
Molecular Mechanisms of Hypertensive Nephropathy: Renoprotective Effect of Losartan through Hsp70

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
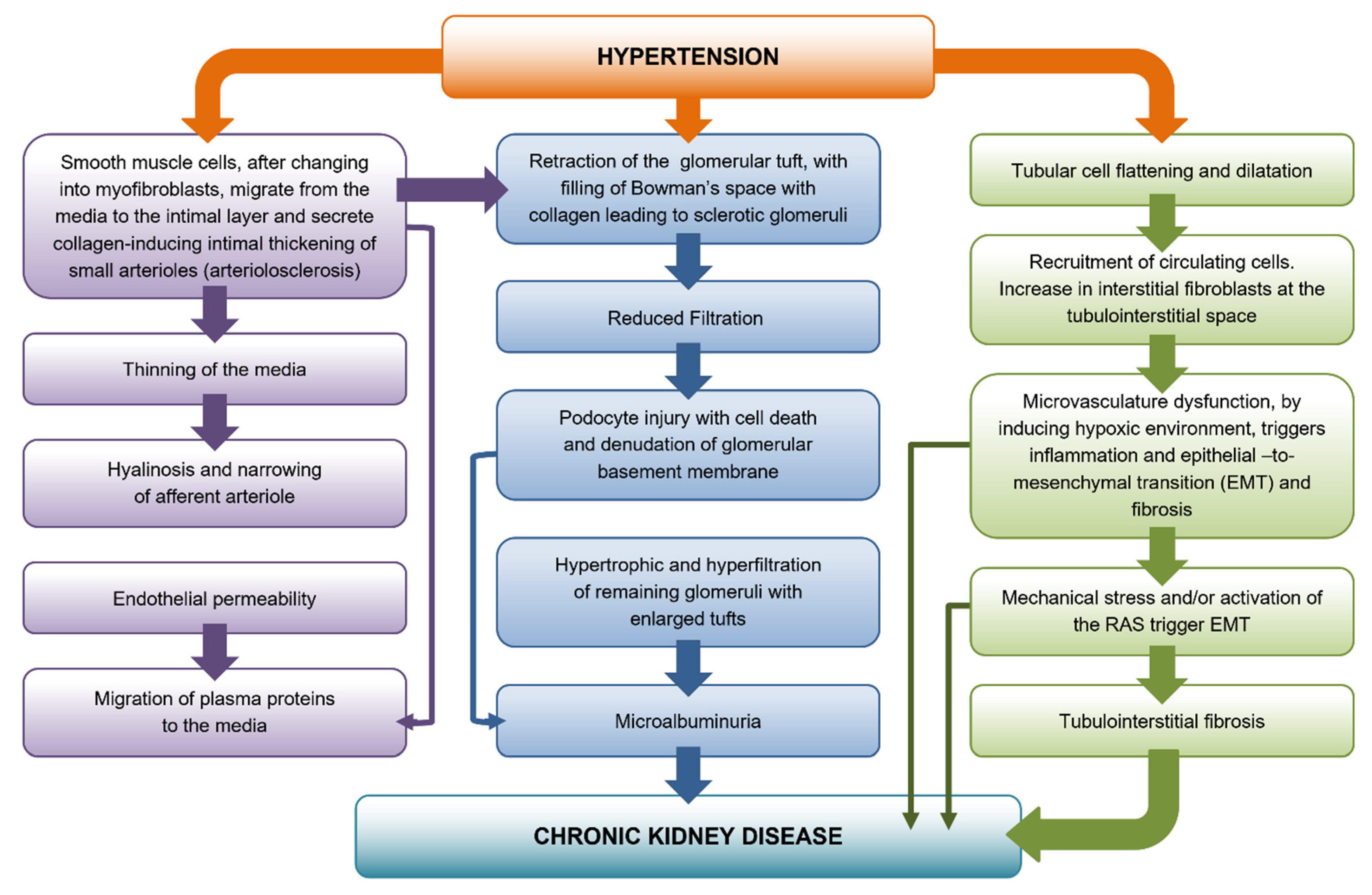
2. Development of Preglomerular Vascular Lesions
Afferent Arteriolar Hyalinosis
3. Glomerular Damage
4. Podocyte Depletion
5. Tubulointerstitial Fibrosis
Angiotensin II and Tubulointerstitial Fibrosis
6. Involvement of Heat Shock Protein 70 in the Antioxidative Action of Losartan in Proximal Tubule Cells
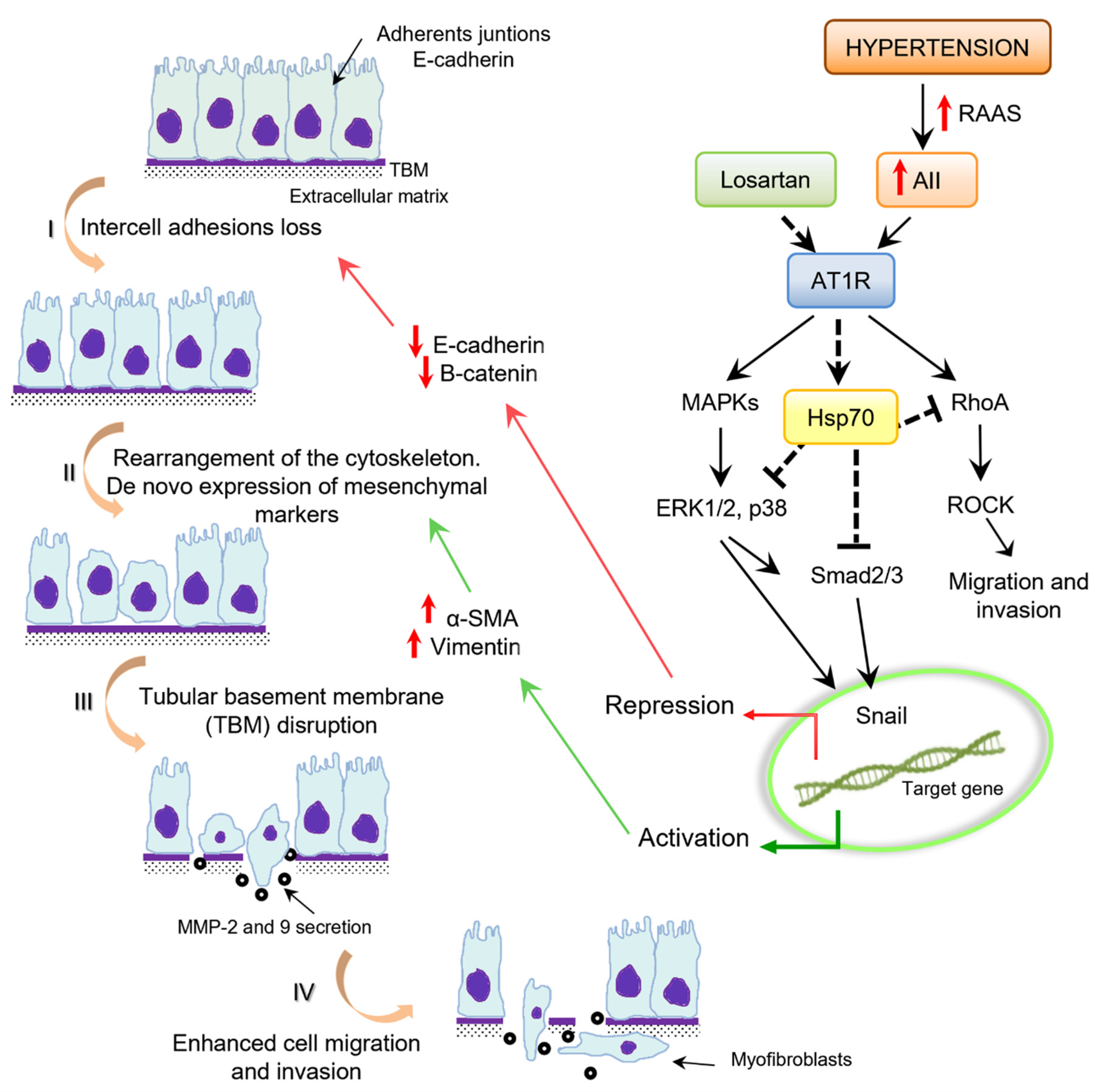
7. Epithelial–Mesenchymal Transition
8. Angiotensin II and Epithelial–Mesenchymal Transition
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ESRD | end-stage renal disease |
| BP | blood pressure |
| CKD | chronic kidney disease |
| RAS | renin–angiotensin system |
| EMT | epithelial–mesenchymal transition |
| Hsp 70 | heat shock proteins 70 KDa |
| ECM | extracellular matrix |
| GBM | glomerular basement membrane |
| FSGS | focal segmental glomerulosclerosis |
| RBF | renal blood flow |
| GFR | glomerular filtration rate |
| PGC | glomerular capillary hydraulic pressure |
| TIF | tubulointerstitial fibrosis |
| PTECs | proximal tubular epithelial cells |
| ROS | reactive oxygen species |
| AT1R | angiotensin II type 1 receptor |
| SHR | spontaneously hypertensive rats |
| VDR | vitamin D receptor |
| TGFβRI | serine/threonine kinase receptor of TGF-β/Smad signaling |
References
- Udani, S.; Lazich, I.; Bakris, G.L. Epidemiology of hypertensive kidney disease. Nat. Rev. Nephrol. 2011, 7, 11–21. [Google Scholar] [CrossRef]
- Seccia, T.M.; Caroccia, B.; Calo, L.A. Hypertensive nephropathy. Moving from classic to emerging pathogenetic mechanisms. J. Hypertens. 2017, 35, 205–212. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Rodriguez-Vita, J.; Sanchez-Lopez, E.; Carvajal, G.; Egido, J. TGF-beta signaling in vascular fibrosis. Cardiovasc Res. 2007, 74, 196–206. [Google Scholar] [CrossRef] [Green Version]
- Bakin, A.V.; Rinehart, C.; Tomlinson, A.K.; Arteaga, C.L. p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migration. J. Cell Sci. 2002, 115, 3193–3206. [Google Scholar] [CrossRef]
- Patel, S.; Takagi, K.I.; Suzuki, J.; Imaizumi, A.; Kimura, T.; Mason, R.M.; Kamimura, T.; Zhang, Z. RhoGTPase activation is a key step in renal epithelial mesenchymal transdifferentiation. J. Am. Soc. Nephrol. 2005, 16, 1977–1984. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, J.L.; Kobori, H.; Li, X.C.; Satou, R.; Katsurada, A.; Navar, L.G. Augmentation of angiotensinogen expression in the proximal tubule by intracellular angiotensin II via AT1a/MAPK/NF-small ka, CyrillicB signaling pathways. Am. J. Physiol. Ren. Physiol. 2016, 310, F1103–F1112. [Google Scholar] [CrossRef] [Green Version]
- Carvajal, G.; Rodriguez-Vita, J.; Rodrigues-Diez, R.; Sanchez-Lopez, E.; Ruperez, M.; Cartier, C.; Esteban, V.; Ortiz, A.; Egido, J.; Mezzano, S.A.; et al. Angiotensin II activates the Smad pathway during epithelial mesenchymal transdifferentiation. Kidney Int. 2008, 74, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 4–19. [Google Scholar] [CrossRef]
- Zuiderweg, E.R.; Hightower, L.E.; Gestwicki, J.E. The remarkable multivalency of the Hsp70 chaperones. Cell Stress Chaperones 2017, 22, 173–189. [Google Scholar] [CrossRef] [Green Version]
- Tracy, R.E.; Malcom, G.T.; Oalmann, M.C.; Qureshi, U.; Ishii, T.; Velez-Duran, M. Renal microvascular features of hypertension in Japan, Guatemala, and the United States. Arch. Pathol. Lab. Med. 1992, 116, 50–55. [Google Scholar]
- Hill, G.S. Hypertensive nephrosclerosis. Curr. Opin. Nephrol. Hypertens. 2008, 17, 266–270. [Google Scholar] [CrossRef]
- Alsaad, K.O.; Herzenberg, A.M. Distinguishing diabetic nephropathy from other causes of glomerulosclerosis: An update. J. Clin. Pathol. 2007, 60, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Kremers, W.K.; Denic, A.; Lieske, J.C.; Alexander, M.P.; Kaushik, V.; Elsherbiny, H.E.; Chakkera, H.A.; Poggio, E.D.; Rule, A.D. Distinguishing age-related from disease-related glomerulosclerosis on kidney biopsy: The Aging Kidney Anatomy study. Nephrol. Dial. Transpl. 2015, 30, 2034–2039. [Google Scholar] [CrossRef] [Green Version]
- Hamet, P.; Thorin-Trescases, N.; Moreau, P.; Dumas, P.; Tea, B.S.; de Blois, D.; Kren, V.; Pravenec, M.; Kunes, J.; Sun, Y.; et al. Workshop: Excess growth and apoptosis: Is hypertension a case of accelerated aging of cardiovascular cells? Hypertension 2001, 37, 760–766. [Google Scholar] [CrossRef] [Green Version]
- Cirillo, M.; Stellato, D.; Laurenzi, M.; Panarelli, W.; Zanchetti, A.; De Santo, N.G. Pulse pressure and isolated systolic hypertension: Association with microalbuminuria. The GUBBIO Study Collaborative Research Group. Kidney Int. 2000, 58, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Hill, G.S.; Heudes, D.; Bariety, J. Morphometric study of arterioles and glomeruli in the aging kidney suggests focal loss of autoregulation. Kidney Int. 2003, 63, 1027–1036. [Google Scholar] [CrossRef] [Green Version]
- Hill, G.S.; Heudes, D.; Jacquot, C.; Gauthier, E.; Bariety, J. Morphometric evidence for impairment of renal autoregulation in advanced essential hypertension. Kidney Int. 2006, 69, 823–831. [Google Scholar] [CrossRef] [Green Version]
- Marcantoni, C.; Ma, L.J.; Federspiel, C.; Fogo, A.B. Hypertensive nephrosclerosis in African Americans versus Caucasians. Kidney Int. 2002, 62, 172–180. [Google Scholar] [CrossRef] [Green Version]
- Bidani, A.K.; Griffin, K.A.; Williamson, G.; Wang, X.; Loutzenhiser, R. Protective importance of the myogenic response in the renal circulation. Hypertension 2009, 54, 393–398. [Google Scholar] [CrossRef]
- Renal disease caused by hypertension. In Heptinstall’s Pathology of the Kidney; Jennette, J.C.; Olson, J.L.; Schwartz, M.M.; Silva, F.G. (Eds.) Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2006; pp. 937–990. [Google Scholar]
- Appel, L.J.; Wright, J.T., Jr.; Greene, T.; Agodoa, L.Y.; Astor, B.C.; Bakris, G.L.; Cleveland, W.H.; Charleston, J.; Contreras, G.; Faulkner, M.L.; et al. Intensive blood-pressure control in hypertensive chronic kidney disease. N. Engl. J. Med. 2010, 363, 918–929. [Google Scholar] [CrossRef] [Green Version]
- Appel, L.J.; Wright, J.T., Jr.; Greene, T.; Kusek, J.W.; Lewis, J.B.; Wang, X.; Lipkowitz, M.S.; Norris, K.C.; Bakris, G.L.; Rahman, M.; et al. Long-term effects of renin-angiotensin system-blocking therapy and a low blood pressure goal on progression of hypertensive chronic kidney disease in African Americans. Arch. Intern. Med. 2008, 168, 832–839. [Google Scholar] [CrossRef] [Green Version]
- Genovese, G.; Friedman, D.J.; Ross, M.D.; Lecordier, L.; Uzureau, P.; Freedman, B.I.; Bowden, D.W.; Langefeld, C.D.; Oleksyk, T.K.; Uscinski Knob, A.L.; et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010, 329, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Kopp, J.B.; Smith, M.W.; Nelson, G.W.; Johnson, R.C.; Freedman, B.I.; Bowden, D.W.; Oleksyk, T.; McKenzie, L.M.; Kajiyama, H.; Ahuja, T.S.; et al. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat. Genet. 2008, 40, 1175–1184. [Google Scholar] [CrossRef] [Green Version]
- Marcantoni, C.; Fogo, A.B. A perspective on arterionephrosclerosis: From pathology to potential pathogenesis. J. Nephrol. 2007, 20, 518–524. [Google Scholar]
- Meyrier, A. Nephrosclerosis: A term in quest of a disease. Nephron 2015, 129, 276–282. [Google Scholar] [CrossRef]
- Meyrier, A. Nephrosclerosis: Update on a centenarian. Nephrol. Dial. Transpl. 2015, 30, 1833–1841. [Google Scholar] [CrossRef] [Green Version]
- Kriz, W.; Lemley, K.V. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J. Am. Soc. Nephrol. 2015, 26, 258–269. [Google Scholar] [CrossRef] [Green Version]
- Cellesi, F.; Li, M.; Rastaldi, M.P. Podocyte injury and repair mechanisms. Curr. Opin. Nephrol. Hypertens. 2015, 24, 239–244. [Google Scholar] [CrossRef]
- Drumond, M.C.; Deen, W.M. Structural determinants of glomerular hydraulic permeability. Am. J. Physiol. 1994, 266, F1–F12. [Google Scholar] [CrossRef]
- Kriz, W.; Shirato, I.; Nagata, M.; LeHir, M.; Lemley, K.V. The podocyte’s response to stress: The enigma of foot process effacement. Am. J. Physiol. Ren. Physiol. 2013, 304, F333–F347. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, A.; Chowdhury, M.A.; Venkatareddy, M.P.; Wang, S.Q.; Nishizono, R.; Suzuki, T.; Wickman, L.T.; Wiggins, J.E.; Muchayi, T.; Fingar, D.; et al. Growth-dependent podocyte failure causes glomerulosclerosis. J. Am. Soc. Nephrol. 2012, 23, 1351–1363. [Google Scholar] [CrossRef] [Green Version]
- Petermann, A.; Floege, J. Podocyte damage resulting in podocyturia: A potential diagnostic marker to assess glomerular disease activity. Nephron Clin. Pr. 2007, 106, c61–c66. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W. The pathogenesis of ‘classic’ focal segmental glomerulosclerosis-lessons from rat models. Nephrol. Dial. Transpl. 2003, 18 (Suppl. 6), vi39–vi44. [Google Scholar] [CrossRef]
- Farris, A.B.; Colvin, R.B. Renal interstitial fibrosis: Mechanisms and evaluation. Curr. Opin. Nephrol. Hypertens. 2012, 21, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.C.; Tang, T.T.; Lv, L.L.; Lan, H.Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef]
- Freedman, B.I.; Cohen, A.H. Hypertension-attributed nephropathy: What’s in a name? Nat. Rev. Nephrol. 2016, 12, 27–36. [Google Scholar] [CrossRef]
- Fine, L.G.; Norman, J.T. Chronic hypoxia as a mechanism of progression of chronic kidney diseases: From hypothesis to novel therapeutics. Kidney Int. 2008, 74, 867–872. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Ning, X.; Zhang, Y.; Lu, Y.; Nie, Y.; Han, S.; Liu, L.; Du, R.; Xia, L.; He, L.; et al. Hypoxia-inducible factor-1alpha induces Twist expression in tubular epithelial cells subjected to hypoxia, leading to epithelial-to-mesenchymal transition. Kidney Int. 2009, 75, 1278–1287. [Google Scholar] [CrossRef] [Green Version]
- Eddy, A.A. Progression in chronic kidney disease. Adv. Chronic Kidney Dis. 2005, 12, 353–365. [Google Scholar] [CrossRef]
- Leonard, M.O.; Cottell, D.C.; Godson, C.; Brady, H.R.; Taylor, C.T. The role of HIF-1 alpha in transcriptional regulation of the proximal tubular epithelial cell response to hypoxia. J. Biol. Chem. 2003, 278, 40296–40304. [Google Scholar] [CrossRef] [Green Version]
- Hill, P.; Shukla, D.; Tran, M.G.; Aragones, J.; Cook, H.T.; Carmeliet, P.; Maxwell, P.H. Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2008, 19, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norman, J.T.; Fine, L.G. Intrarenal oxygenation in chronic renal failure. Clin. Exp. Pharm. Physiol. 2006, 33, 989–996. [Google Scholar] [CrossRef]
- Nangaku, M. Chronic hypoxia and tubulointerstitial injury: A final common pathway to end-stage renal failure. J. Am. Soc. Nephrol. 2006, 17, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Nangaku, M.; Eckardt, K.U. Hypoxia and the HIF system in kidney disease. J. Mol. Med. 2007, 85, 1325–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyman, S.N.; Khamaisi, M.; Rosen, S.; Rosenberger, C. Renal parenchymal hypoxia, hypoxia response and the progression of chronic kidney disease. Am. J. Nephrol. 2008, 28, 998–1006. [Google Scholar] [CrossRef]
- Choi, Y.J.; Chakraborty, S.; Nguyen, V.; Nguyen, C.; Kim, B.K.; Shim, S.I.; Suki, W.N.; Truong, L.D. Peritubular capillary loss is associated with chronic tubulointerstitial injury in human kidney: Altered expression of vascular endothelial growth factor. Hum. Pathol. 2000, 31, 1491–1497. [Google Scholar] [CrossRef]
- Gill, P.S.; Wilcox, C.S. NADPH oxidases in the kidney. Antioxid Redox Signal. 2006, 8, 1597–1607. [Google Scholar] [CrossRef]
- Hitomi, H.; Kiyomoto, H.; Nishiyama, A. Angiotensin II and oxidative stress. Curr. Opin. Cardiol. 2007, 22, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Seikaly, M.G.; Arant, B.S., Jr.; Seney, F.D., Jr. Endogenous angiotensin concentrations in specific intrarenal fluid compartments of the rat. J. Clin. Investig. 1990, 86, 1352–1357. [Google Scholar] [CrossRef] [Green Version]
- Zuo, L.; Ushio-Fukai, M.; Hilenski, L.L.; Alexander, R.W. Microtubules regulate angiotensin II type 1 receptor and Rac1 localization in caveolae/lipid rafts: Role in redox signaling. Arter. Thromb. Vasc. Biol. 2004, 24, 1223–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocanegra, V.; Manucha, W.; Pena, M.R.; Cacciamani, V.; Valles, P.G. Caveolin-1 and Hsp70 interaction in microdissected proximal tubules from spontaneously hypertensive rats as an effect of Losartan. J. Hypertens. 2010, 28, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wei, C.C.; Wu, S.J.; Chenier, I.; Zhang, S.L.; Filep, J.G.; Ingelfinger, J.R.; Chan, J.S. Apocynin attenuates tubular apoptosis and tubulointerstitial fibrosis in transgenic mice independent of hypertension. Kidney Int. 2009, 75, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, J.L.; Carretero, O.A.; Li, X.C. Effects of AT1 receptor-mediated endocytosis of extracellular Ang II on activation of nuclear factor-κB in proximal tubule cells. Ann. N. Y. Acad. Sci. 2006, 1091, 336–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezzano, S.A.; Ruiz-Ortega, M.; Egido, J. Angiotensin II and renal fibrosis. Hypertension 2001, 38, 635–638. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.K.; Williams, H.; Li, S.C.H.; Fletcher, J.P.; Medbury, H.J. Monocyte inflammatory profile is specific for individuals and associated with altered blood lipid levels. Atherosclerosis 2017, 263, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Dogan, A.; Oylumlu, M. Increased monocyte-to-HDL cholesterol ratio is related to cardiac syndrome X. Acta Cardiol. 2017, 72, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Gembillo, G.; Siligato, R.; Cernaro, V.; Satta, E.; Conti, G.; Salvo, A.; Romeo, A.; Calabrese, V.; Sposito, G.; Ferlazzo, G.; et al. Monocyte to HDL ratio: A novel marker of resistant hypertension in CKD patients. Int. Urol. Nephrol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, Z.; Cao, K.; Fang, D.; Wang, F.; Bi, G.; Yang, J.; He, Y.; Wu, J.; Wei, Y.; et al. Adjunctive therapy with statins reduces residual albuminuria/proteinuria and provides further renoprotection by downregulating the angiotensin II-AT1 pathway in hypertensive nephropathy. J. Hypertens. 2017, 35, 1442–1456. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ma, J.; Zhang, X.; Fan, Y.; Wang, L. Protective role of the vitamin D receptor. Cell Immunol. 2012, 279, 160–166. [Google Scholar] [CrossRef]
- Rachez, C.; Freedman, L.P. Mechanisms of gene regulation by vitamin D(3) receptor: A network of coactivator interactions. Gene 2000, 246, 9–21. [Google Scholar] [CrossRef]
- Chandel, N.; Ayasolla, K.; Wen, H.; Lan, X.; Haque, S.; Saleem, M.A.; Malhotra, A.; Singhal, P.C. Vitamin D receptor deficit induces activation of renin angiotensin system via SIRT1 modulation in podocytes. Exp. Mol. Pathol. 2017, 102, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Gembillo, G.; Cernaro, V.; Salvo, A.; Siligato, R.; Laudani, A.; Buemi, M.; Santoro, D. Role of Vitamin D Status in Diabetic Patients with Renal Disease. Medicina 2019, 55, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husain, K.; Ferder, L.; Mizobuchi, M.; Finch, J.; Slatopolsky, E. Combination therapy with paricalcitol and enalapril ameliorates cardiac oxidative injury in uremic rats. Am. J. Nephrol. 2009, 29, 465–472. [Google Scholar] [CrossRef]
- Dong, J.; Wong, S.L.; Lau, C.W.; Lee, H.K.; Ng, C.F.; Zhang, L.; Yao, X.; Chen, Z.Y.; Vanhoutte, P.M.; Huang, Y. Calcitriol protects renovascular function in hypertension by down-regulating angiotensin II type 1 receptors and reducing oxidative stress. Eur. Heart J. 2012, 33, 2980–2990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seccia, T.M.; Belloni, A.S.; Guidolin, D.; Sticchi, D.; Nussdorfer, G.G.; Pessina, A.C.; Rossi, G.P. The renal antifibrotic effects of angiotensin-converting enzyme inhibition involve bradykinin B2 receptor activation in angiotensin II-dependent hypertension. J. Hypertens. 2006, 24, 1419–1427. [Google Scholar] [CrossRef]
- Seccia, T.M.; Maniero, C.; Belloni, A.S.; Guidolin, D.; Pothen, P.; Pessina, A.C.; Rossi, G.P. Role of angiotensin II, endothelin-1 and L-type calcium channel in the development of glomerular, tubulointerstitial and perivascular fibrosis. J. Hypertens. 2008, 26, 2022–2029. [Google Scholar] [CrossRef]
- Jiang, F.; Yang, J.; Zhang, Y.; Dong, M.; Wang, S.; Zhang, Q.; Liu, F.F.; Zhang, K.; Zhang, C. Angiotensin-converting enzyme 2 and angiotensin 1–7: Novel therapeutic targets. Nat. Rev. Cardiol. 2014, 11, 413–426. [Google Scholar] [CrossRef]
- Tikellis, C.; Bernardi, S.; Burns, W.C. Angiotensin-converting enzyme 2 is a key modulator of the renin-angiotensin system in cardiovascular and renal disease. Curr. Opin. Nephrol. Hypertens. 2011, 20, 62–68. [Google Scholar] [CrossRef]
- Yang, F.; Chung, A.C.; Huang, X.R.; Lan, H.Y. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: The role of Smad3. Hypertension 2009, 54, 877–884. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Huang, X.R.; Lan, H.Y. Smad3 mediates ANG II-induced hypertensive kidney disease in mice. Am. J. Physiol. Ren. Physiol. 2012, 302, F986–F997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Huang, X.R.; Li, A.G.; Liu, F.; Li, J.H.; Truong, L.D.; Wang, X.J.; Lan, H.Y. Signaling mechanism of TGF-beta1 in prevention of renal inflammation: Role of Smad7. J. Am. Soc. Nephrol. 2005, 16, 1371–1383. [Google Scholar] [CrossRef]
- Liu, G.X.; Li, Y.Q.; Huang, X.R.; Wei, L.H.; Zhang, Y.; Feng, M.; Meng, X.M.; Chen, H.Y.; Shi, Y.J.; Lan, H.Y. Smad7 inhibits AngII-mediated hypertensive nephropathy in a mouse model of hypertension. Clin. Sci. 2014, 127, 195–208. [Google Scholar] [CrossRef]
- Liu, G.X.; Li, Y.Q.; Huang, X.R.; Wei, L.; Chen, H.Y.; Shi, Y.J.; Heuchel, R.L.; Lan, H.Y. Disruption of Smad7 promotes ANG II-mediated renal inflammation and fibrosis via Sp1-TGF-beta/Smad3-NF.kappaB-dependent mechanisms in mice. PLoS ONE 2013, 8, e53573. [Google Scholar]
- Liu, Z.; Huang, X.R.; Chen, H.Y.; Fung, E.; Liu, J.; Lan, H.Y. Deletion of Angiotensin-Converting Enzyme-2 Promotes Hypertensive Nephropathy by Targeting Smad7 for Ubiquitin Degradation. Hypertension 2017, 70, 822–830. [Google Scholar] [CrossRef]
- Yang, F.; Huang, X.R.; Chung, A.C.; Hou, C.C.; Lai, K.N.; Lan, H.Y. Essential role for Smad3 in angiotensin II-induced tubular epithelial-mesenchymal transition. J. Pathol. 2010, 221, 390–401. [Google Scholar] [CrossRef]
- Ng, Y.Y.; Hou, C.C.; Wang, W.; Huang, X.R.; Lan, H.Y. Blockade of NFkappaB activation and renal inflammation by ultrasound-mediated gene transfer of Smad7 in rat remnant kidney. Kidney Int. 2005, 94, S83–S91. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.; Yang, F.; Huang, X.R.; Meng, J.; Chen, J.; Bader, M.; Penninger, J.M.; Fung, E.; Yu, X.Q.; Lan, H.Y. Dual deficiency of angiotensin-converting enzyme-2 and Mas receptor enhances angiotensin II-induced hypertension and hypertensive nephropathy. J. Cell Mol. Med. 2020, 24, 13093–13103. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Zhou, Y.; Huang, H. MiR-101a ameliorates AngII-mediated hypertensive nephropathy by blockade of TGFβ/Smad3 and NF-κB signalling in a mouse model of hypertension. Clin. Exp. Pharm. Physiol. 2019, 46, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Kalmar, B.; Greensmith, L. Induction of heat shock proteins for protection against oxidative stress. Adv. Drug Deliv. Rev. 2009, 61, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Assimon, V.A.; Gillies, A.T.; Rauch, J.N.; Gestwicki, J.E. Hsp70 protein complexes as drug targets. Curr. Pharm. Des. 2013, 19, 404–417. [Google Scholar] [CrossRef] [Green Version]
- O’neill, S.; Harrison, E.M.; Ross, J.A.; Wigmore, S.J.; Hughes, J. Heat-shock proteins and acute ischaemic kidney injury. Nephron Exp. Nephrol. 2014, 126, 167–174. [Google Scholar] [CrossRef]
- Chebotareva, N.; Bobkova, I.; Shilov, E. Heat shock proteins and kidney disease: Perspectives of HSP therapy. Cell Stress Chaperones 2017, 22, 319–343. [Google Scholar] [CrossRef] [PubMed]
- Schlecht, R.; Erbse, A.H.; Bukau, B.; Mayer, M.P. Mechanics of Hsp70 chaperones enables differential interaction with client proteins. Nat. Struct. Mol. Biol. 2011, 18, 345–351. [Google Scholar] [CrossRef]
- Valles, P.G.; Bocanegra, V.; Costantino, V.V.; Gil Lorenzo, A.F.; Benardon, M.E.; Cacciamani, V. The renal antioxidative effect of losartan involves heat shock protein 70 in proximal tubule cells. Cell Stress Chaperones 2020, 25, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Anwar, A.; Siegel, D.; Kepa, J.K.; Ross, D. Interaction of the molecular chaperone Hsp70 with human NAD(P)H: Quinone oxidoreductase 1. J. Biol. Chem. 2002, 277, 14060–14067. [Google Scholar] [CrossRef] [Green Version]
- Wickner, S.; Maurizi, M.R.; Gottesman, S. Posttranslational quality control: Folding, refolding, and degrading proteins. Science 1999, 286, 1888–1893. [Google Scholar] [CrossRef]
- Demand, J.; Alberti, S.; Patterson, C.; Hohfeld, J. Cooperation of a ubiquitin domain protein and an E3 ubiquitin ligase during chaperone/proteasome coupling. Curr. Biol. 2001, 11, 1569–1577. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Cao, Z.; Xu, X.; van Meir, E.G.; Lambeth, J.D. Homologs of gp91phox: Cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 2001, 269, 131–140. [Google Scholar] [CrossRef]
- Gil Lorenzo, A.F.; Costantino, V.V.; Appiolaza, M.L.; Cacciamani, V.; Benardon, M.E.; Bocanegra, V.; Valles, P.G. Heat Shock Protein 70 and CHIP Promote Nox4 Ubiquitination and Degradation within the Losartan Antioxidative Effect in Proximal Tubule Cells. Cell Physiol. Biochem. 2015, 36, 2183–2197. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Hills, C.E.; Squires, P.E. TGF-beta1-induced epithelial-to-mesenchymal transition and therapeutic intervention in diabetic nephropathy. Am. J. Nephrol. 2010, 31, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues-Diez, R.; Carvajal-Gonzalez, G.; Sanchez-Lopez, E.; Rodriguez-Vita, J.; Rodrigues, D.R.; Selgas, R.; Ortiz, A.; Egido, J.; Mezzano, S.; Ruiz-Ortega, M. Pharmacological modulation of epithelial mesenchymal transition caused by angiotensin II. Role of ROCK and MAPK pathways. Pharm. Res. 2008, 25, 2447–2461. [Google Scholar] [CrossRef]
- Seccia, T.M.; Caroccia, B.; Gioco, F.; Piazza, M.; Buccella, V.; Guidolin, D.; Guerzoni, E.; Montini, B.; Petrelli, L.; Pagnin, E.; et al. Endothelin-1 Drives Epithelial-Mesenchymal Transition in Hypertensive Nephroangiosclerosis. J. Am. Heart Assoc. 2016, 5, e003888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ortega, M.; Egido, J. Angiotensin II modulates cell growth-related events and synthesis of matrix proteins in renal interstitial fibroblasts. Kidney Int. 1997, 52, 1497–1510. [Google Scholar] [CrossRef] [Green Version]
- Border, W.A.; Noble, N.A. Interactions of transforming growth factor-beta and angiotensin II in renal fibrosis. Hypertension 1998, 31, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.J.; Alpers, C.E.; Yoshimura, A.; Lombardi, D.; Pritzl, P.; Floege, J.; Schwartz, S.M. Renal injury from angiotensin II-mediated hypertension. Hypertension 1992, 19, 464–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, W.C.; Thomas, M.C. Angiotensin II and its role in tubular epithelial to mesenchymal transition associated with chronic kidney disease. Cells Tissues Organs 2011, 193, 74–84. [Google Scholar] [CrossRef]
- Costantino, V.V.; Bocanegra, V.; Cacciamani, V.; Lorenzo, A.F.G.; Benardon, M.E.; Valles, P.G. Losartan through Hsp70 Avoids Angiotensin II Induced Mesenchymal Epithelial Transition in Proximal Tubule Cells from Spontaneously Hypertensive Rats. Cell Physiol. Biochem. 2019, 53, 713–730. [Google Scholar] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costantino, V.V.; Gil Lorenzo, A.F.; Bocanegra, V.; Vallés, P.G. Molecular Mechanisms of Hypertensive Nephropathy: Renoprotective Effect of Losartan through Hsp70. Cells 2021, 10, 3146. https://doi.org/10.3390/cells10113146
Costantino VV, Gil Lorenzo AF, Bocanegra V, Vallés PG. Molecular Mechanisms of Hypertensive Nephropathy: Renoprotective Effect of Losartan through Hsp70. Cells. 2021; 10(11):3146. https://doi.org/10.3390/cells10113146
Chicago/Turabian StyleCostantino, Valeria Victoria, Andrea Fernanda Gil Lorenzo, Victoria Bocanegra, and Patricia G. Vallés. 2021. "Molecular Mechanisms of Hypertensive Nephropathy: Renoprotective Effect of Losartan through Hsp70" Cells 10, no. 11: 3146. https://doi.org/10.3390/cells10113146