
Walking down Skeletal Muscle Lane: From Inflammasome to Disease

 , , ,
, , ,
Abstract
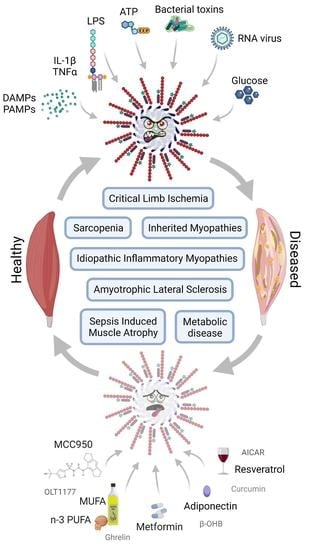
:
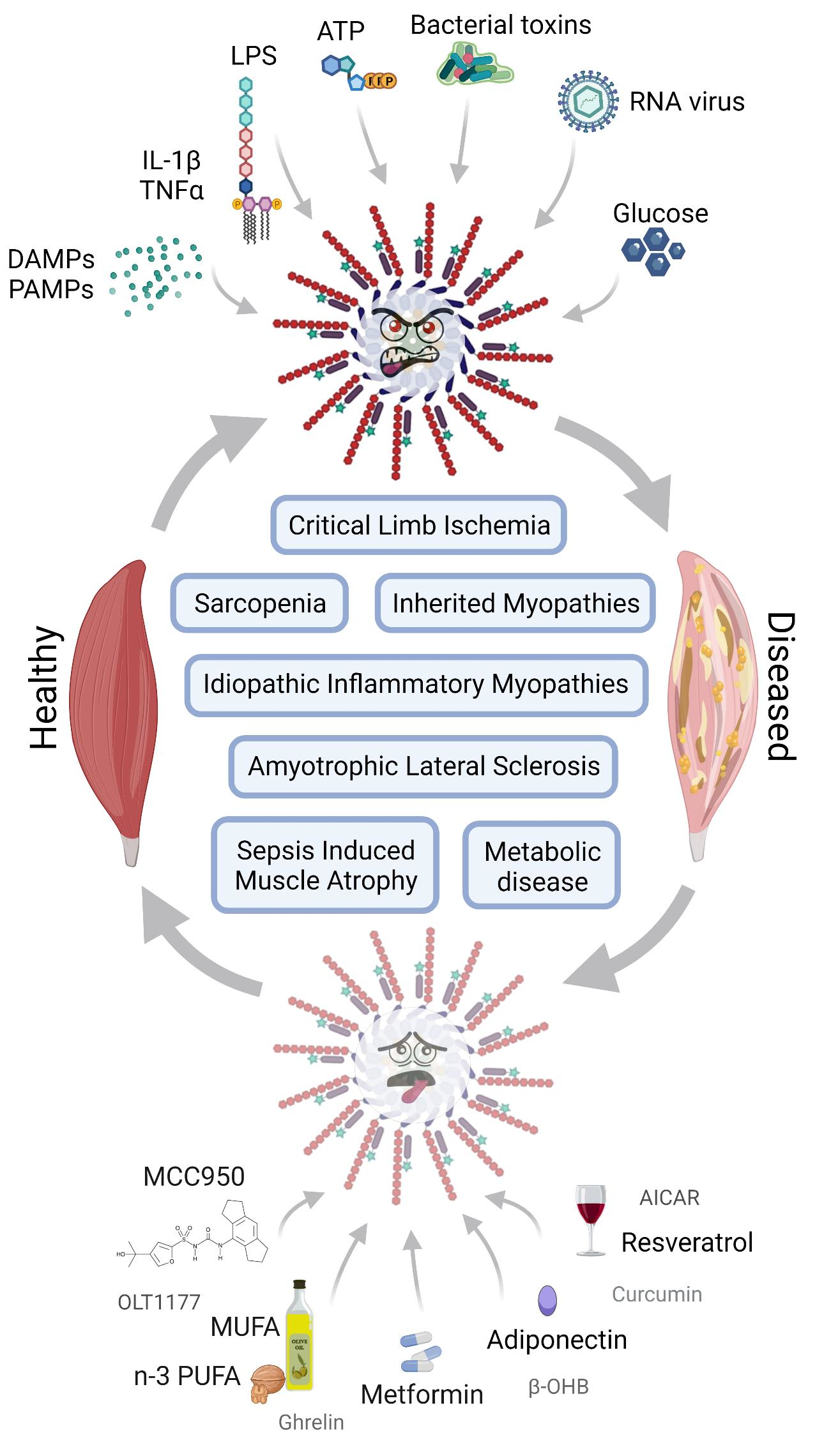
1. The NLRP3 Inflammasome
1.1. NLRP3 Inflammasome Actors
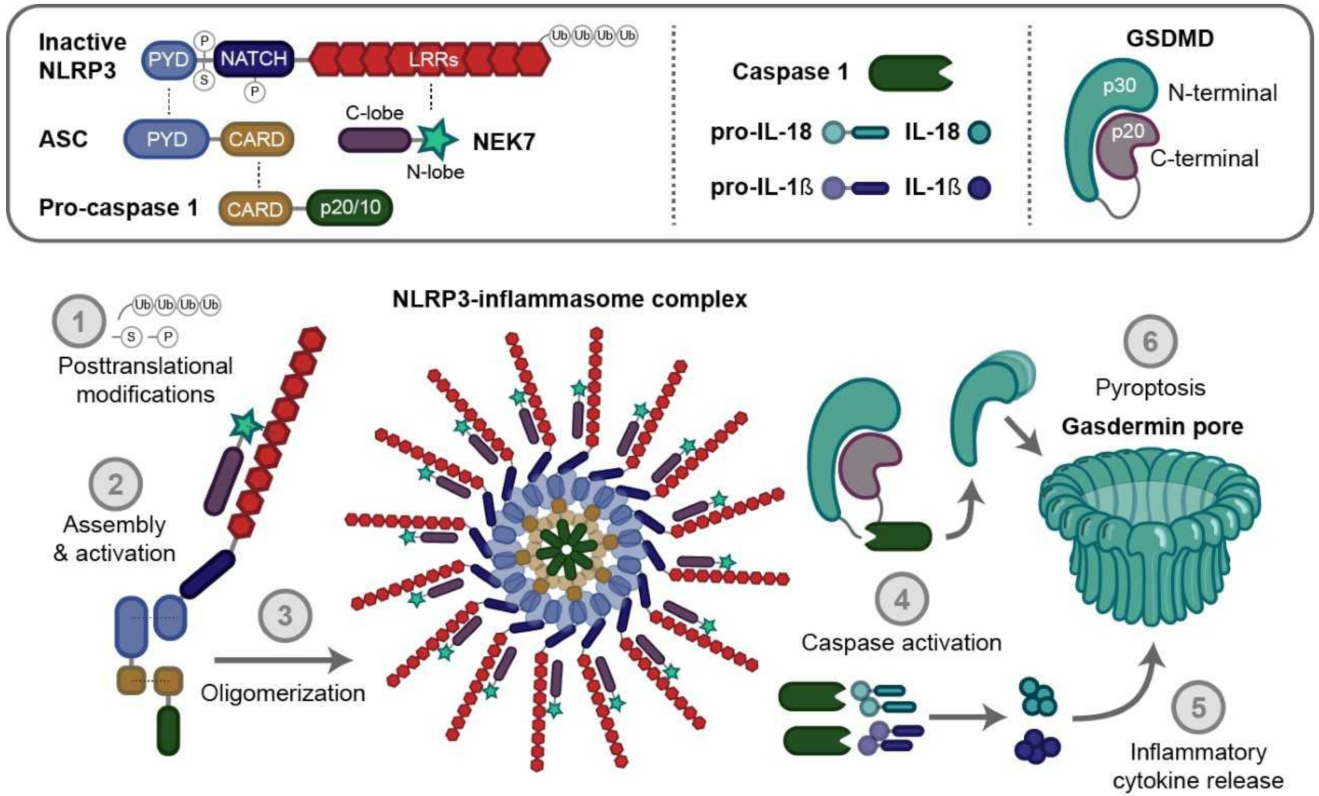
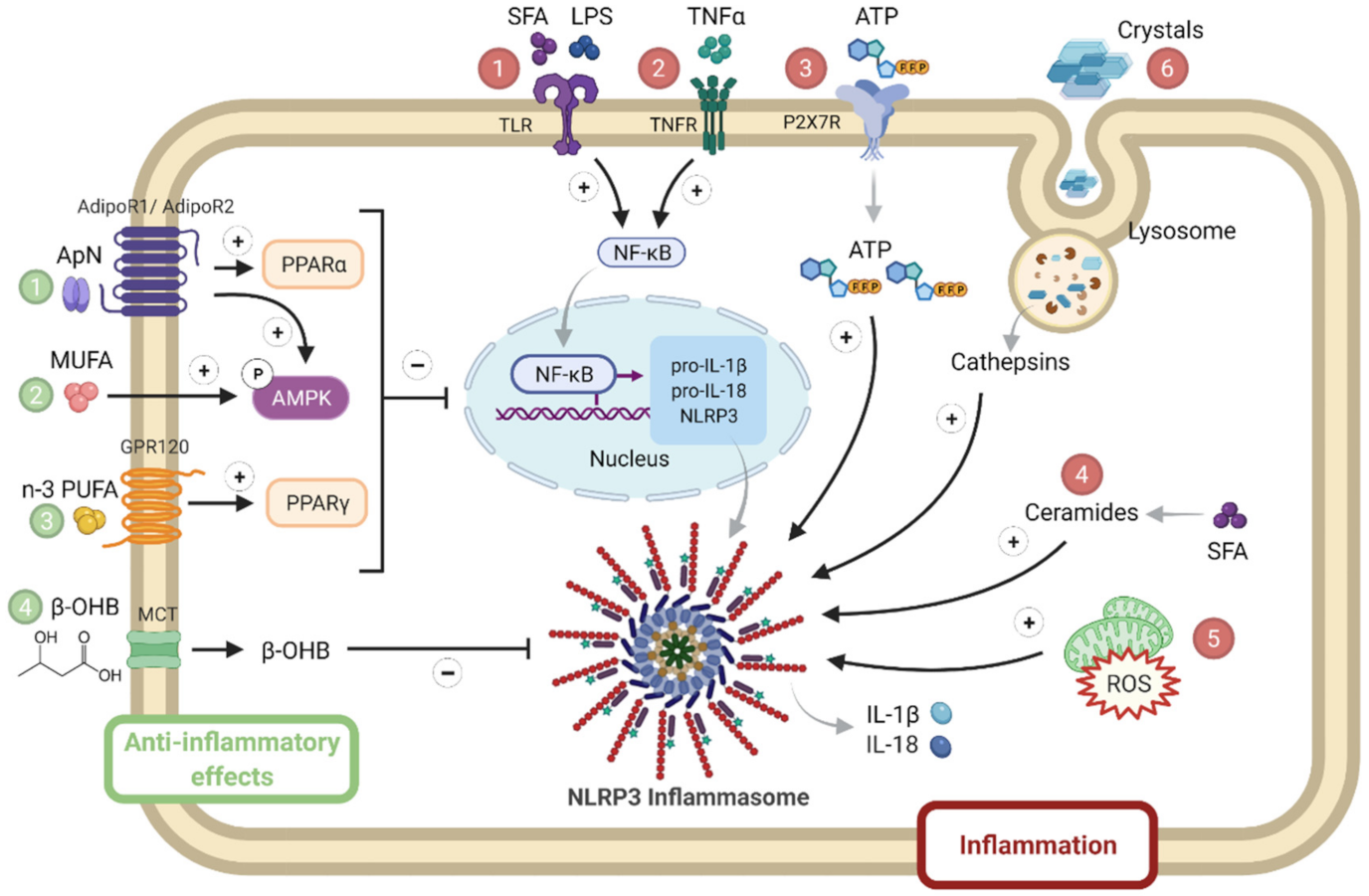
1.2. NLRP3 Activation
1.3. NLRP3 Regulation
2. NLRP3 and Skeletal Muscle
2.1. Introduction
2.2. NLRP3 and Skeletal Muscle Diseases
2.2.1. Metabolic Disorders
2.2.2. Muscle in Aging/Sarcopenia
2.2.3. Critical Limb Ischemia
2.2.4. Sepsis Induced Muscle Atrophy
2.2.5. Inherited Myopathies
2.2.6. Acquired Myopathies
2.2.7. Amyotrophic Lateral Sclerosis
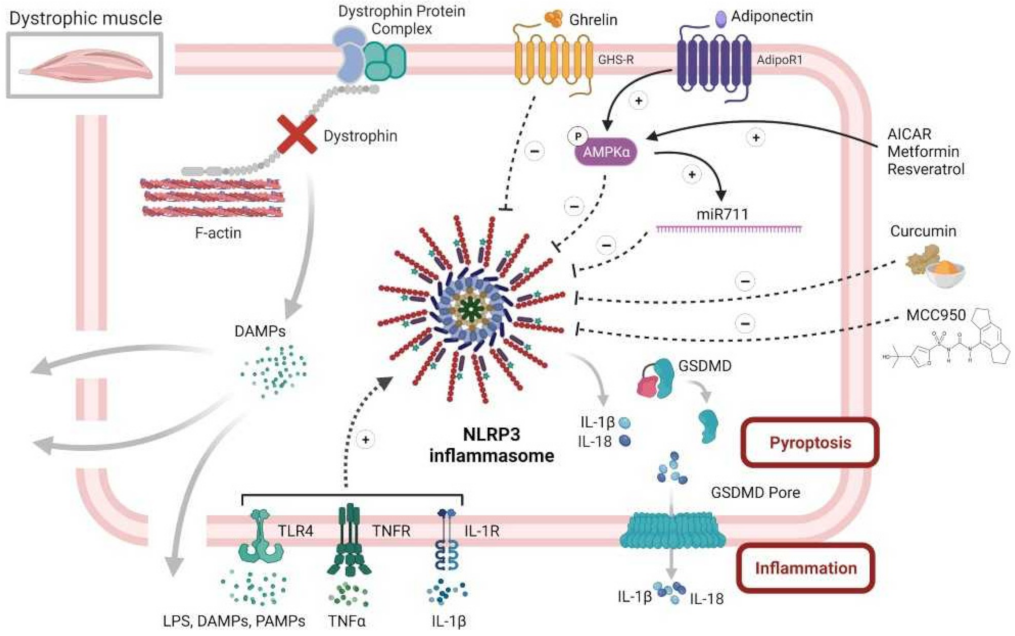
3. Therapeutic Perspective Targeting NLRP3
3.1. NLRP3 Direct Inhibitors
3.1.1. Inflammatory Disorders
3.1.2. Skeletal Muscle Disorders

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | Target Site | Inhibitory Effect | Tested on SM | Diseases | Clinical Trials | References |
|---|---|---|---|---|---|---|
| Cy-09 | NLRP3 NACHT Domain | NLRP3 ATPase activity | − | Gout, T2D, CAPS | − | [172] |
| MCC950 | + | Multiple sclerosis, CAPS, Ischemia, CLI, VCP, DMD | − | [60,79,173] | ||
| MNS | − | Inflammatory diseases | − | [171] | ||
| OLT1177 | + | Arthritis, CAPS, Gout | Arthritis, Phase IICovid19, Phase II | [130] | ||
| Oridonin | NLRP3 oligomerization | − | T2D, gout | - | [176] | |
| Tranilast | − | Gout, T2D, CAPS | CAPS, Phase II | [175] | ||
| β-OHB | Unknown | +/* | ALS | ALS, Phase II | [97,132] | |
| ZYIL1 | − | − | Phase I | No publication | ||
| INF39 | NLRP3 ATPase activity and oligomerization | − | Inflammatory bowel disease | − | [177] |
3.2. NLRP3 Indirect Inhibitors
3.2.1. NLRP3 Upstream Inhibitors in Skeletal Muscle
| Agent | Mechanism of Action | Effect | Relevant Clinical Trial | References |
|---|---|---|---|---|
| Adiponectin | Activation of AMPK signaling pathway | Reduction in NF-κB activity leading to downregulation of NLRP3 and proinflammatory cytokine expression | − | [80,138] |
| AICAR | − | [143] | ||
| Resveratrol | CHFC, Phase II Metabolic Syndrome, Phase II | [142] | ||
| Oleic acid (MUFA) | − | [102] | ||
| Metformin | Activation of AMPK pathway and inhibition of TLR4 signaling pathway | Commercialised for T2D, Phase III | [140] | |
| BBG | Inhibition of P2X7R pathway/K+ outflow | − | [60] | |
| Glyburide | Commercialised for T2D | |||
| Carbenoxolone | Decreased phosphorylation of IκBα | − | [100] | |
| Triptolide | − | [129] | ||
| Curcumin | Decreased phosphorylation of IKKα-IKKβ | T2D, Phase IV | [145,184] | |
| Ghrelin | Inhibition of JAK2-STAT3 and p38 MAPK signaling pathway | − | [135] | |
| Melatonin | Induction of SIRT1 deacetylase activity through RORα-dependent mechanisms | − | [115,183] | |
| Shikonin | Inhibition of PKM2 | Downregulation of NLRP3 and proinflammatory cytokine expression by unknown mechanism | − | [150] |
3.2.2. NLRP3 Downstream Inhibitors in Skeletal Muscle
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-Beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Gurung, P.; Kanneganti, T.-D. Novel Roles for Caspase-8 in IL-1β and Inflammasome Regulation. Am. J. Pathol. 2015, 185, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Cao, X. Cellular and Molecular Regulation of Innate Inflammatory Responses. Cell. Mol. Immunol. 2016, 13, 711–721. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Compan, V.; Martín-Sánchez, F.; Baroja-Mazo, A.; López-Castejón, G.; Gomez, A.I.; Verkhratsky, A.; Brough, D.; Pelegrín, P. Apoptosis-Associated Speck-like Protein Containing a CARD Forms Specks but Does Not Activate Caspase-1 in the Absence of NLRP3 during Macrophage Swelling. J. Immunol. 2015, 194, 1261–1273. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Bu, C.H.; Hildebrand, S.; et al. NLRP3 Activation and Mitosis Are Mutually Exclusive Events Coordinated by NEK7, a New Inflammasome Component. Nat. Immunol. 2016, 17, 250–258. [Google Scholar] [CrossRef]
- Kandli, M.; Feige, E.; Chen, A.; Kilfin, G.; Motro, B. Isolation and Characterization of Two Evolutionarily Conserved Murine Kinases (Nek6 and nek7) Related to the Fungal Mitotic Regulator, NIMA. Genomics 2000, 68, 187–196. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase Functions in Cell Death and Disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, New and Emerging Functions of Caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef] [Green Version]
- Pandey, A.; Shen, C.; Feng, S.; Man, S.M. Cell Biology of Inflammasome Activation. Trends Cell Biol. 2021, 31, 924–939. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J. The Interleukin-1 receptor/Toll-like Receptor Superfamily: 10 Years of Progress. Immunol. Rev. 2008, 226, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Moreno-García, L.; Miana-Mena, F.J.; Moreno-Martínez, L.; de la Torre, M.; Lunetta, C.; Tarlarini, C.; Zaragoza, P.; Calvo, A.C.; Osta, R. Inflammasome in ALS Skeletal Muscle: As a Potential Biomarker. Int. J. Mol. Sci. 2021, 22, 2523. [Google Scholar] [CrossRef]
- Arend, W.P.; Palmer, G.; Gabay, C. IL-1, IL-18, and IL-33 Families of Cytokines. Immunol. Rev. 2008, 223, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Wawrocki, S.; Druszczynska, M.; Kowalewicz-Kulbat, M.; Rudnicka, W. Interleukin 18 (IL-18) as a Target for Immune Intervention. Acta Biochim. Pol. 2016, 63, 59–63. [Google Scholar] [CrossRef] [Green Version]
- Kuriakose, T.; Kanneganti, T.-D. Gasdermin D Flashes an Exit Signal for IL-1. Immunity 2018, 48, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- He, W.-T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D Is an Executor of Pyroptosis and Required for Interleukin-1β Secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M. Emerging Inflammasome Effector Mechanisms. Nat. Rev. Immunol. 2011, 11, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Junior, E.S.; Morandini, A.C. Gasdermin: A New Player to the Inflammasome Game. Biomed. J. 2017, 40, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Baroja-Mazo, A.; Martín-Sánchez, F.; Gomez, A.I.; Martínez, C.M.; Amores-Iniesta, J.; Compan, V.; Barberà-Cremades, M.; Yagüe, J.; Ruiz-Ortiz, E.; Antón, J.; et al. The NLRP3 Inflammasome Is Released as a Particulate Danger Signal That Amplifies the Inflammatory Response. Nat. Immunol. 2014, 15, 738–748. [Google Scholar] [CrossRef]
- Franklin, B.S.; Bossaller, L.; De Nardo, D.; Ratter, J.M.; Stutz, A.; Engels, G.; Brenker, C.; Nordhoff, M.; Mirandola, S.R.; Al-Amoudi, A.; et al. The Adaptor ASC Has Extracellular and “Prionoid” Activities That Propagate Inflammation. Nat. Immunol. 2014, 15, 727–737. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Gaul, S.; Schaeffer, K.M.; Opitz, L.; Maeder, C.; Kogel, A.; Uhlmann, L.; Kalwa, H.; Wagner, U.; Haas, J.; Behzadi, A.; et al. Extracellular NLRP3 Inflammasome Particles Are Internalized by Human Coronary Artery Smooth Muscle Cells and Induce pro-Atherogenic Effects. Sci. Rep. 2021, 11, 15156. [Google Scholar] [CrossRef]
- Miao, E.A.; Leaf, I.A.; Treuting, P.M.; Mao, D.P.; Dors, M.; Sarkar, A.; Warren, S.E.; Wewers, M.D.; Aderem, A. Caspase-1-Induced Pyroptosis Is an Innate Immune Effector Mechanism against Intracellular Bacteria. Nat. Immunol. 2010, 11, 1136–1142. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Guarda, G.; Zenger, M.; Yazdi, A.S.; Schroder, K.; Ferrero, I.; Menu, P.; Tardivel, A.; Mattmann, C.; Tschopp, J. Differential Expression of NLRP3 among Hematopoietic Cells. J. Immunol. 2011, 186, 2529–2534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanamsagar, R.; Hanke, M.L.; Kielian, T. Toll-like Receptor (TLR) and Inflammasome Actions in the Central Nervous System. Trends Immunol. 2012, 33, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Latz, E. Critical Functions of Priming and Lysosomal Damage for NLRP3 Activation. Eur. J. Immunol. 2010, 40, 620–623. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, I.; Kratz, A.K.; Alexanderson, H.; Patarroyo, M. Decreased Expression of Interleukin-1alpha, Interleukin-1beta, and Cell Adhesion Molecules in Muscle Tissue Following Corticosteroid Treatment in Patients with Polymyositis and Dermatomyositis. Arthritis Rheum. 2000, 43, 336–348. [Google Scholar] [CrossRef]
- Rawat, R.; Cohen, T.V.; Ampong, B.; Francia, D.; Henriques-Pons, A.; Hoffman, E.P.; Nagaraju, K. Inflammasome up-Regulation and Activation in Dysferlin-Deficient Skeletal Muscle. Am. J. Pathol. 2010, 176, 2891–2900. [Google Scholar] [CrossRef]
- Elliott, E.I.; Sutterwala, F.S. Initiation and Perpetuation of NLRP3 Inflammasome Activation and Assembly. Immunol. Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franchi, L.; Muñoz-Planillo, R.; Núñez, G. Sensing and Reacting to Microbes through the Inflammasomes. Nat. Immunol. 2012, 13, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Ozkurede, U.; Kim, Y.-G.; Arindam, C.; Gale, M., Jr.; Silverman, R.H.; Colonna, M.; Akira, S.; et al. Cytosolic Double-Stranded RNA Activates the NLRP3 Inflammasome via MAVS-Induced Membrane Permeabilization and K+ Efflux. J. Immunol. 2014, 193, 4214–4222. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin Activates the Inflammasome in Response to Toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Muto, J.; Taylor, K.R.; Cogen, A.L.; Audish, D.; Bertin, J.; Grant, E.P.; Coyle, A.J.; Misaghi, A.; Hoffman, H.M.; et al. NLRP3/cryopyrin Is Necessary for Interleukin-1beta (IL-1beta) Release in Response to Hyaluronan, an Endogenous Trigger of Inflammation in Response to Injury. J. Biol. Chem. 2009, 284, 12762–12771. [Google Scholar] [CrossRef] [Green Version]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 Inflammasome Is Involved in the Innate Immune Response to Amyloid-Beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dostert, C.; Pétrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate Immune Activation through Nalp3 Inflammasome Sensing of Asbestos and Silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [Green Version]
- Feldmeyer, L.; Keller, M.; Niklaus, G.; Hohl, D.; Werner, S.; Beer, H.-D. The Inflammasome Mediates UVB-Induced Activation and Secretion of Interleukin-1beta by Keratinocytes. Curr. Biol. 2007, 17, 1140–1145. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Y.; Hu, C.; Ding, G.; Zhang, Y.; Huang, S.; Jia, Z.; Zhang, A. Albumin Impairs Renal Tubular Tight Junctions via Targeting the NLRP3 Inflammasome. Am. J. Physiol. Renal Physiol. 2015, 308, F1012–F1019. [Google Scholar] [CrossRef] [Green Version]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P.-Y. Fatty Acid-Induced NLRP3-ASC Inflammasome Activation Interferes with Insulin Signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-Associated Uric Acid Crystals Activate the NALP3 Inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koizumi, Y.; Toma, C.; Higa, N.; Nohara, T.; Nakasone, N.; Suzuki, T. Inflammasome Activation via Intracellular NLRs Triggered by Bacterial Infection. Cell. Microbiol. 2012, 14, 149–154. [Google Scholar] [CrossRef]
- Róg, J.; Oksiejuk, A.; Gosselin, M.R.F.; Brutkowski, W.; Dymkowska, D.; Nowak, N.; Robson, S.; Górecki, D.C.; Zabłocki, K. Dystrophic Mdx Mouse Myoblasts Exhibit Elevated ATP/UTP-Evoked Metabotropic Purinergic Responses and Alterations in Calcium Signalling. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1138–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, C.N.J.; Brutkowski, W.; Lien, C.-F.; Arkle, S.; Lochmüller, H.; Zabłocki, K.; Górecki, D.C. P2X7 Purinoceptor Alterations in Dystrophic Mdx Mouse Muscles: Relationship to Pathology and Potential Target for Treatment. J. Cell. Mol. Med. 2012, 16, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Di, A.; Xiong, S.; Ye, Z.; Malireddi, R.K.S.; Kometani, S.; Zhong, M.; Mittal, M.; Hong, Z.; Kanneganti, T.-D.; Rehman, J.; et al. The TWIK2 Potassium Efflux Channel in Macrophages Mediates NLRP3 Inflammasome-Induced Inflammation. Immunity 2018, 49, 56–65.e4. [Google Scholar] [CrossRef] [Green Version]
- Perregaux, D.; Gabel, C.A. Interleukin-1 Beta Maturation and Release in Response to ATP and Nigericin. Evidence That Potassium Depletion Mediated by These Agents Is a Necessary and Common Feature of Their Activity. J. Biol. Chem. 1994, 269, 15195–15203. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pétrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 Inflammasome Is Triggered by Low Intracellular Potassium Concentration. Cell Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Matsuzawa, A.; Yoshimura, A.; Ichijo, H. The Lysosome Rupture-Activated TAK1-JNK Pathway Regulates NLRP3 Inflammasome Activation. J. Biol. Chem. 2014, 289, 32926–32936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heid, M.E.; Keyel, P.A.; Kamga, C.; Shiva, S.; Watkins, S.C.; Salter, R.D. Mitochondrial Reactive Oxygen Species Induces NLRP3-Dependent Lysosomal Damage and Inflammasome Activation. J. Immunol. 2013, 191, 5230–5238. [Google Scholar] [CrossRef] [Green Version]
- Gurung, P.; Lukens, J.R.; Kanneganti, T.-D. Mitochondria: Diversity in the Regulation of the NLRP3 Inflammasome. Trends Mol. Med. 2015, 21, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Sorbara, M.T.; Girardin, S.E. Mitochondrial ROS Fuel the Inflammasome. Cell Res. 2011, 21, 558–560. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-Interacting Protein Links Oxidative Stress to Inflammasome Activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Tovar, E.; Muriel, P. Molecular Mechanisms That Link Oxidative Stress, Inflammation, and Fibrosis in the Liver. Antioxidants 2020, 9, 1279. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Lan, Z.; Xin, Z.; He, C.; Guo, Z.; Xia, X.; Hu, T. Emerging Insights into Molecular Mechanisms Underlying Pyroptosis and Functions of Inflammasomes in Diseases. J. Cell. Physiol. 2020, 235, 3207–3221. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, W., Jr.; Harton, J.A.; Zhu, X.; Linhoff, M.W.; Ting, J.P.-Y. Cutting Edge: CIAS1/cryopyrin/PYPAF1/NALP3/CATERPILLER 1.1 Is an Inducible Inflammatory Mediator with NF-Kappa B Suppressive Properties. J. Immunol. 2003, 171, 6329–6333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zewinger, S.; Reiser, J.; Jankowski, V.; Alansary, D.; Hahm, E.; Triem, S.; Klug, M.; Schunk, S.J.; Schmit, D.; Kramann, R.; et al. Apolipoprotein C3 Induces Inflammation and Organ Damage by Alternative Inflammasome Activation. Nat. Immunol. 2020, 21, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Nuñez, G. Sterile Inflammation: Sensing and Reacting to Damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, H.M.; Mueller, J.L.; Broide, D.H.; Wanderer, A.A.; Kolodner, R.D. Mutation of a New Gene Encoding a Putative Pyrin-like Protein Causes Familial Cold Autoinflammatory Syndrome and Muckle-Wells Syndrome. Nat. Genet. 2001, 29, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.; Chai, K.; Deng, R. Study of the Correlation between the Noncanonical Pathway of Pyroptosis and Idiopathic Inflammatory Myopathy. Int. Immunopharmacol. 2021, 98, 107810. [Google Scholar] [CrossRef]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszyński, A.; et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruchard, M.; Rebé, C.; Derangère, V.; Togbé, D.; Ryffel, B.; Boidot, R.; Humblin, E.; Hamman, A.; Chalmin, F.; Berger, H.; et al. Corrigendum: The Receptor NLRP3 Is a Transcriptional Regulator of TH2 Differentiation. Nat. Immunol. 2015, 16, 1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Viollet, B.; Terkeltaub, R.; Liu-Bryan, R. AMP-Activated Protein Kinase Suppresses Urate Crystal-Induced Inflammation and Transduces Colchicine Effects in Macrophages. Ann. Rheum. Dis. 2016, 75, 286–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, S.; Srivastava, S.Y.; Brickey, W.J.; Iocca, H.; Toews, A.; Morrison, J.P.; Chen, V.S.; Gris, D.; Matsushima, G.K.; Ting, J.P.-Y. The Inflammasome Sensor, NLRP3, Regulates CNS Inflammation and Demyelination via Caspase-1 and Interleukin-18. J. Neurosci. 2010, 30, 15811–15820. [Google Scholar] [CrossRef]
- Ozaki, E.; Campbell, M.; Doyle, S.L. Targeting the NLRP3 Inflammasome in Chronic Inflammatory Diseases: Current Perspectives. J. Inflamm. Res. 2015, 8, 15–27. [Google Scholar]
- Spalinger, M.R.; Kasper, S.; Gottier, C.; Lang, S.; Atrott, K.; Vavricka, S.R.; Scharl, S.; Raselli, T.; Frey-Wagner, I.; Gutte, P.M.; et al. NLRP3 Tyrosine Phosphorylation Is Controlled by Protein Tyrosine Phosphatase PTPN22. J. Clin. Investig. 2016, 126, 4388. [Google Scholar] [CrossRef] [Green Version]
- Shenoy, A.R.; Wellington, D.A.; Kumar, P.; Kassa, H.; Booth, C.J.; Cresswell, P.; MacMicking, J.D. GBP5 Promotes NLRP3 Inflammasome Assembly and Immunity in Mammals. Science 2012, 336, 481–485. [Google Scholar] [CrossRef]
- Lu, B.; Nakamura, T.; Inouye, K.; Li, J.; Tang, Y.; Lundbäck, P.; Valdes-Ferrer, S.I.; Olofsson, P.S.; Kalb, T.; Roth, J.; et al. Novel Role of PKR in Inflammasome Activation and HMGB1 Release. Nature 2012, 488, 670–674. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Okamura, H.; Hiroshima, Y.; Abe, K.; Kido, J.-I.; Shinohara, Y.; Ozaki, K. PKR Induces the Expression of NLRP3 by Regulating the NF-κB Pathway in Porphyromonas Gingivalis-Infected Osteoblasts. Exp. Cell Res. 2017, 354, 57–64. [Google Scholar] [CrossRef]
- Lang, T.; Lee, J.P.W.; Elgass, K.; Pinar, A.A.; Tate, M.D.; Aitken, E.H.; Fan, H.; Creed, S.J.; Deen, N.S.; Traore, D.A.K.; et al. Macrophage Migration Inhibitory Factor Is Required for NLRP3 Inflammasome Activation. Nat. Commun. 2018, 9, 2223. [Google Scholar] [CrossRef]
- Li, X.; Thome, S.; Ma, X.; Amrute-Nayak, M.; Finigan, A.; Kitt, L.; Masters, L.; James, J.R.; Shi, Y.; Meng, G.; et al. MARK4 Regulates NLRP3 Positioning and Inflammasome Activation through a Microtubule-Dependent Mechanism. Nat. Commun. 2017, 8, 15986. [Google Scholar] [CrossRef] [Green Version]
- Mayor, A.; Martinon, F.; De Smedt, T.; Pétrilli, V.; Tschopp, J. A Crucial Function of SGT1 and HSP90 in Inflammasome Activity Links Mammalian and Plant Innate Immune Responses. Nat. Immunol. 2007, 8, 497–503. [Google Scholar] [CrossRef]
- Piippo, N.; Korhonen, E.; Hytti, M.; Skottman, H.; Kinnunen, K.; Josifovska, N.; Petrovski, G.; Kaarniranta, K.; Kauppinen, A. Hsp90 Inhibition as a Means to Inhibit Activation of the NLRP3 Inflammasome. Sci. Rep. 2018, 8, 6720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Almeida, L.; Khare, S.; Misharin, A.V.; Patel, R.; Ratsimandresy, R.A.; Wallin, M.C.; Perlman, H.; Greaves, D.R.; Hoffman, H.M.; Dorfleutner, A.; et al. The PYRIN Domain-Only Protein POP1 Inhibits Inflammasome Assembly and Ameliorates Inflammatory Disease. Immunity 2015, 43, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atianand, M.K.; Harton, J.A. Uncoupling of Pyrin-Only Protein 2 (POP2)-Mediated Dual Regulation of NF-κB and the Inflammasome. J. Biol. Chem. 2011, 286, 40536–40547. [Google Scholar] [CrossRef] [Green Version]
- Ratsimandresy, R.A.; Chu, L.H.; Khare, S.; de Almeida, L.; Gangopadhyay, A.; Indramohan, M.; Misharin, A.V.; Greaves, D.R.; Perlman, H.; Dorfleutner, A.; et al. The PYRIN Domain-Only Protein POP2 Inhibits Inflammasome Priming and Activation. Nat. Commun. 2017, 8, 15556. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Li, Y.; Schmidt, F.I.; Yin, Q.; Chen, S.; Fu, T.-M.; Tong, A.B.; Ploegh, H.L.; Mao, Y.; Wu, H. Molecular Basis of Caspase-1 Polymerization and Its Inhibition by a New Capping Mechanism. Nat. Struct. Mol. Biol. 2016, 23, 416–425. [Google Scholar] [CrossRef] [Green Version]
- Nalbandian, A.; Khan, A.A.; Srivastava, R.; Llewellyn, K.J.; Tan, B.; Shukr, N.; Fazli, Y.; Kimonis, V.E.; BenMohamed, L. Activation of the NLRP3 Inflammasome Is Associated with Valosin-Containing Protein Myopathy. Inflammation 2017, 40, 21–41. [Google Scholar] [CrossRef] [Green Version]
- Boursereau, R.; Abou-Samra, M.; Lecompte, S.; Noel, L.; Brichard, S.M. Downregulation of the NLRP3 Inflammasome by Adiponectin Rescues Duchenne Muscular Dystrophy. BMC Biol. 2018, 16, 33. [Google Scholar] [CrossRef] [Green Version]
- Lecompte, S.; Abou-Samra, M.; Boursereau, R.; Noel, L.; Brichard, S.M. Skeletal Muscle Secretome in Duchenne Muscular Dystrophy: A Pivotal Anti-Inflammatory Role of Adiponectin. Cell. Mol. Life Sci. 2017, 74, 2487–2501. [Google Scholar] [CrossRef] [Green Version]
- Abou-Samra, M.; Selvais, C.M.; Boursereau, R.; Lecompte, S.; Noel, L.; Brichard, S.M. AdipoRon, a New Therapeutic Prospect for Duchenne Muscular Dystrophy. J. Cachexia Sarcopenia Muscle 2020, 11, 518–533. [Google Scholar] [CrossRef] [PubMed]
- Pillon, N.J.; Krook, A. Innate Immune Receptors in Skeletal Muscle Metabolism. Exp. Cell Res. 2017, 360, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Hise, A.G.; Tomalka, J.; Ganesan, S.; Patel, K.; Hall, B.A.; Brown, G.D.; Fitzgerald, K.A. An Essential Role for the NLRP3 Inflammasome in Host Defense against the Human Fungal Pathogen Candida Albicans. Cell Host Microbe 2009, 5, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Dupaul-Chicoine, J.; Yeretssian, G.; Doiron, K.; Bergstrom, K.S.B.; McIntire, C.R.; LeBlanc, P.M.; Meunier, C.; Turbide, C.; Gros, P.; Beauchemin, N.; et al. Control of Intestinal Homeostasis, Colitis, and Colitis-Associated Colorectal Cancer by the Inflammatory Caspases. Immunity 2010, 32, 367–378. [Google Scholar] [CrossRef] [Green Version]
- Gimenez, F.; Bhela, S.; Dogra, P.; Harvey, L.; Varanasi, S.K.; Jaggi, U.; Rouse, B.T. The Inflammasome NLRP3 Plays a Protective Role against a Viral Immunopathological Lesion. J. Leukoc. Biol. 2016, 99, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhao, G.; Yan, J.; Xu, R.; Che, C.; Zheng, H.; Zhu, G.; Zhang, J. Pannexin 1 Channels Contribute to IL-1β Expression via NLRP3/Caspase-1 Inflammasome in Keratitis. Curr. Eye Res. 2019, 44, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Molecular Biological and Clinical Understanding of the Pathophysiology and Treatments of Hyperuricemia and Its Association with Metabolic Syndrome, Cardiovascular Diseases and Chronic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 9221. [Google Scholar] [CrossRef] [PubMed]
- Jorquera, G.; Russell, J.; Monsalves-Álvarez, M.; Cruz, G.; Valladares-Ide, D.; Basualto-Alarcón, C.; Barrientos, G.; Estrada, M.; Llanos, P. NLRP3 Inflammasome: Potential Role in Obesity Related Low-Grade Inflammation and Insulin Resistance in Skeletal Muscle. Int. J. Mol. Sci. 2021, 22, 3254. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 Inflammasome Instigates Obesity-Induced Inflammation and Insulin Resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Ralston, J.C.; Lyons, C.L.; Kennedy, E.B.; Kirwan, A.M.; Roche, H.M. Fatty Acids and NLRP3 Inflammasome-Mediated Inflammation in Metabolic Tissues. Annu. Rev. Nutr. 2017, 37, 77–102. [Google Scholar] [CrossRef]
- Haneklaus, M.; O’Neill, L.A.J. NLRP3 at the Interface of Metabolism and Inflammation. Immunol. Rev. 2015, 265, 53–62. [Google Scholar] [CrossRef]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 Links Innate Immunity and Fatty Acid-Induced Insulin Resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Barra, N.G.; Henriksbo, B.D.; Anhê, F.F.; Schertzer, J.D. The NLRP3 Inflammasome Regulates Adipose Tissue Metabolism. Biochem. J. 2020, 477, 1089–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in Gut Microbiota Control Metabolic Endotoxemia-Induced Inflammation in High-Fat Diet-Induced Obesity and Diabetes in Mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, B.C.; Gordillo, R.; Scherer, P.E. The Role of Ceramides in Diabetes and Cardiovascular Disease Regulation of Ceramides by Adipokines. Front. Endocrinol. 2020, 11, 569250. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The Ketone Metabolite β-Hydroxybutyrate Blocks NLRP3 Inflammasome-Mediated Inflammatory Disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and Insulin Resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef]
- Hull, C.; Dekeryte, R.; Buchanan, H.; Kamli-Salino, S.; Robertson, A.; Delibegovic, M.; Platt, B. NLRP3 Inflammasome Inhibition with MCC950 Improves Insulin Sensitivity and Inflammation in a Mouse Model of Frontotemporal Dementia. Neuropharmacology 2020, 180, 108305. [Google Scholar] [CrossRef]
- Chen, Y.; Qian, Q.; Yu, J. Carbenoxolone Ameliorates Insulin Sensitivity in Obese Mice Induced by High Fat Diet via Regulating the IκB-α/NF-κB Pathway and NLRP3 Inflammasome. Biomed. Pharmacother. 2019, 115, 108868. [Google Scholar] [CrossRef]
- Cho, K.-A.; Kang, P.B. PLIN2 Inhibits Insulin-Induced Glucose Uptake in Myoblasts through the Activation of the NLRP3 Inflammasome. Int. J. Mol. Med. 2015, 36, 839–844. [Google Scholar] [CrossRef] [Green Version]
- Kien, C.L.; Bunn, J.Y.; Fukagawa, N.K.; Anathy, V.; Matthews, D.E.; Crain, K.I.; Ebenstein, D.B.; Tarleton, E.K.; Pratley, R.E.; Poynter, M.E. Lipidomic Evidence That Lowering the Typical Dietary Palmitate to Oleate Ratio in Humans Decreases the Leukocyte Production of Proinflammatory Cytokines and Muscle Expression of Redox-Sensitive Genes. J. Nutr. Biochem. 2015, 26, 1599–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Jentoft, A.J.; Sayer, A.A. Sarcopenia. Lancet 2019, 393, 2636–2646. [Google Scholar] [CrossRef]
- Ogawa, S.; Yakabe, M.; Akishita, M. Age-Related Sarcopenia and Its Pathophysiological Bases. Inflamm. Regen. 2016, 36, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Ghani, M.A.; DeFronzo, R.A. Pathogenesis of Insulin Resistance in Skeletal Muscle. J. Biomed. Biotechnol. 2010, 2010, 476279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, J.J.; Esser, K.A. Anabolic and Catabolic Pathways Regulating Skeletal Muscle Mass. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 230–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riuzzi, F.; Sorci, G.; Arcuri, C.; Giambanco, I.; Bellezza, I.; Minelli, A.; Donato, R. Cellular and Molecular Mechanisms of Sarcopenia: The S100B Perspective. J. Cachexia Sarcopenia Muscle 2018, 9, 1255–1268. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Hyttinen, J.M.T.; Kauppinen, A.; Kaarniranta, K. Context-Dependent Regulation of Autophagy by IKK-NF-κB Signaling: Impact on the Aging Process. Int. J. Cell Biol. 2012, 2012, 849541. [Google Scholar] [CrossRef] [Green Version]
- Yuk, J.-M.; Silwal, P.; Jo, E.-K. Inflammasome and Mitophagy Connection in Health and Disease. Int. J. Mol. Sci. 2020, 21, 4714. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Kou, X.; Yang, Y.; Chen, N. MicroRNA-Regulated Proinflammatory Cytokines in Sarcopenia. Mediat. Inflamm. 2016, 2016, 1438686. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A New Immune-Metabolic Viewpoint for Age-Related Diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Cordero, M.D.; Williams, M.R.; Ryffel, B. AMP-Activated Protein Kinase Regulation of the NLRP3 Inflammasome during Aging. Trends Endocrinol. Metab. 2018, 29, 8–17. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 Inflammasome Links Systemic Low-Grade Inflammation to Functional Decline in Aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef] [Green Version]
- McBride, M.J.; Foley, K.P.; D’Souza, D.M.; Li, Y.E.; Lau, T.C.; Hawke, T.J.; Schertzer, J.D. The NLRP3 Inflammasome Contributes to Sarcopenia and Lower Muscle Glycolytic Potential in Old Mice. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E222–E232. [Google Scholar] [CrossRef] [Green Version]
- Sayed, R.K.A.; Fernández-Ortiz, M.; Diaz-Casado, M.E.; Aranda-Martínez, P.; Fernández-Martínez, J.; Guerra-Librero, A.; Escames, G.; López, L.C.; Alsaadawy, R.M.; Acuña-Castroviejo, D. Lack of NLRP3 Inflammasome Activation Reduces Age-Dependent Sarcopenia and Mitochondrial Dysfunction, Favoring the Prophylactic Effect of Melatonin. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.M.; Nabavi, S.F.; Sureda, A.; Xiao, J.; Dehpour, A.R.; Shirooie, S.; Silva, A.S.; Baldi, A.; Khan, H.; Daglia, M. Anti-Inflammatory Effects of Melatonin: A Mechanistic Review. Crit. Rev. Food Sci. Nutr. 2019, 59, S4–S16. [Google Scholar] [CrossRef] [PubMed]
- Sayed, R.K.A.; Fernández-Ortiz, M.; Diaz-Casado, M.E.; Rusanova, I.; Rahim, I.; Escames, G.; López, L.C.; Mokhtar, D.M.; Acuña-Castroviejo, D. The Protective Effect of Melatonin Against Age-Associated, Sarcopenia-Dependent Tubular Aggregate Formation, Lactate Depletion, and Mitochondrial Changes. J. Gerontol. Ser. A 2018, 73, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Sayed, R.K.; Fernández-Ortiz, M.; Fernández-Martínez, J.; Aranda Martínez, P.; Guerra-Librero, A.; Rodríguez-Santana, C.; de Haro, T.; Escames, G.; Acuña-Castroviejo, D.; Rusanova, I. The Impact of Melatonin and NLRP3 Inflammasome on the Expression of microRNAs in Aged Muscle. Antioxidants 2021, 10, 524. [Google Scholar] [CrossRef]
- Annex, B.H. Therapeutic Angiogenesis for Critical Limb Ischaemia. Nat. Rev. Cardiol. 2013, 10, 387–396. [Google Scholar] [CrossRef]
- Albadawi, H.; Oklu, R.; Cormier, N.R.; O’Keefe, R.M.; Heaton, J.T.; Kobler, J.B.; Austen, W.G.; Watkins, M.T. Hind Limb Ischemia-Reperfusion Injury in Diet-Induced Obese Mice. J. Surg. Res. 2014, 190, 683–691. [Google Scholar] [CrossRef] [Green Version]
- Elshaer, S.; Mohamed, I.; Coucha, M.; Altantawi, S.; Eldahshan, W.; Bartasi, M.; Shanab, A.; Lorys, R.; El-Remessy, A. Deletion of TXNIP Mitigates High-Fat Diet-Impaired Angiogenesis and Prevents Inflammation in a Mouse Model of Critical Limb Ischemia. Antioxidants 2017, 6, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, B.; Zhang, Y.; Liang, C.; Liu, B.; Ding, F.; Wang, Y.; Zhu, B.; Zhao, R.; Yu, X.-Y.; Li, Y. Stem Cell-Derived Exosomes Prevent Pyroptosis and Repair Ischemic Muscle Injury through a Novel exosome/circHIPK3/ FOXO3a Pathway. Theranostics 2020, 10, 6728–6742. [Google Scholar] [CrossRef] [PubMed]
- Jais, A.; Einwallner, E.; Sharif, O.; Gossens, K.; Lu, T.T.-H.; Soyal, S.M.; Medgyesi, D.; Neureiter, D.; Paier-Pourani, J.; Dalgaard, K.; et al. Heme Oxygenase-1 Drives Metaflammation and Insulin Resistance in Mouse and Man. Cell 2014, 158, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Jia, L.; Wang, Y.; Ji, Y.; Chen, J.; Ma, H.; Lin, X.; Zhang, Y.; Li, W.; Ni, H.; et al. Heme Oxygenase-1 in Macrophages Impairs the Perfusion Recovery After Hindlimb Ischemia by Suppressing Autolysosome-Dependent Degradation of NLRP3. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1710–1723. [Google Scholar] [CrossRef]
- Puthucheary, Z.A.; Rawal, J.; McPhail, M.; Connolly, B.; Ratnayake, G.; Chan, P.; Hopkinson, N.S.; Padhke, R.; Dew, T.; Sidhu, P.S.; et al. Acute Skeletal Muscle Wasting in Critical Illness. JAMA 2013, 310, 1591. [Google Scholar] [CrossRef] [Green Version]
- Wollersheim, T.; Woehlecke, J.; Krebs, M.; Hamati, J.; Lodka, D.; Luther-Schroeder, A.; Langhans, C.; Haas, K.; Radtke, T.; Kleber, C.; et al. Dynamics of Myosin Degradation in Intensive Care Unit-Acquired Weakness during Severe Critical Illness. Intensive Care Med. 2014, 40, 528–538. [Google Scholar] [CrossRef]
- Valentine, R.J.; Jefferson, M.A.; Kohut, M.L.; Eo, H. Imoxin Attenuates LPS-Induced Inflammation and MuRF1 Expression in Mouse Skeletal Muscle. Physiol Rep. 2018, 6, e13941. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Lu, K.; Wang, Y.; Chen, M.; Zhang, F.; Shen, H.; Yao, D.; Gong, K.; Zhang, Z. Triptolide Attenuates Pressure Overload-Induced Myocardial Remodeling in Mice via the Inhibition of NLRP3 Inflammasome Expression. Biochem. Biophys. Res. Commun. 2017, 485, 69–75. [Google Scholar] [CrossRef]
- Fang, W.-Y.; Tseng, Y.-T.; Lee, T.-Y.; Fu, Y.-C.; Chang, W.-H.; Lo, W.-W.; Lin, C.-L.; Lo, Y.-C. Triptolide Prevents LPS-Induced Skeletal Muscle Atrophy via Inhibiting NF-κB/TNF-α and Regulating Protein Synthesis/degradation Pathway. Br. J. Pharmacol. 2021, 178, 2998–3016. [Google Scholar] [CrossRef]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A.; et al. OLT1177, a β-Sulfonyl Nitrile Compound, Safe in Humans, Inhibits the NLRP3 Inflammasome and Reverses the Metabolic Cost of Inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef] [Green Version]
- Huang, N.; Kny, M.; Riediger, F.; Busch, K.; Schmidt, S.; Luft, F.C.; Slevogt, H.; Fielitz, J. Deletion of Nlrp3 Protects from Inflammation-Induced Skeletal Muscle Atrophy. Intensive Care Med. Exp. 2017, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, H.H.; Rittig, N.; Johannsen, M.; Møller, A.B.; Jørgensen, J.O.; Jessen, N.; Møller, N. Effects of 3-Hydroxybutyrate and Free Fatty Acids on Muscle Protein Kinetics and Signaling during LPS-Induced Inflammation in Humans: Anticatabolic Impact of Ketone Bodies. Am. J. Clin. Nutr. 2018, 108, 857–867. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-Approved Disulfiram Inhibits Pyroptosis by Blocking Gasdermin D Pore Formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef]
- González-Jamett, A.M.; Bevilacqua, J.A.; Díaz, A.M.C. Hereditary Myopathies. In Muscle Cell and Tissue—Current Status of Research Field; BoD—Books on Demand: Norderstedt, Germany, 2018. [Google Scholar]
- Chang, L.; Niu, F.; Chen, J.; Cao, X.; Liu, Z.; Bao, X.; Xu, Y. Ghrelin Improves Muscle Function in Dystrophin-Deficient Mdx Mice by Inhibiting NLRP3 Inflammasome Activation. Life Sci. 2019, 232, 116654. [Google Scholar] [CrossRef]
- Aoki, M. LGMD2B (dysferlin deficiency). Ryoikibetsu Shokogun Shirizu 2001, 35, 84–87. Available online: https://www.scopus.com/record/display.uri?eid=2-s2.0-0035237626&origin=inward&txGid=e48ffe062c960be6c2825a0d55ca7952 (accessed on 12 October 2021).
- Sun, X.; Qiu, H. Valosin-Containing Protein, a Calcium-Associated ATPase Protein, in Endoplasmic Reticulum and Mitochondrial Function and Its Implications for Diseases. Int. J. Mol. Sci. 2020, 21, 3842. [Google Scholar] [CrossRef]
- Abou-Samra, M.; Lecompte, S.; Schakman, O.; Noel, L.; Many, M.C.; Gailly, P.; Brichard, S.M. Involvement of Adiponectin in the Pathogenesis of Dystrophinopathy. Skelet. Muscle 2015, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boursereau, R.; Abou-Samra, M.; Lecompte, S.; Noel, L.; Brichard, S.M. New Targets to Alleviate Skeletal Muscle Inflammation: Role of microRNAs Regulated by Adiponectin. Sci. Rep. 2017, 7, 43437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.; Hui, T.; Chen, J.; Yu, Z.; Ren, D.; Zou, S.; Wang, S.; Fei, E.; Jiao, H.; Lai, X. Metformin Increases Sarcolemma Integrity and Ameliorates Neuromuscular Deficits in a Murine Model of Duchenne Muscular Dystrophy. Front. Physiol. 2021, 12, 642908. [Google Scholar] [CrossRef] [PubMed]
- Ljubicic, V.; Burt, M.; Lunde, J.A.; Jasmin, B.J. Resveratrol Induces Expression of the Slow, Oxidative Phenotype in Mdx Mouse Muscle Together with Enhanced Activity of the SIRT1-PGC-1α Axis. Am. J. Physiol. Cell Physiol. 2014, 307, C66–C82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastin, J.; Djouadi, F. Resveratrol and Myopathy. Nutrients 2016, 8, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dial, A.G.; Ng, S.Y.; Manta, A.; Ljubicic, V. The Role of AMPK in Neuromuscular Biology and Disease. Trends Endocrinol. Metab. 2018, 29, 300–312. [Google Scholar] [CrossRef]
- Pradhan, G.; Samson, S.L.; Sun, Y. Ghrelin: Much More than a Hunger Hormone. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 619–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Chen, C.; Shen, Y.; Zhu, C.-H.; Wang, G.; Wang, X.-C.; Chen, H.-Q.; Zhu, M.-S. Curcumin Alleviates Dystrophic Muscle Pathology in Mdx Mice. Mol. Cells 2008, 25, 531–537. [Google Scholar] [PubMed]
- Dubuisson, N.; Abou-Samra, M.; Davis, M.; Noel, L.; Selvais, C.; Brichard, S. DMD—ANIMAL MODELS: EP. 88 Inflammasome Inhibitors for the Treatment of Muscular Dystrophies. Neuromuscul. Disord. 2021, 31, S76. [Google Scholar]
- Dalakas, M.C. Inflammatory Muscle Diseases. N. Engl. J. Med. 2015, 373, 393–394. [Google Scholar] [CrossRef] [Green Version]
- Dobloug, C.; Garen, T.; Bitter, H.; Stjärne, J.; Stenseth, G.; Grøvle, L.; Sem, M.; Gran, J.T.; Molberg, Ø. Prevalence and Clinical Characteristics of Adult Polymyositis and Dermatomyositis; Data from a Large and Unselected Norwegian Cohort. Ann. Rheum. Dis. 2015, 74, 1551–1556. [Google Scholar] [CrossRef] [Green Version]
- Henriques-Pons, A.; Nagaraju, K. Nonimmune Mechanisms of Muscle Damage in Myositis: Role of the Endoplasmic Reticulum Stress Response and Autophagy in the Disease Pathogenesis. Curr. Opin. Rheumatol. 2009, 21, 581–587. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Xiao, Y.; Zhou, B.; Gao, S.; Li, L.; Zhao, L.; Chen, W.; Dai, B.; Li, Q.; Duan, H.; et al. PKM2-Dependent Glycolysis Promotes Skeletal Muscle Cell Pyroptosis by Activating the NLRP3 Inflammasome in Dermatomyositis/polymyositis. Rheumatology 2021, 60, 2177–2189. [Google Scholar] [CrossRef]
- Kang, J.; Feng, D.; Yang, F.; Tian, X.; Han, W.; Jia, H. Comparison of Rapamycin and Methylprednisolone for Treating Inflammatory Muscle Disease in a Murine Model of Experimental Autoimmune Myositis. Exp. Ther. Med. 2020, 20, 219–226. [Google Scholar] [CrossRef]
- Tucci, M.; Quatraro, C.; Dammacco, F.; Silvestris, F. Interleukin-18 Overexpression as a Hallmark of the Activity of Autoimmune Inflammatory Myopathies. Clin. Exp. Immunol. 2006, 146, 21–31. [Google Scholar] [CrossRef]
- Yin, X.; Han, G.-C.; Jiang, X.-W.; Shi, Q.; Pu, C.-Q. Increased Expression of the NOD-like Receptor Family, Pyrin Domain Containing 3 Inflammasome in Dermatomyositis and Polymyositis Is a Potential Contributor to Their Pathogenesis. Chin. Med. J. 2016, 129, 1047–1052. [Google Scholar] [CrossRef]
- Chinoy, H.; Li, C.K.-C.; Platt, H.; Fertig, N.; Varsani, H.; Gunawardena, H.; Betteridge, Z.; Oddis, C.V.; McHugh, N.J.; Wedderburn, L.R.; et al. Genetic Association Study of NF-κB Genes in UK Caucasian Adult and Juvenile Onset Idiopathic Inflammatory Myopathy. Rheumatology 2012, 51, 794–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhou, Y.; Wang, Q. Multiple Values of F-FDG PET/CT in Idiopathic Inflammatory Myopathy. Clin. Rheumatol. 2017, 36, 2297–2305. [Google Scholar] [CrossRef]
- Xie, M.; Yu, Y.; Kang, R.; Zhu, S.; Yang, L.; Zeng, L.; Sun, X.; Yang, M.; Billiar, T.R.; Wang, H.; et al. PKM2-Dependent Glycolysis Promotes NLRP3 and AIM2 Inflammasome Activation. Nat. Commun. 2016, 7, 13280. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Appel, S.H. Immune-Mediated Mechanisms in the Pathoprogression of Amyotrophic Lateral Sclerosis. J. Neuroimmune Pharmacol. 2013, 8, 888–899. [Google Scholar] [CrossRef]
- Debye, B.; Schmülling, L.; Zhou, L.; Rune, G.; Beyer, C.; Johann, S. Neurodegeneration and NLRP3 Inflammasome Expression in the Anterior Thalamus of SOD1(G93A) ALS Mice. Brain Pathol. 2018, 28, 14–27. [Google Scholar] [CrossRef]
- Heitzer, M.; Kaiser, S.; Kanagaratnam, M.; Zendedel, A.; Hartmann, P.; Beyer, C.; Johann, S. Administration of 17β-Estradiol Improves Motoneuron Survival and Down-Regulates Inflammasome Activation in Male SOD1(G93A) ALS Mice. Mol. Neurobiol. 2017, 54, 8429–8443. [Google Scholar] [CrossRef]
- Kadhim, H.; Deltenre, P.; Martin, J.-J.; Sébire, G. In-Situ Expression of Interleukin-18 and Associated Mediators in the Human Brain of sALS Patients: Hypothesis for a Role for Immune-Inflammatory Mechanisms. Med. Hypotheses 2016, 86, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.; Jørgensen, L.H.; Bech, R.D.; Frandsen, U.; Schrøder, H.D. Skeletal Muscle Remodelling as a Function of Disease Progression in Amyotrophic Lateral Sclerosis. BioMed Res. Int. 2016, 2016, 5930621. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, S.; Esch, E.; Hartmann, P.; Goswami, A.; Nikolin, S.; Weis, J.; Beyer, C.; Johann, S. Expression Profile of Pattern Recognition Receptors in Skeletal Muscle of SOD1 Amyotrophic Lateral Sclerosis (ALS) Mice and Sporadic ALS Patients. Neuropathol. Appl. Neurobiol. 2018, 44, 606–627. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Naito, S. Tissue-Specific mRNA Expression Profiles of Human Toll-like Receptors and Related Genes. Biol. Pharm. Bull. 2005, 28, 886–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuis, L.; Gonzalez de Aguilar, J.-L.; Echaniz-Laguna, A.; Eschbach, J.; Rene, F.; Oudart, H.; Halter, B.; Huze, C.; Schaeffer, L.; Bouillaud, F.; et al. Muscle Mitochondrial Uncoupling Dismantles Neuromuscular Junction and Triggers Distal Degeneration of Motor Neurons. PLoS ONE 2009, 4, e5390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, J.G.; Ferrier, A.; Kothary, R. More than a Bystander: The Contributions of Intrinsic Skeletal Muscle Defects in Motor Neuron Diseases. Front. Physiol. 2013, 4, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzano, R.; Toivonen, J.M.; Moreno-Martínez, L.; de la Torre, M.; Moreno-García, L.; López-Royo, T.; Molina, N.; Zaragoza, P.; Calvo, A.C.; Osta, R. What Skeletal Muscle Has to Say in Amyotrophic Lateral Sclerosis: Implications for Therapy. Br. J. Pharmacol. 2021, 178, 1279–1297. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, X.; Miao, Y.; Wang, Y.; Wang, H.; Cheng, Z.; Wang, X.; Jing, X.; Jia, L.; Dai, L.; et al. NLRP3 Regulates Macrophage M2 Polarization through up-Regulation of IL-4 in Asthma. Biochem. J. 2018, 475, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Horsley, V.; Jansen, K.M.; Mills, S.T.; Pavlath, G.K. IL-4 Acts as a Myoblast Recruitment Factor during Mammalian Muscle Growth. Cell 2003, 113, 483–494. [Google Scholar] [CrossRef] [Green Version]
- Deora, V.; Lee, J.D.; Albornoz, E.A.; McAlary, L.; Jagaraj, C.J.; Robertson, A.A.B.; Atkin, J.D.; Cooper, M.A.; Schroder, K.; Yerbury, J.J.; et al. The Microglial NLRP3 Inflammasome Is Activated by Amyotrophic Lateral Sclerosis Proteins. Glia 2020, 68, 407–421. [Google Scholar] [CrossRef]
- Coll, R.C.; O’Neill, L.; Schroder, K. Questions and Controversies in Innate Immune Research: What Is the Physiological Role of NLRP3? Cell Death Discov. 2016, 2, 16019. [Google Scholar] [CrossRef]
- He, Y.; Varadarajan, S.; Muñoz-Planillo, R.; Burberry, A.; Nakamura, Y.; Núñez, G. 3,4-Methylenedioxy-β-Nitrostyrene Inhibits NLRP3 Inflammasome Activation by Blocking Assembly of the Inflammasome. J. Biol. Chem. 2014, 289, 1142–1150. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a Selective and Direct NLRP3 Inhibitor to Treat Inflammatory Disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef] [Green Version]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 Directly Targets the NLRP3 ATP-Hydrolysis Motif for Inflammasome Inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef]
- Duncan, J.A.; Bergstralh, D.T.; Wang, Y.; Willingham, S.B.; Ye, Z.; Zimmermann, A.G.; Ting, J.P.-Y. Cryopyrin/NALP3 Binds ATP/dATP, Is an ATPase, and Requires ATP Binding to Mediate Inflammatory Signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 8041–8046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Jiang, H.; Chen, Y.; Wang, X.; Yang, Y.; Tao, J.; Deng, X.; Liang, G.; Zhang, H.; Jiang, W.; et al. Tranilast Directly Targets NLRP3 to Treat Inflammasome-Driven Diseases. EMBO Mol. Med. 2018, 10, e8689. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W.; et al. Oridonin Is a Covalent NLRP3 Inhibitor with Strong Anti-Inflammasome Activity. Nat. Commun. 2018, 9, 2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Lv, Q.; Zheng, M.; Sun, H.; Shi, F. NLRP3 Inflammasome Inhibitor INF39 Attenuated NLRP3 Assembly in Macrophages. Int. Immunopharmacol. 2021, 92, 107358. [Google Scholar] [CrossRef]
- Brydges, S.D.; Mueller, J.L.; McGeough, M.D.; Pena, C.A.; Misaghi, A.; Gandhi, C.; Putnam, C.D.; Boyle, D.L.; Firestein, G.S.; Horner, A.A.; et al. Inflammasome-Mediated Disease Animal Models Reveal Roles for Innate but Not Adaptive Immunity. Immunity 2009, 30, 875–887. [Google Scholar] [CrossRef] [Green Version]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A Small-Molecule Inhibitor of the NLRP3 Inflammasome for the Treatment of Inflammatory Diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adinolfi, E.; Giuliani, A.L.; De Marchi, E.; Pegoraro, A.; Orioli, E.; Di Virgilio, F. The P2X7 Receptor: A Main Player in Inflammation. Biochem. Pharmacol. 2018, 151, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tian, G.; Quan, Y.; Li, J.; Wang, X.; Wu, W.; Li, M.; Liu, X. Inhibition of P2X7 Purinergic Receptor Ameliorates Cardiac Fibrosis by Suppressing NLRP3/IL-1β Pathway. Oxid. Med. Cell. Longev. 2020, 2020, 7956274. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Mueller, J.L.; Vitari, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide Inhibits the Cryopyrin/Nalp3 Inflammasome. J. Cell Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef] [Green Version]
- García, J.A.; Volt, H.; Venegas, C.; Doerrier, C.; Escames, G.; López, L.C.; Acuña-Castroviejo, D. Disruption of the NF-κB/NLRP3 Connection by Melatonin Requires Retinoid-Related Orphan Receptor-α and Blocks the Septic Response in Mice. FASEB J. 2015, 29, 3863–3875. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, A.; Rostamirad, A.; Seyyedebrahimi, S.; Meshkani, R. Curcumin Ameliorates Palmitate-Induced Inflammation in Skeletal Muscle Cells by Regulating JNK/NF-kB Pathway and ROS Production. Inflammopharmacology 2018, 26, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Qin, A.; Huang, H.; Zhou, P.; Zhang, C.; Liu, N.; Li, S.; Wen, G.; Zhang, C.; Dong, W.; et al. Shikonin Extracted from Medicinal Chinese Herbs Exerts Anti-Inflammatory Effect via Proteasome Inhibition. Eur. J. Pharmacol. 2011, 658, 242–247. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, R.P.; Birks, J.S.; Mani, V.; Biasiolli, L.; Robson, M.D.; L’Allier, P.L.; Gingras, M.-A.; Alie, N.; McLaughlin, M.A.; Basson, C.T.; et al. Arterial Effects of Canakinumab in Patients with Atherosclerosis and Type 2 Diabetes or Glucose Intolerance. J. Am. Coll. Cardiol. 2016, 68, 1769–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, K.S.; Yates, D.P.; Kramer, C.M.; Feller, A.; Mahling, P.; Colin, L.; Clough, T.; Wang, T.; LaPerna, L.; Patel, A.; et al. A Randomized, Placebo-Controlled Trial of Canakinumab in Patients with Peripheral Artery Disease. Vasc. Med. 2019, 24, 414–421. [Google Scholar] [CrossRef]
- Nicholls, M. CANTOS: One Year on: When the Findings of the CANTOS Trial Were Unveiled at ESC Congress 2017 in Barcelona, There Was a Sense of Hope That a New Era in Preventive Cardiology Was Set to Begin. One Year On, CANTOS Principle Investigator and Study Author Paul M. Ridker Discusses Whether That Optimism Is Being Realized. Eur. Heart J. 2018, 39, 3989–3990. [Google Scholar] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubuisson, N.; Versele, R.; Davis-López de Carrizosa, M.A.; Selvais, C.M.; Brichard, S.M.; Abou-Samra, M. Walking down Skeletal Muscle Lane: From Inflammasome to Disease. Cells 2021, 10, 3023. https://doi.org/10.3390/cells10113023
Dubuisson N, Versele R, Davis-López de Carrizosa MA, Selvais CM, Brichard SM, Abou-Samra M. Walking down Skeletal Muscle Lane: From Inflammasome to Disease. Cells. 2021; 10(11):3023. https://doi.org/10.3390/cells10113023
Chicago/Turabian StyleDubuisson, Nicolas, Romain Versele, María A. Davis-López de Carrizosa, Camille M. Selvais, Sonia M. Brichard, and Michel Abou-Samra. 2021. "Walking down Skeletal Muscle Lane: From Inflammasome to Disease" Cells 10, no. 11: 3023. https://doi.org/10.3390/cells10113023