
A Multi-Factorial Observational Study on Sequential Fecal Microbiota Transplant in Patients with Medically Refractory Clostridioides difficile Infection

 , ,
, ,  , , , , , ,
, , , , , ,  , , and add
Show full author list
, , and add
Show full author list
Abstract
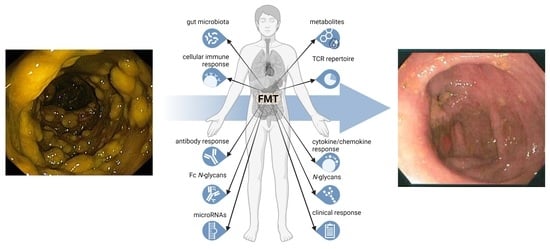
:
1. Introduction
2. Materials and Methods
2.1. Study Cohort, Treatment Regimen and Outcome Definitions
2.2. FMT Preparation
2.3. Multiomics Studies
2.4. 16S rRNA Gene Sequencing
2.5. Metabolomic Analysis
2.6. Serum N-Glycome Analysis
2.7. IgG Fc N-Glycopeptides Analysis
2.8. RT-qPCR for miRNAs
2.9. Multiplex ELISA for Profiling Cytokine and Multi-Isotype Antibody Responses
2.10. Antigen-Specific Microarray
2.11. Toxin Neutralization Assay
2.12. Isolation and Freezing of Peripheral Blood Mononuclear Cells
2.13. Immunostaining via Flow Cytometry
2.14. RNA Isolation, TCR Library Preparation and Sequencing
2.15. TCR Data Analysis
2.16. Statistical Analysis
3. Results
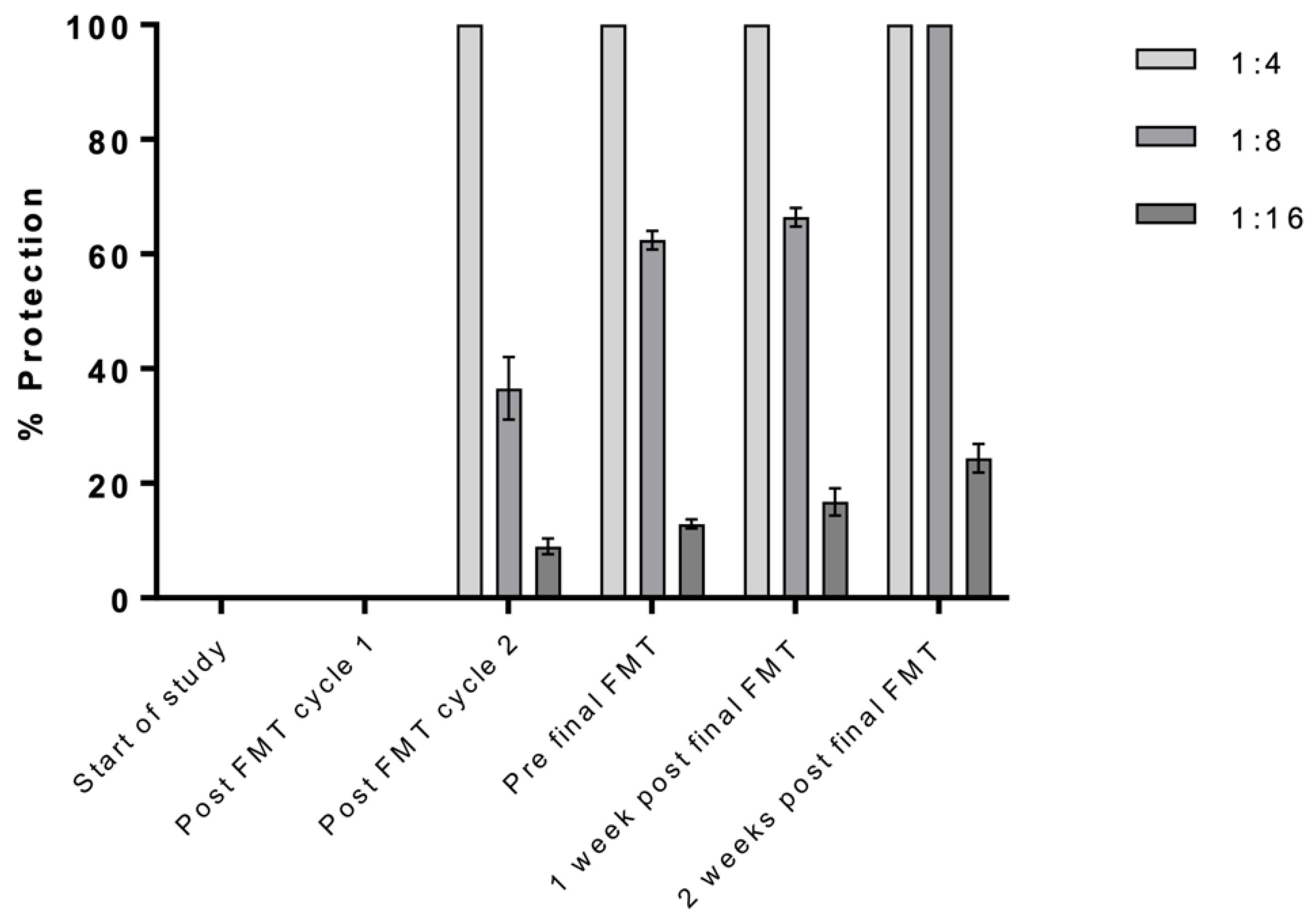
3.1. Clinical Outcomes
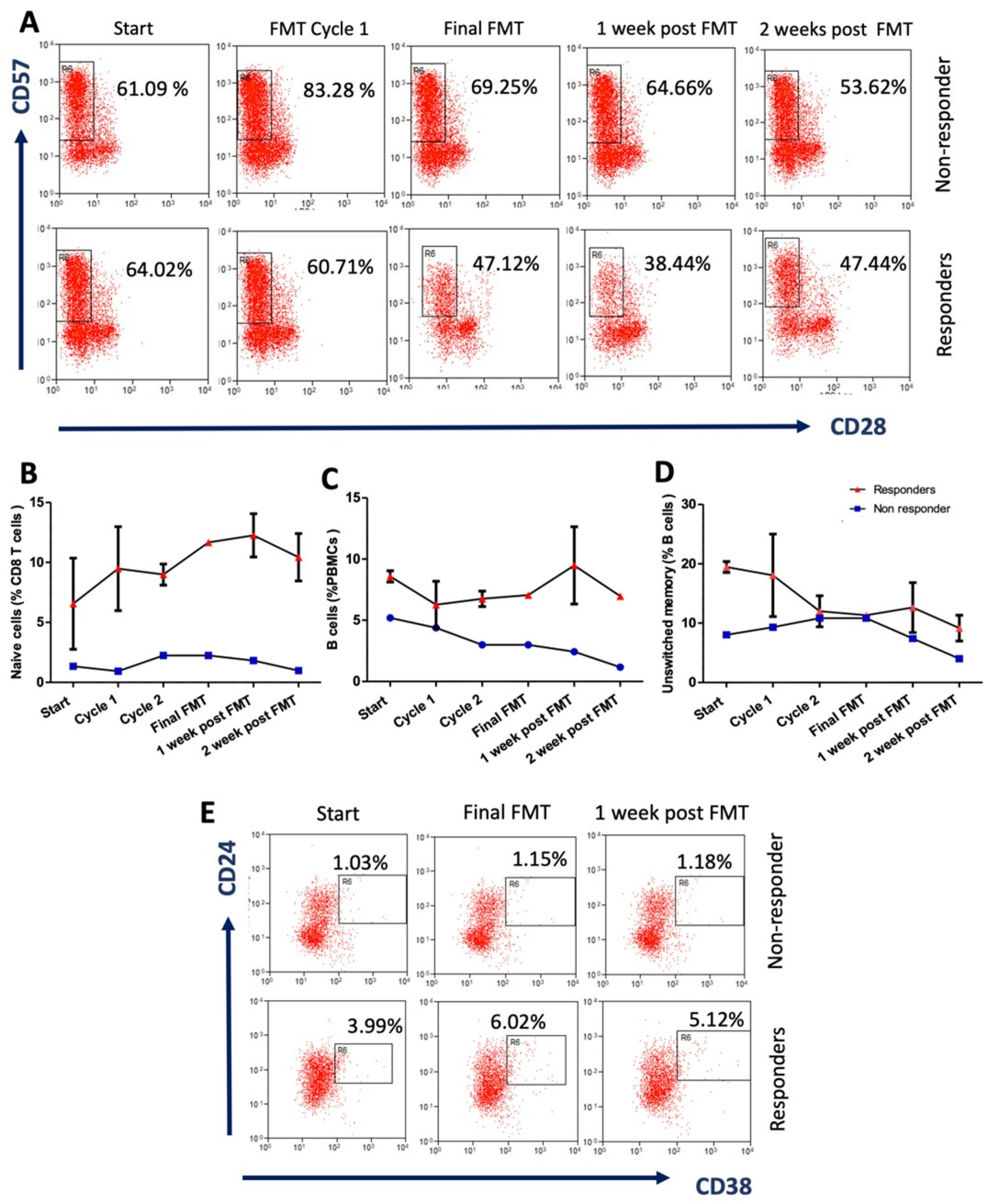
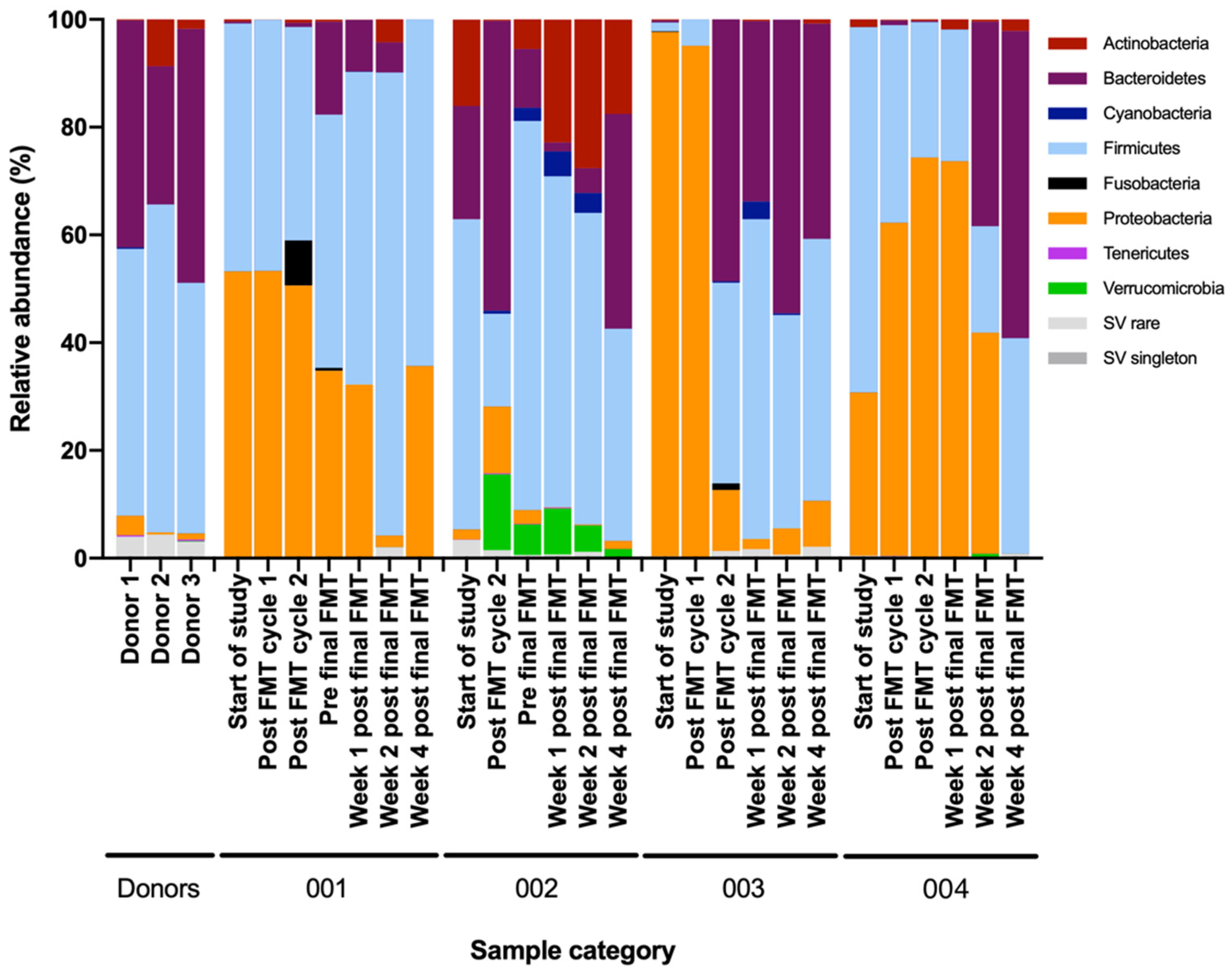
3.2. Extensive Multi-Analyte Changes Occur with Sequential FMT
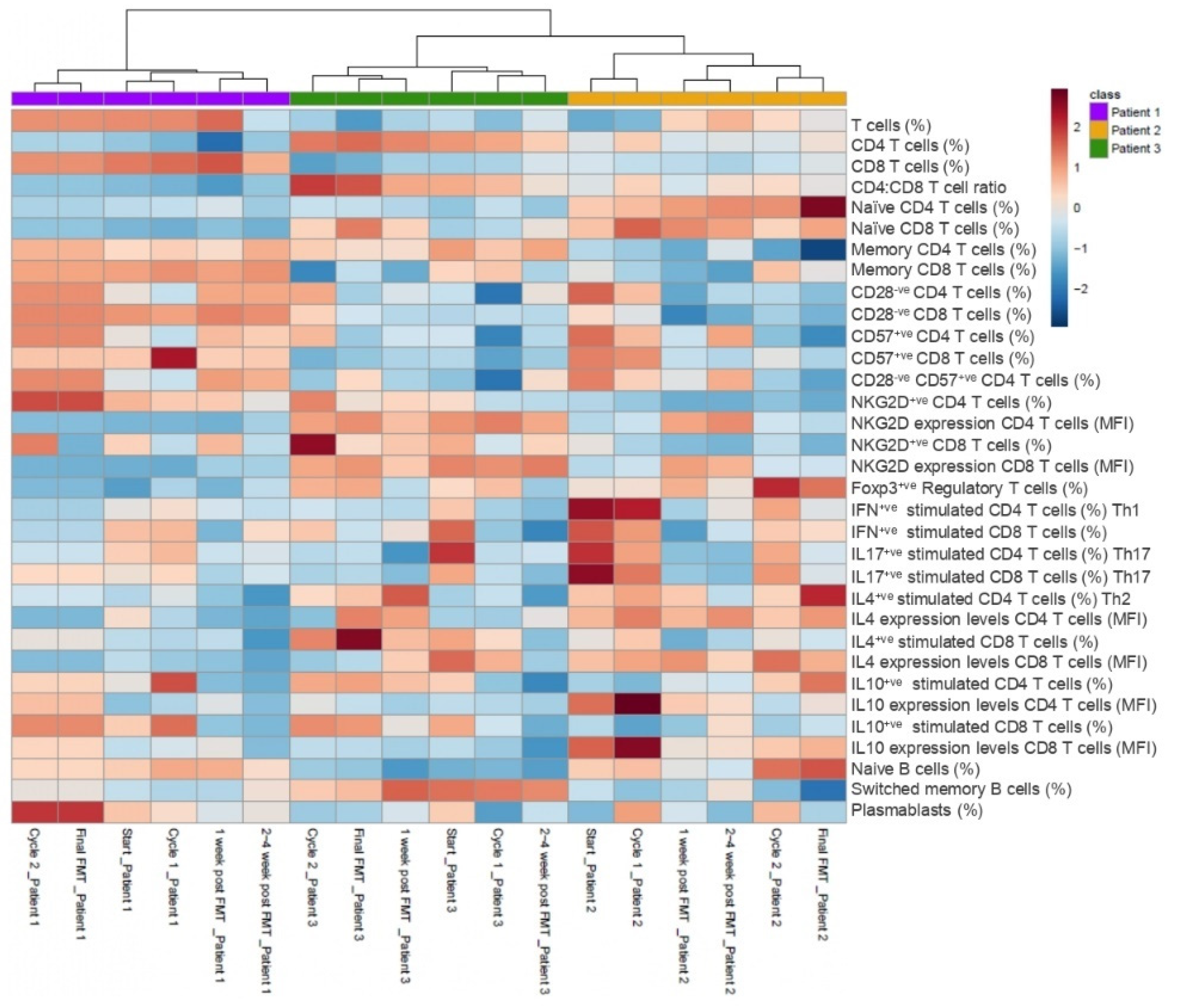
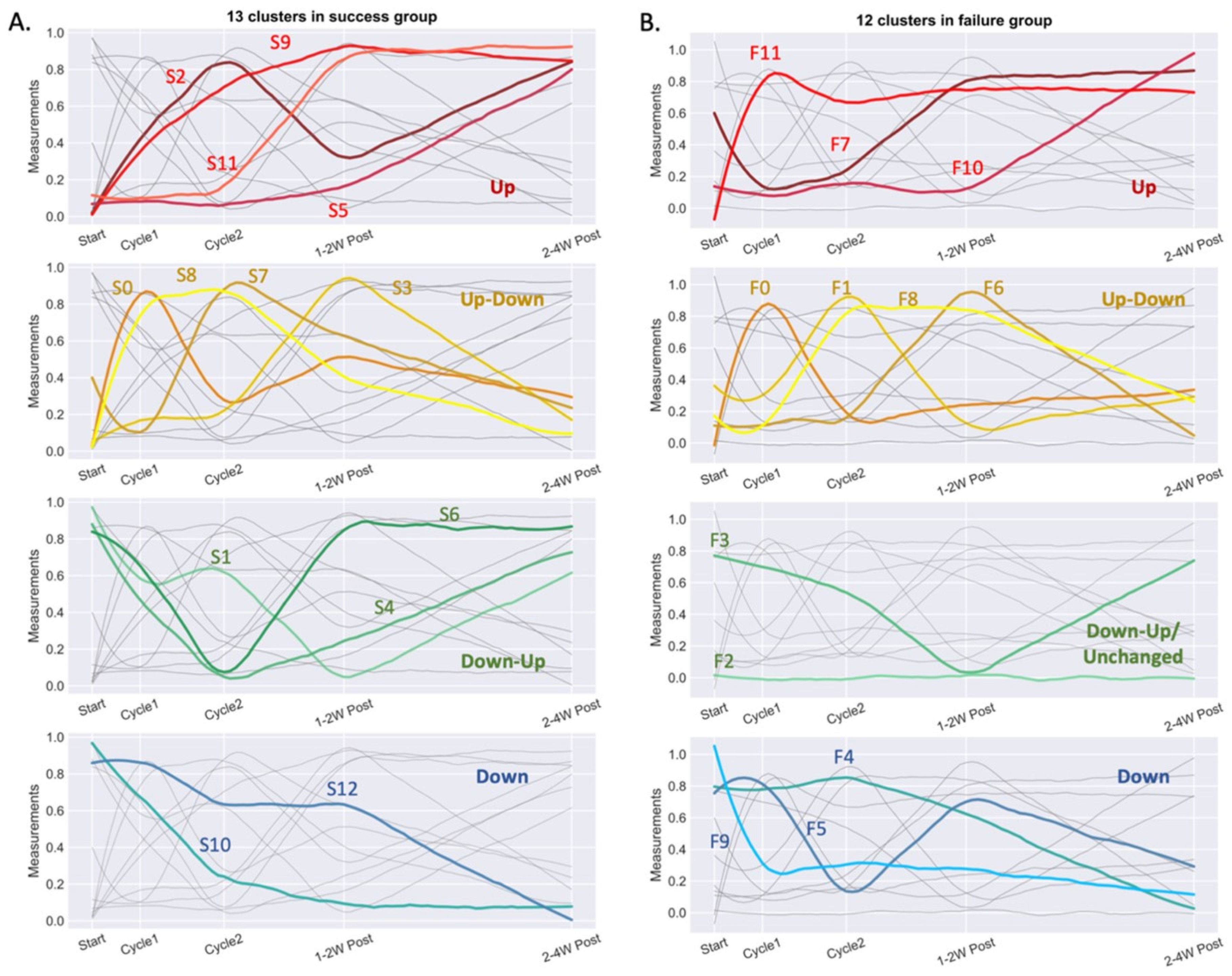
3.3. Multiomics Longitudinal Patterns Possibly Associated with FMT Response
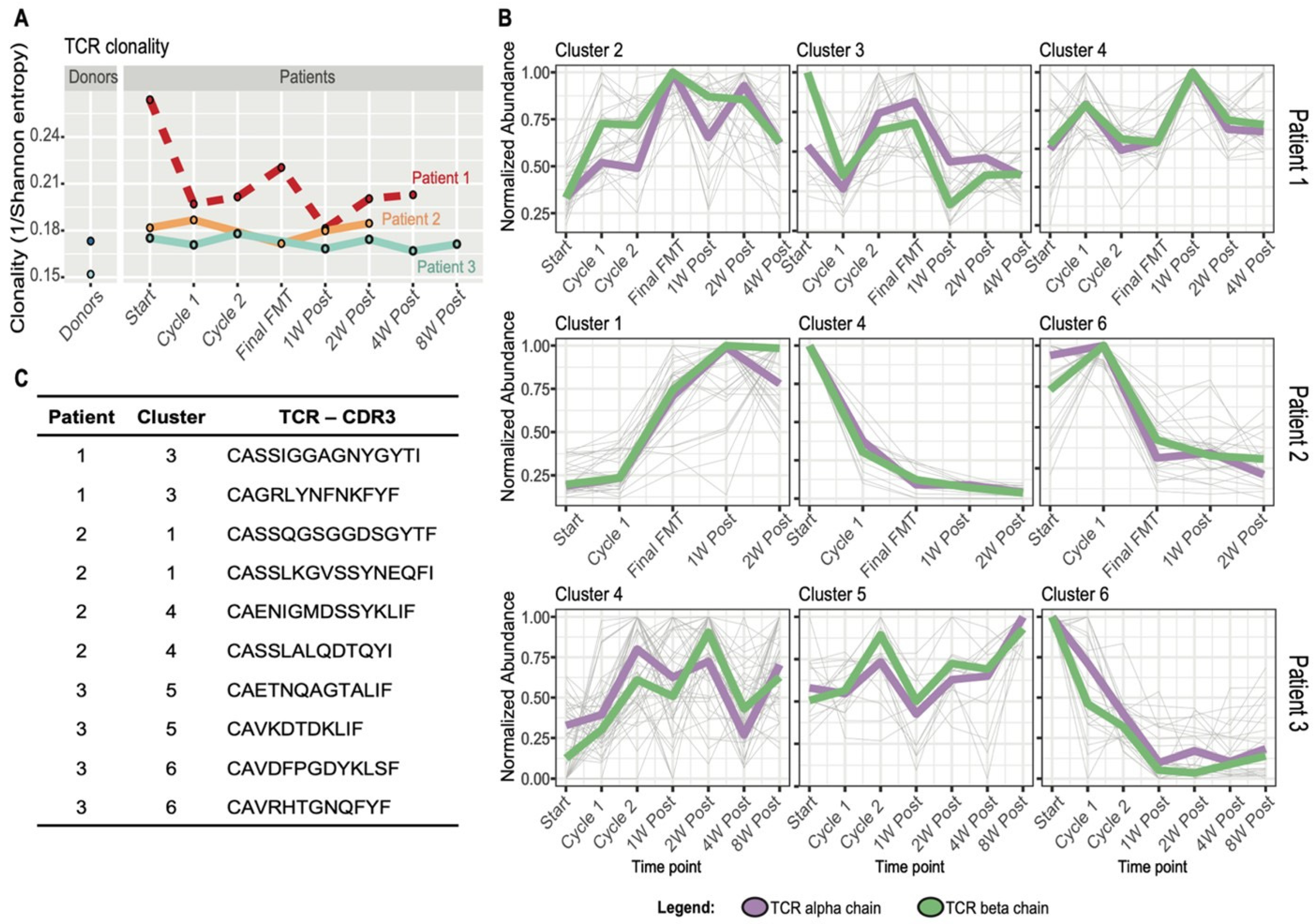
3.4. FMT Impact on T Cell Receptor Repertoire and Multiomics Integration
3.5. Temporal Correlation among Features: A Closer Look at T Cell Immunosenescence Signatures, Gut Microbiome and Immunometabolic Features
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gravel, D.; Miller, M.; Simor, A.; Taylor, G.; Gardam, M.; McGeer, A.; Hutchinson, J.; Moore, D.; Kelly, S.; Boyd, D.; et al. Health Care–AssociatedClostridium difficileInfection in Adults Admitted to Acute Care Hospitals in Canada: A Canadian Nosocomial Infection Surveillance Program Study. Clin. Infect. Dis. 2009, 48, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Kyne, L.; Hamel, M.B.; Polavaram, R.; Kelly, C.P. Health Care Costs and Mortality Associated with Nosocomial Diarrhea Due to Clostridium difficile. Clin. Infect. Dis. 2002, 34, 346–353. [Google Scholar] [CrossRef]
- McDonald, L.C.; Gerding, D.N.; Johnson, S.; Bakken, J.S.; Carroll, K.C.; E Coffin, S.; Dubberke, E.R.; Garey, K.W.; Gould, C.V.; Kelly, C.; et al. Clinical Practice Guidelines for Clostridium difficile Infection in Adults and Children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin. Infect. Dis. 2018, 66, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.S.; Monaghan, T.M.; Wilcox, M.H. Clostridium difficile infection: Epidemiology, diagnosis and understanding transmission. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Monaghan, T.M. New Perspectives in Clostridium difficile Disease Pathogenesis. Infect. Dis. Clin. N. Am. 2015, 29, 1–11. [Google Scholar] [CrossRef]
- Baunwall, S.M.D.; Lee, M.M.; Eriksen, M.K.; Mullish, B.H.; Marchesi, J.R.; Dahlerup, J.F.; Hvas, C.L. Faecal microbiota transplantation for recurrent Clostridioides difficile infection: An updated systematic review and meta-analysis. EClinicalMedicine 2020, 29–30, 100642. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Sipe, B.; Cheng, Y.-W.; Phelps, E.; Rogers, N.; Sagi, S.; Bohm, M.; Xu, H.; Kassam, Z. Fecal microbiota transplant in severe and severe-complicated Clostridium difficile: A promising treatment approach. Gut Microbes 2016, 8, 289–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weingarden, A.R.; Hamilton, M.J.; Sadowsky, M.J.; Khoruts, A. Resolution of Severe Clostridium difficile Infection Following Sequential Fecal Microbiota Transplantation. J. Clin. Gastroenterol. 2013, 47, 735–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alukal, J.; Dutta, S.K.; Surapaneni, B.K.; Le, M.; Tabbaa, O.; Phillips, L.; Mattar, M.C.; Philips, L. Safety and efficacy of fecal microbiota transplant in 9 critically ill patients with severe and complicated Clostridium difficile infection with impending colectomy. J. Dig. Dis. 2019, 20, 301–307. [Google Scholar] [CrossRef]
- Ianiro, G.; Masucci, L.; Quaranta, G.; Simonelli, C.; Lopetuso, L.R.; Sanguinetti, M.; Gasbarrini, A.; Cammarota, G. Randomised clinical trial: Faecal microbiota transplantation by colonoscopy plus vancomycin for the treatment of severe refractory Clostridium difficile infection-single versus multiple infusions. Aliment. Pharmacol. Ther. 2018, 48, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Khoruts, A.; Staley, C.; Sadowsky, M.J. Faecal microbiota transplantation for Clostridioides difficile: Mechanisms and pharmacology. Nat. Rev. Gastroenterol. Hepatol. 2020, 18, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, T.; Mullish, B.H.; Patterson, J.; Wong, G.K.; Marchesi, J.R.; Xu, H.; Jilani, T.; Kao, D. Effective fecal microbiota transplantation for recurrent Clostridioides difficile infection in humans is associated with increased signalling in the bile acid-farnesoid X receptor-fibroblast growth factor pathway. Gut Microbes 2018, 10, 142–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weingarden, A.R.; Chen, C.; Bobr, A.; Yao, D.; Lu, Y.; Nelson, V.M.; Sadowsky, M.; Khoruts, A. Microbiota transplantation restores normal fecal bile acid composition in recurrentClostridium difficileinfection. Am. J. Physiol. Liver Physiol. 2014, 306, G310–G319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullish, B.H.; McDonald, J.A.K.; Pechlivanis, A.; Allegretti, J.R.; Kao, D.; Barker, G.F.; Kapila, D.; Petrof, E.O.; Joyce, S.A.; Gahan, C.; et al. Microbial bile salt hydrolases mediate the efficacy of faecal microbiota transplant in the treatment of recurrent Clostridioides difficile infection. Gut 2019, 68, 1791–1800. [Google Scholar] [CrossRef] [Green Version]
- Allegretti, J.R.; Kearney, S.M.; Li, N.; Bogart, E.; Bullock, K.; Gerber, G.K.; Bry, L.; Clish, C.; Alm, E.J.; Korzenik, J. RecurrentClostridium difficileinfection associates with distinct bile acid and microbiome profiles. Aliment. Pharmacol. Ther. 2016, 43, 1142–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, C.; McKenney, P.T.; Konstantinovsky, D.; Isaeva, O.; Schizas, M.; Verter, J.; Mai, C.; Jin, W.-B.; Guo, C.-J.; Violante, S.; et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 2020, 581, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Seekatz, A.M.; Theriot, C.M.; Rao, K.; Chang, Y.-M.; Freeman, A.E.; Kao, J.Y.; Young, V.B. Restoration of short chain fatty acid and bile acid metabolism following fecal microbiota transplantation in patients with recurrent Clostridium difficile infection. Anaerobe 2018, 53, 64–73. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.; Mullish, B.; Pechlivanis, A.; Liu, Z.; Brignardello, J.; Kao, D.; Holmes, E.; Li, J.; Clarke, T.B.; Thursz, M.; et al. Inhibiting Growth of Clostridioides difficile by Restoring Valerate, Produced by the Intestinal Microbiota. Gastroenterology 2018, 155, 1495–1507.e15. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly-Y, M.; Glickman, J.N.; Garrett, W.S. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Monaghan, T.M.; Pučić-Baković, M.; Vučković, F.; Lee, C.; Kao, D.; Wójcik, I.; Kliček, F.; Polytarchou, C.; Roach, B.; Louie, T.; et al. Decreased Complexity of Serum N-glycan Structures Associates with Successful Fecal Microbiota Transplantation for Recurrent Clostridioides difficile Infection. Gastroenterology 2019, 157, 1676–1678.e3. [Google Scholar] [CrossRef]
- Abhyankar, M.M.; Ma, J.Z.; Scully, K.W.; Nafziger, A.J.; Frisbee, A.L.; Saleh, M.M.; Madden, G.R.; Hays, A.R.; Poulter, M.; Petri, W.A. Immune Profiling To Predict Outcome of Clostridioides difficile Infection. mBio 2020, 11, e00905-20. [Google Scholar] [CrossRef] [PubMed]
- Rees, W.; Steiner, T.S. Adaptive immune response toClostridium difficileinfection: A perspective for prevention and therapy. Eur. J. Immunol. 2018, 48, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Monaghan, T.; Robins, A.; Knox, A.; Sewell, H.F.; Mahida, Y.R. Circulating Antibody and Memory B-Cell Responses to C. difficile Toxins A and B in Patients with C. difficile-Associated Diarrhoea, Inflammatory Bowel Disease and Cystic Fibrosis. PLoS ONE 2013, 8, e74452. [Google Scholar] [CrossRef] [Green Version]
- Devera, T.S.; Lang, G.A.; Lanis, J.M.; Rampuria, P.; Gilmore, C.L.; James, J.A.; Ballard, J.D.; Lang, M.L. Memory B Cells Encode Neutralizing Antibody Specific for Toxin B from the Clostridium difficile Strains VPI 10463 and NAP1/BI/027 but with Superior Neutralization of VPI 10463 Toxin B. Infect. Immun. 2016, 84, 194–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amani, S.A.; Shadid, T.; Ballard, J.D.; Lang, M.L. Clostridioides difficile Infection Induces an Inferior IgG Response to That Induced by Immunization and Is Associated with a Lack of T Follicular Helper Cell and Memory B Cell Expansion. Infect. Immun. 2020, 88, e00829-19. [Google Scholar] [CrossRef]
- Kao, D.; Roach, B.; Silva, M.; Beck, P.; Rioux, K.; Kaplan, G.G.; Chang, H.-J.; Coward, S.; Goodman, K.J.; Xu, H.; et al. Effect of Oral Capsule- vs. Colonoscopy-Delivered Fecal Microbiota Transplantation on Recurrent Clostridium difficile Infection: A Randomized Clinical Trial. JAMA 2017, 318, 1985–1993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullish, B.H.; Pechlivanis, A.; Barker, G.F.; Thursz, M.R.; Marchesi, J.; McDonald, J.A. Functional microbiomics: Evaluation of gut microbiota-bile acid metabolism interactions in health and disease. Methods 2018, 149, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Akmačić, I.T.; Ugrina, I.; Štambuk, J.; Gudelj, I.; Vučković, F.; Lauc, G.; Pučić-Baković, M. High-throughput glycomics: Optimization of sample preparation. Biochemistry 2015, 80, 934–942. [Google Scholar] [CrossRef]
- Šimurina, M.; de Haan, N.; Vučković, F.; Kennedy, N.A.; Štambuk, J.; Falck, D.; Trbojević-Akmačić, I.; Clerc, F.; Razdorov, G.; Khon, A.; et al. Glycosylation of Immunoglobulin G Associates With Clinical Features of Inflammatory Bowel Diseases. Gastroenterology 2018, 154, 1320–1333.e10. [Google Scholar] [CrossRef]
- Monaghan, T.M.; Seekatz, A.M.; Markham, N.O.; Yau, T.O.; Hatziapostolou, M.; Jilani, T.; Christodoulou, N.; Roach, B.; Birli, E.; Pomenya, O.; et al. Fecal Microbiota Transplantation for Recurrent Clostridioides difficile Infection Associates With Functional Alterations in Circulating microRNAs. Gastroenterology 2021, 161, 255–270.e4. [Google Scholar] [CrossRef]
- Negm, O.H.; Hamed, M.R.; Dilnot, E.M.; Shone, C.C.; Marszalowska, I.; Lynch, M.; Loscher, C.E.; Edwards, L.; Tighe, P.; Wilcox, M.H.; et al. Profiling Humoral Immune Responses to Clostridium difficile-Specific Antigens by Protein Microarray Analysis. Clin. Vaccine Immunol. 2015, 22, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Negm, O.H.; MacKenzie, B.; Hamed, M.R.; Ahmad, O.A.J.; Shone, C.C.; Humphreys, D.P.; Acharya, K.R.; Loscher, C.E.; Marszalowska, I.; Lynch, M.; et al. Protective antibodies against Clostridium difficile are present in intravenous immunoglobulin and are retained in humans following its administration. Clin. Exp. Immunol. 2017, 188, 437–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaghan, T.M.; Negm, O.H.; MacKenzie, B.; Hamed, M.R.; Shone, C.C.; Humphreys, D.P.; Acharya, K.R.; Wilcox, M.H. High prevalence of subclass-specific binding and neutralizing antibodies against Clostridium difficile toxins in adult cystic fibrosis sera: Possible mode of immunoprotection against symptomatic C. difficile infection. Clin. Exp. Gastroenterol. 2017, 10, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Duggal, N.A.; Pollock, R.D.; Lazarus, N.R.; Harridge, S.; Lord, J.M. Major features of immunesenescence, including reduced thymic output, are ameliorated by high levels of physical activity in adulthood. Aging Cell 2018, 17, e12750. [Google Scholar] [CrossRef] [Green Version]
- Duggal, N.A.; Upton, J.; Phillips, A.C.; Sapey, E.; Lord, J.M. An age-related numerical and functional deficit in CD19+CD24hiCD38hiB cells is associated with an increase in systemic autoimmunity. Aging Cell 2013, 12, 873–881. [Google Scholar] [CrossRef] [Green Version]
- Pogorelyy, M.V.; Elhanati, Y.; Marcou, Q.; Sycheva, A.L.; Komech, E.A.; Nazarov, V.I.; Britanova, O.V.; Chudakov, D.M.; Mamedov, I.Z.; Lebedev, Y.B.; et al. Persisting fetal clonotypes influence the structure and overlap of adult human T cell receptor repertoires. PLoS Comput. Biol. 2017, 13, e1005572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shugay, M.; Britanova, O.V.; Merzlyak, E.; Turchaninova, M.; Mamedov, I.Z.; Tuganbaev, T.R.; Bolotin, D.; Staroverov, D.; Putintseva, E.; Plevova, K.; et al. Towards error-free profiling of immune repertoires. Nat. Methods 2014, 11, 653–655. [Google Scholar] [CrossRef]
- Bolotin, D.; Poslavsky, S.; Mitrophanov, I.; Shugay, M.; Mamedov, I.Z.; Putintseva, E.; Chudakov, D.M. MiXCR: Software for comprehensive adaptive immunity profiling. Nat. Methods 2015, 12, 380–381. [Google Scholar] [CrossRef]
- Oksanene, J.B.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; et al. R Package Version 2.5-6. Vegan: Community Ecology Package. 2019. Available online: http://cran.r-project.org (accessed on 3 June 2021).
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef]
- Müllner, D. Modern hierachical, agglomerative clustering algorithms. arXiv 2011, arXiv:1109.2378. [Google Scholar]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef] [PubMed]
- Seyda, M.; Quante, M.; Uehara, H.; Slegtenhorst, B.R.; Elkhal, A.; Tullius, S.G. Immunosenescence in renal transplantation: A changing balance of innate and adaptive immunity. Curr. Opin. Organ Transplant. 2015, 20, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Coppé, J.-P.; Lam, E. Cellular Senescence: The Sought or the Unwanted? Trends Mol. Med. 2018, 24, 871–885. [Google Scholar] [CrossRef]
- Larochette, V.; Miot, C.; Poli, C.; Beaumont, E.; Roingeard, P.; Fickenscher, H.; Jeannin, P.; Delneste, Y. IL-26, a Cytokine With Roles in Extracellular DNA-Induced Inflammation and Microbial Defense. Front. Immunol. 2019, 10, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, H.; Sasagawa, S.; Itoh, K. Sodium butyrate induces senescence and inhibits the invasiveness of glioblastoma cells. Oncol. Lett. 2017, 15, 1495–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Luo, Y.-F.; Wang, Y.-S.; Xiao, Y.-L.; Cai, H.-R.; Xie, C.-M. Pseudomonas aeruginosa induces cellular senescence in lung tissue at the early stage of two-hit septic mice. Pathog. Dis. 2018, 76, ftz001. [Google Scholar] [CrossRef]
- Koch, S.; Larbi, A.; Özcelik, D.; Solana, R.; Gouttefangeas, C.; Attig, S.; Wikby, A.; Strindhall, J.; Franceschi, C.; Pawelec, G. Cytomegalovirus Infection: A Driving Force in Human T Cell Immunosenescence. Ann. N. Y. Acad. Sci. 2007, 1114, 23–35. [Google Scholar] [CrossRef]
- Koide, N.; Morikawa, A.; Ito, H.; Sugiyama, T.; Hassan, F.; Islam, S.; Tumurkhuu, G.; Mori, I.; Yoshida, T.; Yokochi, T. Defective responsiveness of CD5+ B1 cells to lipopolysaccharide in cytokine production. J. Endotoxin Res. 2006, 12, 346–351. [Google Scholar] [CrossRef]
- Heath, J.J.; Grant, M.D. The Immune Response Against Human Cytomegalovirus Links Cellular to Systemic Senescence. Cells 2020, 9, 766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couzi, L.; Pitard, V.; Moreau, J.F.; Merville, P.; Dechanet-Merville, J. Direct and Indirect Effects of Cytomegalovirus-Induced gammadelta T Cells after Kidney Transplantation. Front. Immunol. 2015, 6, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egli, A.; Humar, A.; Kumar, D. State-of-the-Art Monitoring of Cytomegalovirus-Specific Cell-Mediated Immunity After Organ Transplant: A Primer for the Clinician. Clin. Infect. Dis. 2012, 55, 1678–1689. [Google Scholar] [CrossRef] [Green Version]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.B.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Thanissery, R.; Winston, J.A.; Theriot, C.M. Inhibition of spore germination, growth, and toxin activity of clinically relevant C. difficile strains by gut microbiota derived secondary bile acids. Anaerobe 2017, 45, 86–100. [Google Scholar] [CrossRef]
- Tam, J.; Icho, S.; Utama, E.; Orrell, K.E.; Gómez-Biagi, R.F.; Theriot, C.M.; Kroh, H.K.; Rutherford, S.A.; Lacy, D.B.; Melnyk, R.A. Intestinal bile acids directly modulate the structure and function of C. difficile TcdB toxin. Proc. Natl. Acad. Sci. USA 2020, 117, 6792–6800. [Google Scholar] [CrossRef] [PubMed]
- Weingarden, A.R.; Dosa, P.; Dewinter, E.; Steer, C.J.; Shaughnessy, M.K.; Johnson, J.R.; Khoruts, A.; Sadowsky, M.J. Changes in Colonic Bile Acid Composition following Fecal Microbiota Transplantation Are Sufficient to Control Clostridium difficile Germination and Growth. PLoS ONE 2016, 11, e0147210. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Gili, L.; McDonald, J.A.K.; Liu, Z.; Kao, D.; Allegretti, J.R.; Monaghan, T.M.; Barker, G.F.; Blanco, J.M.; Williams, H.R.T.; Holmes, E.; et al. Understanding the mechanisms of efficacy of fecal microbiota transplant in treating recurrent Clostridioides difficile infection and beyond: The contribution of gut microbial-derived metabolites. Gut Microbes 2020, 12, 1810531. [Google Scholar] [CrossRef]
- Sinha, S.R.; Haileselassie, Y.; Nguyen, L.P.; Tropini, C.; Wang, M.; Becker, L.S.; Sim, D.; Jarr, K.; Spear, E.T.; Singh, G.; et al. Dysbiosis-Induced Secondary Bile Acid Deficiency Promotes Intestinal Inflammation. Cell Host Microbe 2020, 27, 659–670.e5. [Google Scholar] [CrossRef]
- Glauben, R.; Batra, A.; Fedke, I.; Zeitz, M.; Lehr, H.A.; Leoni, F.; Mascagni, P.; Fantuzzi, G.; Dinarello, C.A.; Siegmund, B. Histone Hyperacetylation Is Associated with Amelioration of Experimental Colitis in Mice. J. Immunol. 2006, 176, 5015–5022. [Google Scholar] [CrossRef] [Green Version]
- Gudelj, I.; Lauc, G.; Pezer, M. Immunoglobulin G glycosylation in aging and diseases. Cell. Immunol. 2018, 333, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Johal, S.S.; Lambert, C.; Hammond, J.; James, P.D.; Borriello, S.P.; Mahida, Y.R. Colonic IgA producing cells and macrophages are reduced in recurrent and non-recurrent Clostridiumdifficile associated diarrhoea. J. Clin. Pathol. 2004, 57, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Leslie, J.L.; Vendrov, K.C.; Jenior, M.L.; Young, V.B. The Gut Microbiota Is Associated with Clearance of Clostridium difficile Infection Independent of Adaptive Immunity. mSphere 2019, 4, e00698-18. [Google Scholar] [CrossRef] [Green Version]
- Sutton, T.C.; Lamirande, E.W.; Bock, K.W.; Moore, I.N.; Koudstaal, W.; Rehman, M.; Weverling, G.J.; Goudsmit, J.; Subbarao, K. In Vitro Neutralization Is Not Predictive of Prophylactic Efficacy of Broadly Neutralizing Monoclonal Antibodies CR6261 and CR9114 against Lethal H2 Influenza Virus Challenge in Mice. J. Virol. 2017, 91, e01603-17. [Google Scholar] [CrossRef] [Green Version]
- Bootz, A.; Karbach, A.; Spindler, J.; Kropff, B.; Reuter, N.; Sticht, H.; Winkler, T.H.; Britt, W.J.; Mach, M. Protective capacity of neutralizing and non-neutralizing antibodies against glycoprotein B of cytomegalovirus. PLoS Pathog. 2017, 13, e1006601. [Google Scholar] [CrossRef] [PubMed]
- Steele, J.; Chen, K.; Sun, X.; Zhang, Y.; Wang, H.; Tzipori, S.; Feng, H. Systemic Dissemination of Clostridium difficile Toxins A and B Is Associated With Severe, Fatal Disease in Animal Models. J. Infect. Dis. 2011, 205, 384–391. [Google Scholar] [CrossRef]
- Lawrence, S.J.; Dubberke, E.R.; Johnson, S.; Gerding, D.N. Clostridium difficile—Associated Disease Treatment Response Depends on Definition of Cure. Clin. Infect. Dis. 2007, 45, 1648. [Google Scholar] [CrossRef] [Green Version]
- A Cohen, N.; Miller, T.; Na’Aminh, W.; Hod, K.; Adler, A.; Cohen, D.; Guzner-Gur, H.; Santo, E.; Halpern, Z.; Carmeli, Y.; et al. Clostridium difficile fecal toxin level is associated with disease severity and prognosis. United Eur. Gastroenterol. J. 2018, 6, 773–780. [Google Scholar] [CrossRef] [Green Version]
- Mensen, A.; Oh, Y.; Becker, S.C.; Hemmati, P.G.; Jehn, C.; Westermann, J.; Szyska, M.; Göldner, H.; Dörken, B.; Scheibenbogen, C.; et al. Apoptosis Susceptibility Prolongs the Lack of Memory B Cells in Acute Leukemic Patients After Allogeneic Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2015, 21, 1895–1906. [Google Scholar] [CrossRef] [Green Version]
- Shah, H.B.; Smith, K.; Scott, E.J.; Larabee, J.L.; James, J.A.; Ballard, J.D.; Lang, M.L. Human C. difficile toxin–specific memory B cell repertoires encode poorly neutralizing antibodies. JCI Insight 2020, 5, 138137. [Google Scholar] [CrossRef]
- Beckett, J.; Hester, J.; Issa, F.; Shankar, S. Regulatory B cells in transplantation: Roadmaps to clinic. Transpl. Int. 2020, 33, 1353–1368. [Google Scholar] [CrossRef]
- Burrello, C.; Garavaglia, F.; Cribiù, F.M.; Ercoli, G.; Lopez, G.; Troisi, J.; Colucci, A.; Guglietta, S.; Carloni, S.; Guglielmetti, S.; et al. Therapeutic faecal microbiota transplantation controls intestinal inflammation through IL10 secretion by immune cells. Nat. Commun. 2018, 9, 5184. [Google Scholar] [CrossRef] [PubMed]
- Amrouche, K.; Pers, J.-O.; Jamin, C. Glatiramer Acetate Stimulates Regulatory B Cell Functions. J. Immunol. 2019, 202, 1970–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaiserman, A.M.; Koliada, A.; Marotta, F. Gut microbiota: A player in aging and a target for anti-aging intervention. Ageing Res. Rev. 2017, 35, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Bárcena, C.; Valdés-Mas, R.; Mayoral, P.; Garabaya, C.; Durand, S.; Rodríguez, F.; Fernández-García, M.T.; Salazar, N.; Nogacka, A.M.; Garatachea, N.; et al. Healthspan and lifespan extension by fecal microbiota transplantation into progeroid mice. Nat. Med. 2019, 25, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Stebegg, M.; Silva-Cayetano, A.; Innocentin, S.; Jenkins, T.P.; Cantacessi, C.; Gilbert, C.; Linterman, M.A. Heterochronic faecal transplantation boosts gut germinal centres in aged mice. Nat. Commun. 2019, 10, 2443. [Google Scholar] [CrossRef]
- Schluter, J.; Peled, J.; Taylor, B.P.; Markey, K.A.; Smith, J.A.; Taur, Y.; Niehus, R.; Staffas, A.; Dai, A.; Fontana, E.; et al. The gut microbiota is associated with immune cell dynamics in humans. Nature 2020, 588, 303–307. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Routy, B.; Le Chatelier, E.; DeRosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2017, 359, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuth, T.; van Baarle, D.; van Erpecum, K.J.; Siersema, P.D.; Hoepelman, A.I.M.; Arends, J.E. CD4/CD8 ratio is a promising candidate for non-invasive measurement of liver fibrosis in chronic HCV-monoinfected patients. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1113–1117. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Participant ID | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|---|---|---|---|
| Sex | Male | Female | Female | Male |
| Age | 70 | 61 | 85 | 84 |
| Comorbidities | Chronic pain, NASH cirrhosis (MELD score 9), bariatric Roux-en-Y surgery, chronic obstructive pulmonary disease, depression, atrial fibrillation, hypothyroidism | Congenital blindness in left eye, anxiety | Hypertension, moderate aortic stenosis, oseoarthritis | Hypothyroidism, type 2 diabetes, hypertension, myocardial infarction abdominal aortic aneurysm, benign prostatic hypertrophy, chronic kidney disease, prior laparotomy for diverticulosis and small bowel obstruction |
| Pertinent Medications | Hydromorphone, Flomax, Furosemide, Breo-Ellipta, Synthroid, Apixaban | Temazepam, Citalopram, Gabapentin | Rosuvastatin, Perindopril, Hydrochlorothiazide | Lipitor, Fenofibrate, Synthroid, Lopressor, Flomax |
| Treatment outcome | Failure | Success | Success | Success |
| Number of prior CDI | 4 | 1 | None | None |
| FMT prior to study enrolment | >5 | None | None | None |
| CDI Severity | Fulminant | Fulminant | Severe | Fulminant |
| Anti-CDI Antibiotics during treatment cycles (Total Days) | Fidaxomicin (16 days) | Fidaxomicin (18 days) | Fidaxomicin (11 days) | Metronidazole IV and Vancomycin PO (25 days) |
| Number of treatment cycles * | 2 | 2 | 2 | 5 FMTs by colonoscopy |
| Feature | Category | p-Value | Fold Change (log2) (Succ/Fail) | Mean Value (Responders) | |
|---|---|---|---|---|---|
| Features with higher mean in FMT responders | |||||
| Naïve:memory CD8 T cell ratio | Flow cytometry | 0.0007 | 2.8982 | 0.1109 | 0.0149 |
| Naïve CD8 T cells (%) | Flow cytometry | 0.0005 | 2.6972 | 9.4820 | 1.4620 |
| miR-451a | Serum microRNA | 0.0040 | 2.3704 | 2.2922 | 0.4433 |
| Regulatory B cells; Bregs (%) | Flow cytometry | 0.0008 | 2.1593 | 4.2080 | 0.9420 |
| Toxin B IgG * | Antigen-specific antibody panel | 0.0471 | 1.4260 | 8.7778 | 3.2667 |
| Total B cell (%) | Flow cytometry | 0.0007 | 1.3481 | 7.6880 | 3.0200 |
| miR-16 | Serum microRNA | 0.0071 | 1.3251 | 1.5138 | 0.6042 |
| CD4:CD8 T cell ratio | Flow cytometry | 0.0002 | 1.2064 | 1.7049 | 0.7388 |
| IgM | Isotype panel | 0.0395 | 0.9896 | 1.1970 | 0.6028 |
| EMRA CD4 T cells (%) | Flow cytometry | 0.0390 | 0.9057 | 25.0740 | 13.3840 |
| CD28 expression levels on CD4 T cells (MFI) | Flow cytometry | 0.0022 | 0.8997 | 44.6770 | 23.9460 |
| Unswitched memory B cells (%) | Flow cytometry | 0.0106 | 0.8939 | 15.3640 | 8.2680 |
| IL4+ve stimulated CD8 T cells (%) | Flow cytometry | 0.0323 | 0.8806 | 2.0290 | 1.1020 |
| Glycodeoxycholic acid | Stool bile acids | 0.0221 | 0.7647 | 6.4343 | 3.7870 |
| A027 IgM [‘A’ = surface layer proteins (SLP) of ribotype 027] | Antigen-specific antibody panel | 0.0240 | 0.6851 | 5.1609 | 3.2099 |
| Stimulated CD4 T cells IL4 expression levels (MFI) | Flow cytometry | 0.0277 | 0.6786 | 10.0230 | 6.2620 |
| IL4+ve stimulated CD4 T cells (%) | Flow cytometry | 0.0451 | 0.6227 | 2.5160 | 1.6340 |
| Total CD4 T cells (%) | Flow cytometry | 0.0001 | 0.4822 | 55.8040 | 39.9500 |
| CD28 expression levels on CD4 T cells (MFI) | Flow cytometry | 0.0001 | 0.4684 | 28.6430 | 20.7020 |
| Total memory B cells (%) | Flow cytometry | 0.0241 | 0.4349 | 51.7350 | 38.2700 |
| IgGII1H5N4F1S1: IgG2&3 glycopeptide with digalactosylated and monosialylated glycan with core fucose | IgG glycoprofiling | 0.0289 | 0.3593 | 7.8840 | 6.1460 |
| Stimulated CD8 T cells IL4 expression levels (MFI) | Flow cytometry | 0.0030 | 0.3565 | 2.3670 | 1.8488 |
| IgGII1H4N4F1: IgG2&3 glycopeptide with monogalactosylated glycan with core fucose | IgG glycoprofiling | 0.0059 | 0.3545 | 14.2693 | 11.1605 |
| CD28 expression levels on CD8 T cells (MFI) | Flow cytometry | 0.0315 | 0.3306 | 23.2890 | 18.5200 |
| IgGIV1H5N4F1: IgG4 glycopeptide with digalactosylated glycan with core fucose | IgG glycoprofiling | 0.0471 | 0.2910 | 4.0918 | 3.3443 |
| Monosialylated glycans | Serum glycan traits | 0.0014 | 0.2556 | 16.6257 | 13.9260 |
| NKG2D expression levels on CD4 T cells (MFI) | Flow cytometry | 0.0003 | 0.2181 | 5.2907 | 4.5483 |
| IgGI1H4N4F1: IgG1 glycopeptide with monogalactosylated glycan with core fucose | IgG glycoprofiling | 0.0140 | 0.1917 | 20.8725 | 18.2761 |
| Candida IgM | Antigen-specific antibody panel | 0.0009 | 0.1841 | 7.2038 | 6.3409 |
| Senescent CD4 T cells NKG2D expression levels (MFI) | Flow cytometry | 0.0011 | 0.1781 | 5.2329 | 4.6252 |
| Digalactosylated glycans | Serum glycan traits | 0.0042 | 0.1467 | 54.6643 | 49.3780 |
| MMP-1: matrix metalloproteinase-1 | Inflammation panel | 0.0072 | 0.1443 | 7.3383 | 6.6399 |
| Low-branching glycans | Serum glycan traits | 0.0155 | 0.1114 | 72.3240 | 66.9480 |
| Features with higher mean in FMT non-responder | |||||
| Acidaminococcaceae | Family | 0.0212 | −3.6351 | 0.1625 | 2.0193 |
| Phascolarctobacterium | Genus | 0.0212 | −3.6351 | 0.1625 | 2.0193 |
| Enterobacteriaceae_unclassified | Genus | 0.0013 | −2.2113 | 1.0058 | 4.6577 |
| Pseudocitrobacter | Genus | 0.0080 | −2.2079 | 0.7009 | 3.2383 |
| Enterococcaceae | Family | 0.0035 | −1.8467 | 1.4509 | 5.2185 |
| Enterococcus | Genus | 0.0035 | −1.8467 | 1.4509 | 5.2185 |
| L001 IgA (‘L’ = lysates of ribotype 001) | Antigen-specific antibody panel | 0.0000 | −1.5667 | 24.6667 | 73.0667 |
| 3-alpha-hydroxy-7,12-diketocholanic acid | Stool bile acids | 0.0189 | −1.4326 | 2.0780 | 5.6094 |
| CMV IgG | Antigen-specific antibody panel | 0.0000 | −1.0003 | 3350.2467 | 6702.0667 |
| Toxin B IgA* | Antigen-specific antibody panel | 0.0194 | −0.9586 | 1.5437 | 3.0000 |
| A001 IgA [‘A’ = surface layer proteins (SLP) of ribotype 001] | Antigen-specific antibody panel | 0.0381 | −0.8841 | 2.1246 | 3.9212 |
| 3 dehydrocholic acid | Stool bile acids | 0.0082 | −0.8481 | 4.0741 | 7.3340 |
| Beta muricholic acid | Stool bile acids | 0.0004 | −0.8238 | 4.3596 | 7.7168 |
| A027 IgG [‘A’ = surface layer proteins (SLP) of ribotype 027] | Antigen-specific antibody panel | 0.0499 | −0.7879 | 3.1054 | 5.3618 |
| CD28−ve T cells (%) | Flow cytometry | 0.0000 | −0.7757 | 35.0250 | 59.9640 |
| sTNF-R1: soluble tumor necrosis factor receptor-1 | Inflammation panel | 0.0019 | −0.7465 | 3079.3947 | 5166.5200 |
| IL-26 | Inflammation panel | 0.0267 | −0.7191 | 1044.0507 | 1718.7220 |
| Integrin+ve dendritic cells (%) | Flow cytometry | 0.0000 | −0.7071 | 2.0863 | 3.4059 |
| sTNF-R2 | Inflammation panel | 0.0150 | −0.6979 | 1274.3983 | 2067.2920 |
| Total CD8 T cells (%) | Flow cytometry | 0.0000 | −0.6956 | 33.6670 | 54.5240 |
| 12 dehydrocholic acid | Stool bile acids | 0.0135 | −0.6126 | 6.7393 | 10.3044 |
| Chenodeoxycholic acid | Stool bile acids | 0.0000 | −0.5788 | 8.4395 | 12.6054 |
| CD28−veCD57+ve senescent CD8 T cells (%) | Flow cytometry | 0.0008 | −0.5387 | 46.6120 | 67.7120 |
| Antennary fucosylation | Serum glycan traits | 0.0017 | −0.5289 | 9.8970 | 14.2800 |
| CD28−ve senescent CD8 T cells (%) | Flow cytometry | 0.0002 | −0.4370 | 61.7170 | 83.5540 |
| CD28−veCD57+ve senescent CD4 T cells (%) | Flow cytometry | 0.0497 | −0.4303 | 23.3810 | 31.5060 |
| Tetragalactosylated glycans | Serum glycan traits | 0.0017 | −0.4232 | 4.9430 | 6.6280 |
| Cholic acid-3-sulfate | Stool bile acids | 0.0362 | −0.4122 | 5.5642 | 7.4042 |
| IgGIV1H3N5F1: IgG4 glycopeptide with bisected agalactosylated glycan with core fucose | IgG glycoprofiling | 0.0161 | −0.3958 | 8.3615 | 11.0012 |
| Tetrasialylated glycans | Serum glycan traits | 0.0074 | −0.3942 | 4.2567 | 5.5940 |
| Chenodeoxycholic acid-3-sulfate | Stool bile acids | 0.0412 | −0.3777 | 7.2848 | 9.4649 |
| CD8 effector memory T cells (%) | Flow cytometry | 0.0009 | −0.3580 | 58.0380 | 74.3860 |
| CD57+ve senescent CD8 T cells (%) | Flow cytometry | 0.0290 | −0.3452 | 55.0520 | 69.9360 |
| IgGII1H3N4: IgG2&3 glycopeptide with agalactosylated glycan without core fucose | IgG glycoprofiling | 0.0051 | −0.3291 | 1.5296 | 1.9215 |
| High-branching glycans | Serum glycan traits | 0.0071 | −0.2972 | 25.0977 | 30.8400 |
| IgGII1H4N4: IgG2&3 glycopeptide with monogalactosylated glycan without core fucose | IgG glycoprofiling | 0.0276 | −0.2911 | 3.3512 | 4.1005 |
| Trisialylated glycans | Serum glycan traits | 0.0207 | −0.2873 | 16.2837 | 19.8720 |
| Trigalactosylated glycans | Serum glycan traits | 0.0327 | −0.2646 | 20.1547 | 24.2120 |
| Total T cells (%) | Flow cytometry | 0.0077 | −0.2526 | 57.9910 | 69.0860 |
| IgGII1H4N5S1: IgG2&3 glycopeptide with bisected monogalactosylated and monosialylated glycan without core fucose | IgG glycoprofiling | 0.0458 | −0.2491 | 1.8785 | 2.2325 |
| MMP-2: matrix metalloproteinase-2 | Inflammation panel | 0.0000 | −0.2025 | 9.3344 | 10.7411 |
| Candida IgG | Antigen-specific antibody panel | 0.0001 | −0.1961 | 8.0645 | 9.2384 |
| Total CD8 memory T cells (%) | Flow cytometry | 0.0015 | −0.1777 | 87.0120 | 98.4160 |
| TWEAK/TNFSF12: TNF-like weak inducer of apoptosis/tumor necrosis factor superfamily | Inflammation panel | 0.0033 | −0.1684 | 5.4639 | 6.1403 |
| Integrin expression levels on dendritic cells (MFI) | Flow cytometry | 0.0235 | −0.1316 | 3.7044 | 4.0582 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monaghan, T.M.; Duggal, N.A.; Rosati, E.; Griffin, R.; Hughes, J.; Roach, B.; Yang, D.Y.; Wang, C.; Wong, K.; Saxinger, L.; et al. A Multi-Factorial Observational Study on Sequential Fecal Microbiota Transplant in Patients with Medically Refractory Clostridioides difficile Infection. Cells 2021, 10, 3234. https://doi.org/10.3390/cells10113234
Monaghan TM, Duggal NA, Rosati E, Griffin R, Hughes J, Roach B, Yang DY, Wang C, Wong K, Saxinger L, et al. A Multi-Factorial Observational Study on Sequential Fecal Microbiota Transplant in Patients with Medically Refractory Clostridioides difficile Infection. Cells. 2021; 10(11):3234. https://doi.org/10.3390/cells10113234
Chicago/Turabian StyleMonaghan, Tanya M., Niharika A. Duggal, Elisa Rosati, Ruth Griffin, Jamie Hughes, Brandi Roach, David Y. Yang, Christopher Wang, Karen Wong, Lynora Saxinger, and et al. 2021. "A Multi-Factorial Observational Study on Sequential Fecal Microbiota Transplant in Patients with Medically Refractory Clostridioides difficile Infection" Cells 10, no. 11: 3234. https://doi.org/10.3390/cells10113234