fibrillation. Digoxin comes from Digitalis lanata. BMJ 1996, 312, 912. [Google Scholar] [CrossRef]
- Ehle, M.; Patel, C.; Giugliano, R.P. Digoxin: Clinical highlights: A review of digoxin and its use in contemporary medicine. Crit. Pathw. Cardiol. 2011, 10, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Hundeshagen, P.; Hamacher-Brady, A.; Eils, R.; Brady, N.R. Concurrent detection of autolysosome formation and lysosomal degradation by flow cytometry in a high-content screen for inducers of autophagy. BMC Biol. 2011, 9, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, F.; Xu, X.; Wang, X.; Zhang, B. Regulation of autophagy by Ca2+. Tumour Biol. 2016, 37, 15467–15476. [Google Scholar] [CrossRef] [Green Version]
- Rasheduzzaman, M.; Yin, H.; Park, S.Y. Cardiac glycoside sensitized hepatocellular carcinoma cells to TRAIL via ROS generation, p38MAPK, mitochondrial transition, and autophagy mediation. Mol. Carcinog. 2019, 58, 2040–2051. [Google Scholar] [CrossRef]
- Crezee, T.; Tesselaar, M.H.; Nagarajah, J.; Corver, W.E.; Morreau, J.; Pritchard, C.; Kimura, S.; Kuiper, J.G.; van Engen-van Grunsven, I.; Smit, J.W.A.; et al. Digoxin treatment reactivates in vivo radioactive iodide uptake and correlates with favorable clinical outcome in non-medullary thyroid cancer. Cell. Oncol. (Dordr.) 2021, 44, 611–625. [Google Scholar] [CrossRef]
- Fernández Fernández, Á.; Liu, Y.; Ginet, V.; Shi, M.; Nah, J.; Zou, Z.; Zhou, A.; Posner, B.; Xiao, G.; Tanguy, M.; et al. Interaction between the autophagy protein Beclin 1 and Na+,K+-ATPase during starvation, exercise, and ischemia. JCI Insight 2020, 5, e133282. [Google Scholar] [CrossRef]
- Słabiak-Błaż, N.; Piecha, G. Endogenous mammalian cardiotonic steroids—A new cardiovascular risk factor?—A mini-review. Life 2021, 11, 727. [Google Scholar] [CrossRef]
- Hamlyn, J.M.; Blaustein, M.P. Endogenous ouabain: Recent advances and controversies. Hypertension 2016, 68, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhan, Y.; Xu, R.; Shao, R.; Jiang, J.; Wang, Z. Src mediates extracellular signal-regulated kinase 1/2 activation and autophagic cell death induced by cardiac glycosides in human non-small cell lung cancer cell lines. Mol. Carcinog. 2015, 54, E26–E34. [Google Scholar] [CrossRef]
- Shen, J.-j.; Zhan, Y.-c.; Li, H.-y.; Wang, Z. Ouabain impairs cancer metabolism and activates AMPK-Src signaling pathway in human cancer cell lines. Acta Pharmacol. Sin. 2020, 41, 110–118. [Google Scholar] [CrossRef]
- Trenti, A.; Grumati, P.; Cusinato, F.; Orso, G.; Bonaldo, P.; Trevisi, L. Cardiac glycoside ouabain induces autophagic cell death in non-small cell lung cancer cells via a JNK-dependent decrease of Bcl-2. Biochem. Pharmacol. 2014, 89, 197–209. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Yoon, B.; Zhang, W.C.; Adams, B.D.; Slack, F.J. A high-throughput small molecule screen identifies ouabain as synergistic with miR-34a in killing lung cancer cells. iScience 2020, 23, 100878. [Google Scholar] [CrossRef] [Green Version]
- Song, H.L.; Demirev, A.V.; Kim, N.Y.; Kim, D.H.; Yoon, S.Y. Ouabain activates transcription factor EB and exerts neuroprotection in models of Alzheimer’s disease. Mol. Cell. Neurosci. 2019, 95, 13–24. [Google Scholar] [CrossRef]
- Meng, L.; Wen, Y.; Zhou, M.; Li, J.; Wang, T.; Xu, P.; Ouyang, J. Ouabain induces apoptosis and autophagy in Burkitt’s lymphoma Raji cells. Biomed. Pharmacother. 2016, 84, 1841–1848. [Google Scholar] [CrossRef] [PubMed]
- Nah, J.; Zhai, P.; Huang, C.-Y.; Fernández, Á.F.; Mareedu, S.; Levine, B.; Sadoshima, J. Upregulation of Rubicon promotes autosis during myocardial ischemia/reperfusion injury. J. Clin. Invest. 2020, 130, 2978–2991. [Google Scholar] [CrossRef] [PubMed]
- L’Hote, V.; Courbeyrette, R.; Pinna, G.; Cintrat, J.C.; Le Pavec, G.; Delaunay-Moisan, A.; Mann, C.; Thuret, J.Y. Ouabain and chloroquine trigger senolysis of BRAF-V600E-induced senescent cells by targeting autophagy. Aging Cell 2021, 20, e13447. [Google Scholar] [CrossRef]
- Hsu, I.L.; Chou, C.-Y.; Wu, Y.-Y.; Wu, J.-E.; Liang, C.-H.; Tsai, Y.-T.; Ke, J.-Y.; Chen, Y.-L.; Hsu, K.-F.; Hong, T.-M. Targeting FXYD2 by cardiac glycosides potently blocks tumor growth in ovarian clear cell carcinoma. Oncotarget 2016, 7, 62925–62938. [Google Scholar] [CrossRef]
- Škubník, J.; Bejček, J.; Pavlíčková, V.S.; Rimpelová, S. Repurposing cardiac glycosides: Drugs for heart failure surmounting viruses. Molecules 2021, 26, 5627. [Google Scholar] [CrossRef]
- Mukhopadhyay, R.; Venkatadri, R.; Katsnelson, J.; Arav-Boger, R. Digitoxin suppresses human cytomegalovirus replication via Na+, K+/ATPase α1 subunit-dependent AMP-activated protein kinase and autophagy activation. J. Virol. 2018, 92, e01861-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.A.; Kim, M.-S.; Kim, W.; Um, J.-H.; Shin, Y.-J.; Song, J.-Y.; Jeong, J.-H. Lanatoside C suppressed colorectal cancer cell growth by inducing mitochondrial dysfunction and increased radiation sensitivity by impairing DNA damage repair. Oncotarget 2016, 7, 6074–6087. [Google Scholar] [CrossRef] [Green Version]
- Reddy, D.; Kumavath, R.; Ghosh, P.; Barh, D. Lanatoside C induces G2/M cell cycle arrest and suppresses cancer cell growth by attenuating MAPK, Wnt, JAK-STAT, and PI3K/AKT/mTOR signaling pathways. Biomolecules 2019, 9, 792. [Google Scholar] [CrossRef] [Green Version]
- Reddy, D.; Ghosh, P.; Kumavath, R. Strophanthidin attenuates MAPK, PI3K/AKT/mTOR, and Wnt/beta-Catenin signaling Pathways in human cancers. Front. Oncol. 2020, 9, 1469. [Google Scholar] [CrossRef] [Green Version]
- Reddy, D.; Kumavath, R.; Tan, T.Z.; Ampasala, D.R.; Kumar, A.P. Peruvoside targets apoptosis and autophagy through MAPK Wnt/β-catenin and PI3K/AKT/mTOR signaling pathways in human cancers. Life Sci. 2020, 241, 117147. [Google Scholar] [CrossRef]
- Kaushik, V.; Yakisich, J.S.; Azad, N.; Kulkarni, Y.; Venkatadri, R.; Wright, C.; Rojanasakul, Y.; Iyer, A.K.V. Anti-tumor effects of cardiac glycosides on human lung cancer cells and lung tumorspheres. J. Cell. Physiol. 2017, 232, 2497–2507. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Kim, N.H.; Cho, Y.S.; Lee, H.; Kwon, H.J. Convallatoxin, a dual inducer of autophagy and apoptosis, inhibits angiogenesis in vitro and in vivo. PLoS ONE 2014, 9, e91094. [Google Scholar] [CrossRef]
- Newman, R.A.; Kondo, Y.; Yokoyama, T.; Dixon, S.; Cartwright, C.; Chan, D.; Johansen, M.; Yang, P. Autophagic cell death of human pancreatic tumor cells mediated by oleandrin, a lipid-soluble cardiac glycoside. Integr. Cancer Ther. 2007, 6, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Terzioglu-Usak, S.; Nalli, A.; Elibol, B.; Ozek, E.; Hatiboglu, M.A. AnvirzelTMregulates cell death through inhibiting GSK-3 activity in human U87 glioma cells. Neurol. Res. 2020, 42, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.-R.; Bai, L.-Y.; Chiu, S.-J.; Chiu, C.-F.; Lin, W.-Y.; Hu, J.-L.; Shieh, T.-M. Divaricoside exerts antitumor effects, in part, by modulating Mcl-1 in human oral squamous cell carcinoma cells. Comput. Struct. Biotechnol. J. 2019, 17, 151–159. [Google Scholar] [CrossRef]
- Luo, M.J.; Liu, Y.F.; Liu, N.N.; Shao, W.Q.; Ming, L.J.; Liu, J.; Xie, Y.H. Proscillaridin A inhibits hepatocellular carcinoma progression through inducing mitochondrial damage and autophagy. Acta Biochim. Biophys. Sin. 2021, 53, 19–28. [Google Scholar] [CrossRef]
- Saleem, M.Z.; Alshwmi, M.; Zhang, H.; Din, S.R.U.; Nisar, M.A.; Khan, M.; Alam, S.; Alam, G.; Jin, L.; Ma, T. Inhibition of JNK-mediated autophagy promotes proscillaridin A-induced apoptosis via ROS generation, itracellular Ca+2 oscillation and inhibiting STAT3 signaling in breast cancer cells. Front. Pharmacol. 2020, 11, 01055. [Google Scholar] [CrossRef]
- Schneider, N.F.Z.; Cerella, C.; Lee, J.-Y.; Mazumder, A.; Kim, K.R.; de Carvalho, A.; Munkert, J.; Pádua, R.M.; Kreis, W.; Kim, K.-W.; et al. Cardiac glycoside glucoevatromonoside induces cancer type-specific sell death. Front. Pharmacol. 2018, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Keller, C.W.; Sina, C.; Kotur, M.B.; Ramelli, G.; Mundt, S.; Quast, I.; Ligeon, L.A.; Weber, P.; Becher, B.; Münz, C.; et al. ATG-dependent phagocytosis in dendritic cells drives myelin-specific CD4(+) T cell pathogenicity during CNS inflammation. Proc. Natl. Acad. Sci. USA 2017, 114, E11228–E11237. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cardiac Glycoside | Cell Line | Concentration, Incubation Time | Marker | Autophagy Induced (+)/ Suppressed (−) | Ref. |
|---|---|---|---|---|---|
| Bufalin | HT-29 | 100 nM, 48 h | ↑ LC3-II, ↑ ROS, ↑ JNK2 | + | [75] |
| Huh7 | 40 nM, 12 h | ↑ chemokine receptor 4, ↑ tumor necrosis factor, ↑ JNK, ↑ Beclin 1 | + | [77] | |
| HepG2 | 100 nM, 48 h | ↑ pAMPK, ↓ mTOR, ↑ LC3-II, ↑ Beclin 1 | + | [78] | |
| U87MG | 20–80 nM, 24 h | ↑ CHOP, ↑ GRP78, ↑ pPERK, ↑ p (eIF2α) | + | [79] | |
| LN229 | + | ||||
| SGC7901 | 80 nM, 48 h | ↓ LC3-I, ↑LC3-II, ↓ p62 | + | [80] | |
| BGC823 | + | ||||
| Digoxin | MCF-7 | 100 ng/mL, 6 h | ↑ lysosomal turnover | + | [84] |
| Huh7 | >100 nM, 16 h | ↑ pAMPK | + | [86] | |
| HeLa | >10 nM, 24 h | ↑LC3-II, ↓ p62 | - | [57] | |
| A549 | 50 nM, 24 h | ↑ LC3-II,↑ ATG5, ↑ AMPK, ↓ mTOR | + | [64,91] | |
| H460 | + | ||||
| Ouabain | A549 | 25 nM, 24 h | ↑ LC3-II,↑ ATG5, ↑ AMPK, ↑ Beclin 1, ↓ mTOR | + | [64,91,92] |
| H460 | + | ||||
| A549 | 100 nM, 24 h | ↑ LC3-II, ↑ pULK1, ↑ pJNK | + | [93] | |
| HeLa | 500 nM, 8 h | ↑ LC3-II, ↑ TFEB | + | [95] | |
| Raji | 100 nM, 48 h | ↑ LC3-II, ↑ Beclin 1, ↓ caspase 3, ↓ Bcl2 | + | [96] | |
| Digitoxin | TOV-21G | 5 nM, 48 h | ↑ LC3-II | + | [99] |
| HFF | 30 nM, 24 h | ↑ pAMPK, ↑ pULK1, ↓ mTOR | + | [101] | |
| Lanatoside C | HCT116 | 1 μM, 4 h | ↑ LC3-II, ↑pJNK, ↑ pERK 1/2 | + | [102] |
| A549 | 160 nM, 24 h | ↓ Akt, ↓ mTOR, ↓PI3K, ↓ LC3 | - | [103] | |
| MCF-7 | 1.2 μM, 24 h | - | |||
| HepG2 | 700 nM, 24 h | - | |||
| Huh7 | >100 nM, 16 h | ↑ pAMPK | + | [86] | |
| Strophanthidin | A549 | 1 μM, 24 h | ↓ Akt, ↓ mTOR, ↓PI3K, ↓ LC3 | - | [104] |
| MCF-7 | 2 μM, 24 h | - | |||
| HepG2 | 2.5 μM, 24 h | - | |||
| Peruvoside | A549 | 100 nM, 24 h | ↓ Akt, ↓ mTOR, ↓PI3K, ↓ LC3, MEK1 blocking | - | [105] |
| MCF-7 | - | ||||
| HepG2 | - | ||||
| H460 | 25 nM, 24 h | ↑ pERK, ↑ pMAPK p38, ↑ pAkt | - | [106] | |
| Convallatoxin | H460 | 25 nM, 24h | ↓ pERK, ↓ pMAPK p38 | - | |
| HeLa | 10 nM, 24 h | ↑ LC3-II, ↑ p (S6 kinase beta-1), ↓ mTOR | + | [107] | |
| Oleandrin | PANC-1 | 20 nM, 72 h | ↑ LC3-II | + | [108] |
| PANC-1 | 50 nM, 24 h | ↑ pERK, ↓ pAkt | + | ||
| AnvirzelTM | U87 | 10–250 µg/mL, 48 h | ↑ pERK, ↑ p(mTOR), ↓ pAkt, ↓ pJNK1/2 | + | [109] |
| Divaricoside | SCC2095 | 10–500 nM, 48 h | ↑ LC3-II, ↑ p62 | + | [110] |
| Proscillaridin A | MHCC97H | 50 nM, 24 h | ↑ LC3-II, ↓ mTOR | + | [111] |
| Huh7 | + | ||||
| MCF-7 | >100 nM, 24 h | ↑ LC3-II, ↑pJNK, ↓ p (mTOR), ↓ pAkt | + | [112] | |
| MDA-MB-231 | + | ||||
| Glucoevatromonoside | A549 | 50 nM, 24 h | ↓ LC3-II, ↑LC3-I, ↓ p62, ↓Beclin 1 | - | [113] |
| Neriifolin | RAW 264.7 | 1 µM, 24 h | ↓ LC3-II | - | [114] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Škubník, J.; Svobodová Pavlíčková, V.; Psotová, J.; Rimpelová, S. Cardiac Glycosides as Autophagy Modulators. Cells 2021, 10, 3341. https://doi.org/10.3390/cells10123341
Škubník J, Svobodová Pavlíčková V, Psotová J, Rimpelová S. Cardiac Glycosides as Autophagy Modulators. Cells. 2021; 10(12):3341. https://doi.org/10.3390/cells10123341
Chicago/Turabian StyleŠkubník, Jan, Vladimíra Svobodová Pavlíčková, Jana Psotová, and Silvie Rimpelová. 2021. "Cardiac Glycosides as Autophagy Modulators" Cells 10, no. 12: 3341. https://doi.org/10.3390/cells10123341