
Recent Advances in Understanding the Pathogenesis of Rheumatoid Arthritis: New Treatment Strategies

 , ,
, , 

Abstract
:1. Introduction
2. Auto-Antibodies Involved in RA Pathogenesis and Development
2.1. The Role of RF in Pathogenesis and Its Clinical Significance
2.2. The Role of Anti-Modified Protein Antibodies in RA
2.2.1. Anti-Citrullinated Protein Antibodies (ACPA)
2.2.2. Anti-Carbamylated Protein Antibodies
2.2.3. Anti-PAD4 Antibodies
2.2.4. Anti-b-Raf and Anti-RA33 Antibodies
3. The Role of Epigenetic Modifications and Glycosylation in RA
3.1. DNA Methylation and Demethylation
3.2. Histone Modifications
3.3. Glycosylation
4. Auto-Antibody Cross-Talk and T-Cells in RA
5. The Role of Autophagy and Oxidative Stress in RA
5.1. Autophagy and Apoptosis
5.2. Autophagy in Lymphocyte Homeostasis
5.3. Autophagy and Citrullination
5.4. Oxidative Stress
6. Treatment Strategies in RA
6.1. First-Line Treatment
6.2. New Treatment Strategies
6.2.1. Biological Agents
Targeting TNF by TNF Inhibitors (TNFi) in RA Therapy
Targeting IL by IL Inhibitors in RA Therapy
Targeting Co-Stimulation by Co-Stimulation Blockers in RA Therapy
Targeting CXCL by CXCL Chemokine Inhibitors in RA Therapy
Targeting B-Cells by Anti-B-Cell-Agents in RA Therapy
6.2.2. Synthetical Agents
Targeting Janus-Activated-Kinase (JAK) by JAK Inhibitors in RA Therapy
6.2.3. Cell Therapy
Targeting Immunomodulation by MSC in RA Therapy
Targeting Plasma Cell Depletion in RA Therapy
6.2.4. Combinational Therapy
6.2.5. Targeting Epigenetic Factors in RA Therapy
DNMT and HDAC Inhibitors
6.2.6. Targeting Autophagy in RA Therapy
6.2.7. Treat-to-Target Strategy
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3-MA | 3-Methyladenine |
| Abs | Antibodies |
| ACPA | Anti-Citrullinated Antibodies |
| ACR | American College of Rheumatology |
| ADA | Antidrug Auto-antibodies |
| AKT | Protein Kinase B |
| AMPA | Anti-Modified Protein Antibody |
| Anti-CarP | Antibodies targeting Carbamylated Proteins |
| Anti-RA33 | Antibodies to the heterogeneous nuclear ribonucleoprotein A2/B1 |
| APC | Antigen-Presenting Cell |
| APRIL | Proliferation-Inducing Ligand |
| ATG | Anti-Thymocyte Globulin |
| ATP | Adenosine Triphosphate |
| BAFF | B-cell Activating Factor |
| BCR | B-Cell Receptor |
| CCP | Cyclic Citrullinated Peptide |
| CD | Cluster of Differentiation |
| CD4+ CTL | CD4+ T-cells with Cytotoxic Potential |
| CIA | Collagen Induced Arthritis |
| CII | Collagen type II |
| CMV | Cytomegalovirus |
| COX | Cyclooxygenase |
| CQ | Chloroquine |
| CTLA4 | Cytotoxic T-lymphocyte-associated Protein 4 |
| CXCL | Chemokine Ligand |
| DNA | Deoxyribonucleic Acid |
| DAS | Disease Activity Score |
| DMARDs | Disease-modifying Anti-Rheumatic Drugs |
| DNMT | DNA-Methyltransferase |
| EA2 | Endophilin A2 |
| ELISA | Enzyme Linked Immunosorbent Assay |
| EMA | European Medicines Agency |
| EOMES | Eomesodermin gene |
| ER | Endoplasmic Reticulum |
| EULAR | European League Against Rheumatism |
| EV | Extracellular Vesicle |
| FDA | Food and Drug Administration |
| FLS | Fibroblast-Like Synoviocytes |
| FOXP3 | Forkhead box P3 |
| GC | Glucocorticoid |
| GWAS | Genome Wide Association Studies |
| HAT | Histone Acetyltransferase |
| HCQ | Hydroxy Chloroquine |
| HDAC | Histone Deacetylase |
| HLA | Human Leukocyte Antigen |
| HLA-DRB1 | HLA class II Histocompatibility Antigen DRB1 beta chain |
| IFN | Interferon |
| IL | Interleukin |
| JAK | Janus-Activated Kinase |
| LC3-II | Microtubule-associated proteins 1A/1B light chain 3B |
| LTQ-ESI-MS | Linear ion-trap Electrospray Ionization Mass Spectrometry |
| mAb | Monoclonal Antibody |
| MHC II | Major histocompatibility Complex class II |
| MMP | Matrix-Metallo-Proteinase |
| mRNA | Messenger RNA |
| MSC | Mesenchymal Stem Cell |
| mTORC1 | mTOR complex 1 |
| MTX | Methotrexate |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| NCF1 | Neutrophil Cytosol Factor 1 |
| ncRNA | Noncoding RNA |
| NF-κB | Nuclear Factor kappa B |
| NO | Nitric Oxide |
| NOS2 | Nitric Oxide Synthase 2 |
| NOX2 | NADPH Oxidase 2 |
| NSAID | Nonsteroidal Anti-Inflammatory Drug |
| OA | Osteoarthritis |
| PAD | Peptidyl Arginine Deiminase |
| PBMC | Peripheral Blood Mononuclear Cell |
| PC | Plasma Cell |
| PI3K | Phosphoinositide 3-Kinase |
| Prdm1 | PR domain Zinc finger protein1 |
| PTK | Protein-Tyrosine-Kinase |
| PTPN22 | Protein tyrosine phosphatase, non-receptor type 22 |
| QTL | Quantitative trait Loci |
| RA | Rheumatoid Arthritis |
| RA33 | Heterogenous nuclear ribonucleoprotein A2/B1 |
| RANKL | Receptor Activator of NF-κB Ligand |
| RF | Rheumatoid Factor |
| RNA | Ribonucleic Acid |
| ROS | Reactive Oxygen Species |
| SNP | Single Nucleotide Polymorphism |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| Syk | Spleen tyrosine kinase |
| TACI | Transmembrane Activator and CAML Interactor CD Antigen |
| TCR | T-cell Receptor |
| Tfh | Follicular T-helper Cell |
| TGF | Tumor Growth Factor |
| Th | T-helper Cell |
| TLR | Toll-Like Receptor |
| TNF | Tumor Necrosis Factor |
| TNFi | Tumor Necrosis Factor inhibitor |
| Treg | Regulatory T-Cell |
| TYK2 | Tyrosine kinase 2 |
References
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nature 2003, 423, 356–361. [Google Scholar] [CrossRef] [PubMed]
- De Brito Rocha, S.; Baldo, D.C.; Andrade, L.E.C. Clinical and pathophysiologic relevance of autoantibodies in rheumatoid arthritis. Adv. Rheumatol. 2019, 59, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Farid, S.S.; Azizi, G.; Mirshafiey, A. Anti-citrullinated protein antibodies and their clinical utility in rheumatoid arthritis. Int. J. Rheum. Dis. 2013, 16, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Sieghart, D.; Platzer, A.; Studenic, P.; Alasti, F.; Grundhuber, M.; Swiniarski, S.; Horn, T.; Haslacher, H.; Blüml, S.; Smolen, J. Determination of autoantibody isotypes increases the sensitivity of serodiagnostics in rheumatoid arthritis. Front. Immunol. 2018, 9, 876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, E.A.; Raychaudhuri, S.; Remmers, E.F.; Xie, G.; Eyre, S.; Thomson, B.P.; Li, Y.; Kurreeman, F.A.; Zhernakova, A.; Hinks, A. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 2010, 42, 508. [Google Scholar] [CrossRef]
- Vang, T.; Congia, M.; Macis, M.D.; Musumeci, L.; Orrú, V.; Zavattari, P.; Nika, K.; Tautz, L.; Taskén, K.; Cucca, F. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat. Genet. 2005, 37, 1317–1319. [Google Scholar] [CrossRef]
- Gregersen, P.K.; Silver, J.; Winchester, R.J. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 1987, 30, 1205–1213. [Google Scholar] [CrossRef]
- Dultz, G.; Matheis, N.; Dittmar, M.; Röhrig, B.; Bender, K.; Kahaly, G.J. The protein tyrosine phosphatase non-receptor type 22 C1858T polymorphism is a joint susceptibility locus for immunthyroiditis and autoimmune diabetes. Thyroid 2009, 19, 143–148. [Google Scholar] [CrossRef]
- Bottini, N.; Peterson, E.J. Tyrosine phosphatase PTPN22: Multifunctional regulator of immune signaling, development, and disease. Annu. Rev. Immunol. 2014, 32, 83–119. [Google Scholar] [CrossRef]
- Begovich, A.B.; Carlton, V.E.; Honigberg, L.A.; Schrodi, S.J.; Chokkalingam, A.P.; Alexander, H.C.; Ardlie, K.G.; Huang, Q.; Smith, A.M.; Spoerke, J.M. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am. J. Hum. Genet. 2004, 75, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Rieck, M.; Arechiga, A.; Onengut-Gumuscu, S.; Greenbaum, C.; Concannon, P.; Buckner, J.H. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J. Immunol. 2007, 179, 4704–4710. [Google Scholar] [CrossRef] [Green Version]
- Aho, K.; Palusuo, T.; Kurki, P. Marker antibodies of rheumatoid arthritis: Diagnostic and pathogenetic implications. Semin. Arthritis Rheum. 1994, 23, 379–387. [Google Scholar] [CrossRef]
- Barik, R.R.; Bhatt, L.K. Emerging epigenetic targets in rheumatoid arthritis. Rheumatol. Int. 2021, 41, 2047–2067. [Google Scholar] [CrossRef]
- Gianfrancesco, M.A.; Crowson, C.S. Where There’s Smoke, There’s a Joint: Passive Smoking and Rheumatoid Arthritis. Arthritis Rheumatol. 2021. [Google Scholar] [CrossRef]
- Adami, G.; Viapiana, O.; Rossini, M.; Orsolini, G.; Bertoldo, E.; Giollo, A.; Gatti, D.; Fassio, A. Association between environmental air pollution and rheumatoid arthritis flares. Rheumatology 2021, 60, 4591–4597. [Google Scholar] [CrossRef]
- Esberg, A.; Johansson, L.; Johansson, I.; Dahlqvist, S.R. Oral Microbiota Identifies Patients in Early Onset Rheumatoid Arthritis. Microorganisms 2021, 9, 1657. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Sehgal, A.; Zengin, G.; Brisc, C.; Brisc, M.C.; Munteanu, M.A.; Nistor-Cseppento, D.C.; Bungau, S. The Lipid Paradox as a Metabolic Checkpoint and Its Therapeutic Significance in Ameliorating the Associated Cardiovascular Risks in Rheumatoid Arthritis Patients. Int. J. Mol. Sci. 2020, 21, 9505. [Google Scholar] [CrossRef]
- Ingegnoli, F.; Castelli, R.; Gualtierotti, R. Rheumatoid factors: Clinical applications. Dis. Markers 2013, 35, 727–734. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Kang, E. Autoantibodies in rheumatoid arthritis: Rheumatoid factors and anticitrullinated protein antibodies. QJM Int. J. Med. 2010, 103, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, T.; Arinbjarnarson, S.; Thorsteinsson, J.; Steinsson, K.; Geirsson, A.; Jonsson, H.; Valdimarsson, H. Raised IgA rheumatoid factor (RF) but not IgM RF or IgG RF is associated with extra-articular manifestations in rheumatoid arthritis. Scand. J. Rheumatol. 1995, 24, 372–375. [Google Scholar] [CrossRef]
- Sokolove, J.; Johnson, D.S.; Lahey, L.J.; Wagner, C.A.; Cheng, D.; Thiele, G.M.; Michaud, K.; Sayles, H.; Reimold, A.M.; Caplan, L. Rheumatoid factor as a potentiator of anti–citrullinated protein antibody–mediated inflammation in rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 813–821. [Google Scholar] [CrossRef]
- Reed, E.; Hedström, A.K.; Hansson, M.; Mathsson-Alm, L.; Brynedal, B.; Saevarsdottir, S.; Cornillet, M.; Jakobsson, P.-J.; Holmdahl, R.; Skriner, K. Presence of autoantibodies in “seronegative” rheumatoid arthritis associates with classical risk factors and high disease activity. Arthritis Res. Ther. 2020, 22, 1–11. [Google Scholar] [CrossRef]
- Steen, J.; Forsström, B.; Sahlström, P.; Odowd, V.; Israelsson, L.; Krishnamurthy, A.; Badreh, S.; Mathsson Alm, L.; Compson, J.; Ramsköld, D.; et al. Recognition of Amino Acid Motifs, Rather Than Specific Proteins, by Human Plasma Cell-Derived Monoclonal Antibodies to Posttranslationally Modified Proteins in Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 196–209. [Google Scholar] [CrossRef]
- Kampstra, A.S.B.; Dekkers, J.S.; Volkov, M.; Dorjée, A.L.; Hafkenscheid, L.; Kempers, A.C.; van Delft, M.; Kissel, T.; Reijm, S.; Janssen, G.M.C.; et al. Different classes of anti-modified protein antibodies are induced on exposure to antigens expressing only one type of modification. Ann. Rheum. Dis. 2019, 78, 908–916. [Google Scholar] [CrossRef]
- Suzuki, A.; Yamada, R.; Chang, X.; Tokuhiro, S.; Sawada, T.; Suzuki, M.; Nagasaki, M.; Nakayama-Hamada, M.; Kawaida, R.; Ono, M.; et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat. Genet. 2003, 34, 395–402. [Google Scholar] [CrossRef]
- Mondal, S.; Thompson, P.R. Protein Arginine Deiminases (PADs): Biochemistry and Chemical Biology of Protein Citrullination. Acc. Chem. Res. 2019, 52, 818–832. [Google Scholar] [CrossRef]
- Vossenaar, E.R.; van Venrooij, W.J. Citrullinated proteins: Sparks that may ignite the fire in rheumatoid arthritis. Arthritis Res. 2004, 6, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Tilvawala, R.; Nguyen, S.H.; Maurais, A.J.; Nemmara, V.V.; Nagar, M.; Salinger, A.J.; Nagpal, S.; Weerapana, E.; Thompson, P.R. The Rheumatoid Arthritis-Associated Citrullinome. Cell Chem. Biol. 2018, 25, 691–704.e696. [Google Scholar] [CrossRef] [Green Version]
- Ge, C.; Tong, D.; Liang, B.; Lönnblom, E.; Schneider, N.; Hagert, C.; Viljanen, J.; Ayoglu, B.; Stawikowska, R.; Nilsson, P. Anti-citrullinated protein antibodies cause arthritis by cross-reactivity to joint cartilage. JCI Insight 2017, 2, e93688. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, M.; Takahara, H.; Nishi, Y.; Sugawara, K.; Sato, C. Ca2+-dependent deimination-induced disassembly of intermediate filaments involves specific modification of the amino-terminal head domain. J. Biol. Chem. 1989, 264, 18119–18127. [Google Scholar] [CrossRef]
- Tarcsa, E.; Marekov, L.N.; Mei, G.; Melino, G.; Lee, S.C.; Steinert, P.M. Protein unfolding by peptidylarginine deiminase. Substrate specificity and structural relationships of the natural substrates trichohyalin and filaggrin. J. Biol. Chem. 1996, 271, 30709–30716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundberg, K.; Nijenhuis, S.; Vossenaar, E.R.; Palmblad, K.; van Venrooij, W.J.; Klareskog, L.; Zendman, A.J.; Harris, H.E. Citrullinated proteins have increased immunogenicity and arthritogenicity and their presence in arthritic joints correlates with disease severity. Arthritis Res. 2005, 7, R458–R467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Delft, M.A.M.; Huizinga, T.W.J. An overview of autoantibodies in rheumatoid arthritis. J. Autoimmun. 2020, 110, 102392. [Google Scholar] [CrossRef] [PubMed]
- Jónsson, T.; Steinsson, K.; Jónsson, H.; Geirsson, A.; Thorsteinsson, J.; Valdimarsson, H. Combined elevation of IgM and IgA rheumatoid factor has high diagnostic specificity for rheumatoid arthritis. Rheumatol. Int. 1998, 18, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Willemze, A.; Trouw, L.A.; Toes, R.E.; Huizinga, T.W. The influence of ACPA status and characteristics on the course of RA. Nat. Rev. Rheumatol. 2012, 8, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Valesini, G.; Gerardi, M.C.; Iannuccelli, C.; Pacucci, V.A.; Pendolino, M.; Shoenfeld, Y. Citrullination and autoimmunity. Autoimmun. Rev. 2015, 14, 490–497. [Google Scholar] [CrossRef]
- Ge, C.; Holmdahl, R. The structure, specificity and function of anti-citrullinated protein antibodies. Nat. Rev. Rheumatol. 2019, 15, 503–508. [Google Scholar] [CrossRef]
- Bersellini Farinotti, A.; Wigerblad, G.; Nascimento, D.; Bas, D.B.; Morado Urbina, C.; Nandakumar, K.S.; Sandor, K.; Xu, B.; Abdelmoaty, S.; Hunt, M.A. Cartilage-binding antibodies induce pain through immune complex–mediated activation of neurons. J. Exp. Med. 2019, 216, 1904–1924. [Google Scholar] [CrossRef] [Green Version]
- Arnoux, F.; Mariot, C.; Peen, E.; Lambert, N.C.; Balandraud, N.; Roudier, J.; Auger, I. Peptidyl arginine deiminase immunization induces anticitrullinated protein antibodies in mice with particular MHC types. Proc. Natl. Acad. Sci. USA 2017, 114, E10169–E10177. [Google Scholar] [CrossRef] [Green Version]
- Auger, I.; Balandraud, N.; Massy, E.; Hemon, M.F.; Peen, E.; Arnoux, F.; Mariot, C.; Martin, M.; Lafforgue, P.; Busnel, J.M.; et al. Peptidylarginine Deiminase Autoimmunity and the Development of Anti-Citrullinated Protein Antibody in Rheumatoid Arthritis: The Hapten-Carrier Model. Arthritis Rheumatol. 2020, 72, 903–911. [Google Scholar] [CrossRef]
- Burska, A.N.; Hunt, L.; Boissinot, M.; Strollo, R.; Ryan, B.J.; Vital, E.; Nissim, A.; Winyard, P.G.; Emery, P.; Ponchel, F. Autoantibodies to posttranslational modifications in rheumatoid arthritis. Mediat. Inflamm. 2014, 2014, 492873. [Google Scholar] [CrossRef] [Green Version]
- Mydel, P.; Wang, Z.; Brisslert, M.; Hellvard, A.; Dahlberg, L.E.; Hazen, S.L.; Bokarewa, M. Carbamylation-dependent activation of T cells: A novel mechanism in the pathogenesis of autoimmune arthritis. J. Immunol. 2010, 184, 6882–6890. [Google Scholar] [CrossRef] [Green Version]
- Lo, K.C.; Sullivan, E.; Bannen, R.M.; Jin, H.; Rowe, M.; Li, H.; Pinapati, R.S.; Cartwright, A.J.; Tan, J.C.; Patel, J.; et al. Comprehensive Profiling of the Rheumatoid Arthritis Antibody Repertoire. Arthritis Rheumatol. 2020, 72, 242–250. [Google Scholar] [CrossRef]
- Derksen, V.; Huizinga, T.W.J.; van der Woude, D. The role of autoantibodies in the pathophysiology of rheumatoid arthritis. Semin. Immunopathol. 2017, 39, 437–446. [Google Scholar] [CrossRef]
- Shi, J.; Knevel, R.; Suwannalai, P.; van der Linden, M.P.; Janssen, G.M.; van Veelen, P.A.; Levarht, N.E.; van der Helm-van, A.H.; Cerami, A.; Huizinga, T.W. Autoantibodies recognizing carbamylated proteins are present in sera of patients with rheumatoid arthritis and predict joint damage. Proc. Natl. Acad. Sci. USA 2011, 108, 17372–17377. [Google Scholar] [CrossRef] [Green Version]
- Sahlström, P.; Hansson, M.; Steen, J.; Amara, K.; Titcombe, P.J.; Forsström, B.; Stålesen, R.; Israelsson, L.; Piccoli, L.; Lundberg, K.; et al. Different Hierarchies of Anti-Modified Protein Autoantibody Reactivities in Rheumatoid Arthritis. Arthritis Rheumatol. 2020, 72, 1643–1657. [Google Scholar] [CrossRef]
- Humphreys, J.H.; Verheul, M.K.; Barton, A.; MacGregor, A.J.; Lunt, M.; Toes, R.E.; Symmons, D.P.; Trouw, L.A.; Verstappen, S.M. Anticarbamylated protein antibodies are associated with long-term disability and increased disease activity in patients with early inflammatory arthritis: Results from the Norfolk Arthritis Register. Ann. Rheum. Dis. 2016, 75, 1139–1144. [Google Scholar] [CrossRef]
- Murata, K.; Ito, H.; Hashimoto, M.; Murakami, K.; Watanabe, R.; Tanaka, M.; Yamamoto, W.; Matsuda, S. Fluctuation in anti-cyclic citrullinated protein antibody level predicts relapse from remission in rheumatoid arthritis: KURAMA cohort. Arthritis Res. Ther. 2020, 22, 1–10. [Google Scholar] [CrossRef]
- Shi, J.; van de Stadt, L.A.; Levarht, E.; Huizinga, T.; Toes, R.; Trouw, L.A.; van Schaardenburg, D. Anti Carbamylated Protein Antibodies (anti-CarP) are present in arthralgia patients and predict the development of rheumatoid arthritis. Arthritis Rheum. 2012, 21, 37830. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; van Steenbergen, H.W.; van Nies, J.A.; Levarht, E.W.; Huizinga, T.W.; van der Helm-van Mil, A.H.; Toes, R.E.; Trouw, L.A. The specificity of anti-carbamylated protein antibodies for rheumatoid arthritis in a setting of early arthritis. Arthritis Res. 2015, 17, 339. [Google Scholar] [CrossRef] [Green Version]
- Scinocca, M.; Bell, D.A.; Racapé, M.; Joseph, R.; Shaw, G.; McCormick, J.K.; Gladman, D.D.; Pope, J.; Barra, L.; Cairns, E. Antihomocitrullinated fibrinogen antibodies are specific to rheumatoid arthritis and frequently bind citrullinated proteins/peptides. J. Rheumatol. 2014, 41, 270–279. [Google Scholar] [CrossRef]
- Shi, J.; van de Stadt, L.A.; Levarht, E.N.; Huizinga, T.W.; Hamann, D.; van Schaardenburg, D.; Toes, R.E.; Trouw, L.A. Anti-carbamylated protein (anti-CarP) antibodies precede the onset of rheumatoid arthritis. Ann. Rheum. Dis. 2014, 73, 780–783. [Google Scholar] [CrossRef]
- Darrah, E.; Giles, J.T.; Ols, M.L.; Bull, H.G.; Andrade, F.; Rosen, A. Erosive rheumatoid arthritis is associated with antibodies that activate PAD4 by increasing calcium sensitivity. Sci. Transl. Med. 2013, 5, 186ra65. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Castillo, Z.; Muñoz-Valle, J.F.; Llamas-Covarrubias, M.A. Clinical and immunological aspects of anti-peptidylarginine deiminase type 4 (anti-PAD4) autoantibodies in rheumatoid arthritis. Autoimmun. Rev. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Umeda, N.; Matsumoto, I.; Kawaguchi, H.; Kurashima, Y.; Kondo, Y.; Tsuboi, H.; Ogishima, H.; Suzuki, T.; Kagami, Y.; Sakyu, T. Prevalence of soluble peptidylarginine deiminase 4 (PAD4) and anti-PAD4 antibodies in autoimmune diseases. Clin. Rheumatol. 2016, 35, 1181–1188. [Google Scholar] [CrossRef]
- Auger, I.; Charpin, C.; Balandraud, N.; Martin, M.; Roudier, J. Autoantibodies to PAD4 and BRAF in rheumatoid arthritis. Autoimmun. Rev. 2012, 11, 801–803. [Google Scholar] [CrossRef]
- Hoffmann, A.; Zimmermann, C.A.; Spengler, D. Molecular epigenetic switches in neurodevelopment in health and disease. Front. Behav. Neurosci. 2015, 9, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verheul, M.; Fearon, U.; Trouw, L.; Veale, D. Biomarkers for rheumatoid and psoriatic arthritis. Clin. Immunol. 2015, 161, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Syed Mohamed Suhail, S.M.; Nur Atiqah, I.; Nur Salimi Zakirah, Z.A.; Lailatul Syazwani, Z.; Batis, W.W.; Md Monoto, E.M.; Abdul Wahab, A.; Mohd Shahrir, M.S. Diagnostic performance of anti-RA33 antibody as a serological marker for rheumatoid arthritis. Malays. J. Pathol. 2019, 41, 259–265. [Google Scholar] [PubMed]
- Schuebel, K.; Gitik, M.; Domschke, K.; Goldman, D. Making sense of epigenetics. Int. J. Neuropsychopharmacol. 2016, 19, pyw058. [Google Scholar] [CrossRef] [Green Version]
- Hedrich, C.M.; Tsokos, G.C. Epigenetic mechanisms in systemic lupus erythematosus and other autoimmune diseases. Trends Mol. Med. 2011, 17, 714–724. [Google Scholar] [CrossRef] [Green Version]
- Zan, H.; Casali, P. Epigenetics of peripheral B-cell differentiation and the antibody response. Front. Immunol. 2015, 6, 631. [Google Scholar] [CrossRef] [Green Version]
- Kagohara, L.T.; Stein-O’Brien, G.L.; Kelley, D.; Flam, E.; Wick, H.C.; Danilova, L.V.; Easwaran, H.; Favorov, A.V.; Qian, J.; Gaykalova, D.A. Epigenetic regulation of gene expression in cancer: Techniques, resources and analysis. Brief. Funct. Genom. 2018, 17, 49–63. [Google Scholar] [CrossRef] [Green Version]
- Topper, M.J.; Vaz, M.; Marrone, K.A.; Brahmer, J.R.; Baylin, S.B. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 75–90. [Google Scholar] [CrossRef]
- Ciechomska, M.; Roszkowski, L.; Maslinski, W. DNA methylation as a future therapeutic and diagnostic target in rheumatoid arthritis. Cells 2019, 8, 953. [Google Scholar] [CrossRef] [Green Version]
- Barwick, B.G.; Scharer, C.D.; Martinez, R.J.; Price, M.J.; Wein, A.N.; Haines, R.R.; Bally, A.P.; Kohlmeier, J.E.; Boss, J.M. B cell activation and plasma cell differentiation are inhibited by de novo DNA methylation. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Muto, A.; Shima, H.; Katoh, Y.; Sax, N.; Tajima, S.; Brydun, A.; Ikura, T.; Yoshizawa, N.; Masai, H. Epigenetic regulation of the Blimp-1 gene (Prdm1) in B cells involves Bach2 and histone deacetylase 3. J. Biol. Chem. 2016, 291, 6316–6330. [Google Scholar] [CrossRef] [Green Version]
- Whitaker, J.W.; Shoemaker, R.; Boyle, D.L.; Hillman, J.; Anderson, D.; Wang, W.; Firestein, G.S. An imprinted rheumatoid arthritis methylome signature reflects pathogenic phenotype. Genome Med. 2013, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ekwall, A.K.H.; Whitaker, J.W.; Hammaker, D.; Bugbee, W.D.; Wang, W.; Firestein, G.S. The rheumatoid arthritis risk gene LBH regulates growth in fibroblast-like synoviocytes. Arthritis Rheumatol. 2015, 67, 1193–1202. [Google Scholar] [CrossRef] [Green Version]
- Nakano, K.; Whitaker, J.W.; Boyle, D.L.; Wang, W.; Firestein, G.S. DNA methylome signature in rheumatoid arthritis. Ann. Rheum. Dis. 2013, 72, 110–117. [Google Scholar] [CrossRef]
- Lefèvre, S.; Knedla, A.; Tennie, C.; Kampmann, A.; Wunrau, C.; Dinser, R.; Korb, A.; Schnäker, E.-M.; Tarner, I.H.; Robbins, P.D. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat. Med. 2009, 15, 1414–1420. [Google Scholar] [CrossRef] [Green Version]
- Rhead, B.; Holingue, C.; Cole, M.; Shao, X.; Quach, H.L.; Quach, D.; Shah, K.; Sinclair, E.; Graf, J.; Link, T. Rheumatoid arthritis naive T cells share hypermethylation sites with synoviocytes. Arthritis Rheumatol. 2017, 69, 550–559. [Google Scholar] [CrossRef] [Green Version]
- Nemtsova, M.V.; Zaletaev, D.V.; Bure, I.V.; Mikhaylenko, D.S.; Kuznetsova, E.B.; Alekseeva, E.A.; Beloukhova, M.I.; Deviatkin, A.A.; Lukashev, A.N.; Zamyatnin, A.A., Jr. Epigenetic changes in the pathogenesis of rheumatoid arthritis. Front. Genet. 2019, 10, 570. [Google Scholar] [CrossRef] [Green Version]
- Huber, L.C.; Brock, M.; Hemmatazad, H.; Giger, O.T.; Moritz, F.; Trenkmann, M.; Distler, J.H.; Gay, R.E.; Kolling, C.; Moch, H. Histone deacetylase/acetylase activity in total synovial tissue derived from rheumatoid arthritis and osteoarthritis patients. Arthritis Rheum. 2007, 56, 1087–1093. [Google Scholar] [CrossRef]
- Wada, T.T.; Araki, Y.; Sato, K.; Aizaki, Y.; Yokota, K.; Kim, Y.T.; Oda, H.; Kurokawa, R.; Mimura, T. Aberrant histone acetylation contributes to elevated interleukin-6 production in rheumatoid arthritis synovial fibroblasts. Biochem. Biophys. Res. Commun. 2014, 444, 682–686. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, B.R.; Suh, D.-h.; Bae, D.; Ha, N.; Choi, Y.I.; Yoo, H.J.; Park, J.K.; Lee, E.Y.; Lee, E.B.; Song, Y.W. Therapeutic effect of a novel histone deacetylase 6 inhibitor, CKD-L, on collagen-induced arthritis in vivo and regulatory T cells in rheumatoid arthritis in vitro. Arthritis Res. Ther. 2017, 19, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, J.; Zhou, J. Targeted inhibition of histone deacetylase 6 in inflammatory diseases. Thorac. Cancer 2019, 10, 405–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonelli, M.; Puchner, A.; Göschl, L.; Hayer, S.; Niederreiter, B.; Steiner, G.; Tillmann, K.; Plasenzotti, R.; Podesser, B.; Georgel, P. CCR6 controls autoimmune but not innate immunity-driven experimental arthritis. J. Cell. Mol. Med. 2018, 22, 5278–5285. [Google Scholar] [CrossRef] [PubMed]
- Göschl, L.; Preglej, T.; Boucheron, N.; Saferding, V.; Müller, L.; Platzer, A.; Hirahara, K.; Shih, H.Y.; Backlund, J.; Matthias, P.; et al. Histone deacetylase 1 (HDAC1): A key player of T cell-mediated arthritis. J. Autoimmun. 2020, 108, 102379. [Google Scholar] [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fc-receptors as regulators of immunity. Adv. Immunol. 2007, 96, 179–204. [Google Scholar]
- Albrecht, S.; Unwin, L.; Muniyappa, M.; Rudd, P.M. Glycosylation as a marker for inflammatory arthritis. Cancer Biomark. 2014, 14, 17–28. [Google Scholar] [CrossRef]
- Hafkenscheid, L.; Bondt, A.; Scherer, H.U.; Huizinga, T.W.; Wuhrer, M.; Toes, R.E.; Rombouts, Y. Structural analysis of variable domain glycosylation of anti-citrullinated protein antibodies in rheumatoid arthritis reveals the presence of highly sialylated glycans. Mol. Cell. Proteom. 2017, 16, 278–287. [Google Scholar] [CrossRef] [Green Version]
- Scherer, H.U.; Huizinga, T.W.; Krönke, G.; Schett, G.; Toes, R.E. The B cell response to citrullinated antigens in the development of rheumatoid arthritis. Nat. Rev. Rheumatol. 2018, 14, 157. [Google Scholar] [CrossRef]
- Seeling, M.; Brückner, C.; Nimmerjahn, F. Differential antibody glycosylation in autoimmunity: Sweet biomarker or modulator of disease activity? Nat. Rev. Rheumatol. 2017, 13, 621–630. [Google Scholar] [CrossRef]
- Kempers, A.C.; Hafkenscheid, L.; Scherer, H.U.; Toes, R.E. Variable domain glycosylation of ACPA-IgG: A missing link in the maturation of the ACPA response? Clin. Immunol. 2018, 186, 34–37. [Google Scholar] [CrossRef]
- Li, J.; Hsu, H.C.; Ding, Y.; Li, H.; Wu, Q.; Yang, P.; Luo, B.; Rowse, A.L.; Spalding, D.M.; Bridges, S.L., Jr. Inhibition of Fucosylation Reshapes Inflammatory Macrophages and Suppresses Type II Collagen–Induced Arthritis. Arthritis Rheumatol. 2014, 66, 2368–2379. [Google Scholar] [CrossRef]
- Shinkawa, T.; Nakamura, K.; Yamane, N.; Shoji-Hosaka, E.; Kanda, Y.; Sakurada, M.; Uchida, K.; Anazawa, H.; Satoh, M.; Yamasaki, M. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J. Biol. Chem. 2003, 278, 3466–3473. [Google Scholar] [CrossRef] [Green Version]
- Zauner, G.; Selman, M.H.; Bondt, A.; Rombouts, Y.; Blank, D.; Deelder, A.M.; Wuhrer, M. Glycoproteomic analysis of antibodies. Mol. Cell. Proteom. 2013, 12, 856–865. [Google Scholar] [CrossRef] [Green Version]
- Ohmi, Y.; Ise, W.; Harazono, A.; Takakura, D.; Fukuyama, H.; Baba, Y.; Narazaki, M.; Shoda, H.; Takahashi, N.; Ohkawa, Y. Sialylation converts arthritogenic IgG into inhibitors of collagen-induced arthritis. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Xie, Q.; Wang, Y.; Li, Y. Abberant Immunoglobulin G Glycosylation in Rheumatoid Arthritis by LTQ-ESI-MS. Int. J. Mol. Sci. 2020, 21, 2045. [Google Scholar] [CrossRef] [Green Version]
- Anderluh, M.; Berti, F.; Bzducha-Wróbel, A.; Chiodo, F.; Colombo, C.; Compostella, F.; Durlik, K.; Ferhati, X.; Holmdahl, R.; Jovanovic, D. Emerging glyco-based strategies to steer immune responses. FEBS J. 2021, 288, 4746–4772. [Google Scholar] [CrossRef]
- Firestein, G.S.; McInnes, I.B. Immunopathogenesis of rheumatoid arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J. Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018, 6, 1–14. [Google Scholar] [CrossRef]
- Yap, H.-Y.; Tee, S.Z.-Y.; Wong, M.M.-T.; Chow, S.-K.; Peh, S.-C.; Teow, S.-Y. Pathogenic role of immune cells in rheumatoid arthritis: Implications in clinical treatment and biomarker development. Cells 2018, 7, 161. [Google Scholar] [CrossRef] [Green Version]
- Consortium, W.T.C.C. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef] [Green Version]
- Stastny, P. Association of the B-cell alloantigen DRw4 with rheumatoid arthritis. N. Engl. J. Med. 1978, 298, 869–871. [Google Scholar] [CrossRef]
- Hill, J.A.; Southwood, S.; Sette, A.; Jevnikar, A.M.; Bell, D.A.; Cairns, E. Cutting edge: The conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule. J. Immunol. 2003, 171, 538–541. [Google Scholar] [CrossRef] [Green Version]
- Chemin, K.; Pollastro, S.; James, E.; Ge, C.; Albrecht, I.; Herrath, J.; Gerstner, C.; Tandre, K.; Sampaio Rizzi, T.; Rönnblom, L. A Novel HLA–DRB1* 10: 01–Restricted T Cell Epitope from Citrullinated Type II Collagen Relevant to Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Sidney, J.; Becart, S.; Zhou, M.; Duffy, K.; Lindvall, M.; Moore, E.C.; Moore, E.L.; Rao, T.; Rao, N.; Nielsen, M.; et al. Citrullination only infrequently impacts peptide binding to HLA class II MHC. PLoS ONE 2017, 12, e0177140. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Kawaguchi, T.; Stahl, E.A.; Kurreeman, F.A.; Nishida, N. Meta-analysis identifies nine new loci associated with rheumatoid arthritis in the Japanese population. Nat. Genet. 2012, 44, 511. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef]
- Borst, J.; Ahrends, T.; Bąbała, N.; Melief, C.J.; Kastenmüller, W. CD4+ T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef]
- Cho, B.-A.; Sim, J.H.; Park, J.A.; Kim, H.W.; Yoo, W.-H.; Lee, S.-H.; Lee, D.-S.; Kang, J.S.; Hwang, Y.-I.; Lee, W.J. Characterization of effector memory CD8+ T cells in the synovial fluid of rheumatoid arthritis. J. Clin. Immunol. 2012, 32, 709–720. [Google Scholar] [CrossRef]
- Cascão, R.; Moura, R.A.; Perpétuo, I.; Canhão, H.; Vieira-Sousa, E.; Mourão, A.F.; Rodrigues, A.M.; Polido-Pereira, J.; Queiroz, M.V.; Rosário, H.S. Identification of a cytokine network sustaining neutrophil and Th17 activation in untreated early rheumatoid arthritis. Arthritis Res. Ther. 2010, 12, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Kotake, S.; Udagawa, N.; Takahashi, N.; Matsuzaki, K.; Itoh, K.; Ishiyama, S.; Saito, S.; Inoue, K.; Kamatani, N.; Gillespie, M.T. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Investig. 1999, 103, 1345–1352. [Google Scholar] [CrossRef]
- Coutant, F.; Miossec, P. Altered dendritic cell functions in autoimmune diseases: Distinct and overlapping profiles. Nat. Rev. Rheumatol. 2016, 12, 703–715. [Google Scholar] [CrossRef]
- Arroyo-Villa, I.; Bautista-Caro, M.-B.; Balsa, A.; Aguado-Acin, P.; Nuno, L.; Bonilla-Hernan, M.-G.; Puig-Kröger, A.; Martin-Mola, E.; Miranda-Carus, M.-E. Frequency of Th17 CD4+ T cells in early rheumatoid arthritis: A marker of anti-CCP seropositivity. PLoS ONE 2012, 7, e42189. [Google Scholar] [CrossRef] [Green Version]
- Herrath, J.; Chemin, K.; Albrecht, I.; Catrina, A.I.; Malmström, V. Surface expression of CD39 identifies an enriched Treg-cell subset in the rheumatic joint, which does not suppress IL-17A secretion. Eur. J. Immunol. 2014, 44, 2979–2989. [Google Scholar] [CrossRef]
- Juno, J.A.; van Bockel, D.; Kent, S.J.; Kelleher, A.D.; Zaunders, J.J.; Munier, C. Cytotoxic CD4 T cells—Friend or foe during viral infection? Front. Immunol. 2017, 8, 19. [Google Scholar] [CrossRef]
- Pachnio, A.; Ciaurriz, M.; Begum, J.; Lal, N.; Zuo, J.; Beggs, A.; Moss, P. Cytomegalovirus infection leads to development of high frequencies of cytotoxic virus-specific CD4+ T cells targeted to vascular endothelium. PLoS Pathog. 2016, 12, e1005832. [Google Scholar] [CrossRef] [Green Version]
- Thome, J.J.; Yudanin, N.; Ohmura, Y.; Kubota, M.; Grinshpun, B.; Sathaliyawala, T.; Kato, T.; Lerner, H.; Shen, Y.; Farber, D.L. Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell 2014, 159, 814–828. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, D.; Goronzy, J.J.; Weyand, C.M. CD4+ CD7-CD28-T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. J. Clin. Investig. 1996, 97, 2027–2037. [Google Scholar] [CrossRef]
- Broadley, I.; Pera, A.; Morrow, G.; Davies, K.A.; Kern, F. Expansions of cytotoxic CD4+ CD28− T cells drive excess cardiovascular mortality in rheumatoid arthritis and other chronic inflammatory conditions and are triggered by CMV infection. Front. Immunol. 2017, 8, 195. [Google Scholar] [CrossRef] [Green Version]
- Kouskoff, V.; Korganow, A.-S.; Duchatelle, V.; Degott, C.; Benoist, C.; Mathis, D. Organ-specific disease provoked by systemic autoimmunity. Cell 1996, 87, 811–822. [Google Scholar] [CrossRef] [Green Version]
- Goldschmidt, T.J.; Holmdahl, R. Anti-T cell receptor antibody treatment of rats with established autologous collagen-induced arthritis: Suppression of arthritis without reduction of anti-type II collagen autoantibody levels. Eur. J. Immunol. 1991, 21, 1327–1330. [Google Scholar] [CrossRef]
- Rosenblum, M.D.; Remedios, K.A.; Abbas, A.K. Mechanisms of human autoimmunity. J. Clin. Investig. 2015, 125, 2228–2233. [Google Scholar] [CrossRef] [Green Version]
- Ashouri, J.F.; Hsu, L.-Y.; Yu, S.; Rychkov, D.; Chen, Y.; Cheng, D.A.; Sirota, M.; Hansen, E.; Lattanza, L.; Zikherman, J. Reporters of TCR signaling identify arthritogenic T cells in murine and human autoimmune arthritis. Proc. Natl. Acad. Sci. USA 2019, 116, 18517–18527. [Google Scholar] [CrossRef] [Green Version]
- Norin, U.; Rintisch, C.; Meng, L.; Forster, F.; Ekman, D.; Tuncel, J.; Klocke, K.; Bäcklund, J.; Yang, M.; Bonner, M.Y. Endophilin A2 deficiency protects rodents from autoimmune arthritis by modulating T cell activation. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A. Autophagy as a cell-repair mechanism: Activation of chaperone-mediated autophagy during oxidative stress. Mol. Asp. Med. 2006, 27, 444–454. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Goronzy, J.J.; Weyand, C.M. Autophagy in autoimmune disease. J. Mol. Med. 2015, 93, 707–717. [Google Scholar] [CrossRef] [Green Version]
- Vomero, M.; Barbati, C.; Colasanti, T.; Perricone, C.; Novelli, L.; Ceccarelli, F.; Spinelli, F.R.; Di Franco, M.; Conti, F.; Valesini, G. Autophagy and rheumatoid arthritis: Current knowledges and future perspectives. Front. Immunol. 2018, 9, 1577. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Wang, H.; Wu, Y.; He, Z.; Qin, Y.; Shen, Q. The Autophagy Level Is Increased in the Synovial Tissues of Patients with Active Rheumatoid Arthritis and Is Correlated with Disease Severity. Mediat. Inflamm. 2017, 2017, 7623145. [Google Scholar] [CrossRef]
- Malemud, C.J. Intracellular signaling pathways in rheumatoid arthritis. J. Clin. Cell. Immunol. 2013, 4, 160. [Google Scholar] [CrossRef]
- Bruyn, G.A.; Tate, G.; Caeiro, F.; Maldonado-Cocco, J.; Westhovens, R.; Tannenbaum, H.; Bell, M.; Forre, O.; Bjorneboe, O.; Tak, P.P. Everolimus in patients with rheumatoid arthritis receiving concomitant methotrexate: A 3-month, double-blind, randomised, placebo-controlled, parallel-group, proof-of-concept study. Ann. Rheum. Dis. 2008, 67, 1090–1095. [Google Scholar] [CrossRef]
- Nedjic, J.; Aichinger, M.; Emmerich, J.; Mizushima, N.; Klein, L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 2008, 455, 396–400. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Chen, G.; Zhang, W.; Xu, N.; Zhu, J.Y.; Jia, J.; Sun, Z.J.; Wang, Y.N.; Zhao, Y.F. Autophagy regulates hypoxia-induced osteoclastogenesis through the HIF-1α/BNIP3 signaling pathway. J. Cell. Physiol. 2012, 227, 639–648. [Google Scholar] [CrossRef]
- Li, R.-F.; Chen, G.; Ren, J.-G.; Zhang, W.; Wu, Z.-X.; Liu, B.; Zhao, Y.; Zhao, Y.-F. The adaptor protein p62 is involved in RANKL-induced autophagy and osteoclastogenesis. J. Histochem. Cytochem. 2014, 62, 879–888. [Google Scholar] [CrossRef] [Green Version]
- Montaseri, A.; Giampietri, C.; Rossi, M.; Riccioli, A.; Fattore, A.D.; Filippini, A. The Role of Autophagy in Osteoclast Differentiation and Bone Resorption Function. Biomolecules 2020, 10, 1398. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.-Y.; Beyer, C.; Gießl, A.; Kireva, T.; Scholtysek, C.; Uderhardt, S.; Munoz, L.E.; Dees, C.; Distler, A.; Wirtz, S. Autophagy regulates TNFα-mediated joint destruction in experimental arthritis. Ann. Rheum. Dis. 2013, 72, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Codogno, P. Signalling and autophagy regulation in health, aging and disease. Mol. Asp. Med. 2006, 27, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg-Lerner, A.; Bialik, S.; Simon, H.-U.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009, 16, 966–975. [Google Scholar] [CrossRef]
- Wirawan, E.; Walle, L.V.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef]
- Liu, H.; Pope, R.M. The role of apoptosis in rheumatoid arthritis. Curr. Opin. Pharmacol. 2003, 3, 317–322. [Google Scholar] [CrossRef]
- Korb, A.; Pavenstädt, H.; Pap, T. Cell death in rheumatoid arthritis. Apoptosis 2009, 14, 447–454. [Google Scholar] [CrossRef]
- Pap, T.; Müller-Ladner, U.; Gay, R.E.; Gay, S. Fibroblast biology: Role of synovial fibroblasts in the pathogenesis of rheumatoid arthritis. Arthritis Res. Ther. 2000, 2, 1–7. [Google Scholar]
- Firestein, G.S.; Yeo, M.; Zvaifler, N.J. Apoptosis in rheumatoid arthritis synovium. J. Clin. Investig. 1995, 96, 1631–1638. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.-J.; Han, S.-H.; Kim, D.-S.; Lee, G.-H.; Yoo, W.-H.; Kang, Y.-M.; Choi, J.-Y.; Lee, Y.C.; Park, S.J.; Jeong, S.-K. Autophagy induction and CHOP under-expression promotes survival of fibroblasts from rheumatoid arthritis patients under endoplasmic reticulum stress. Arthritis Res. Ther. 2010, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Xu, P.; Yao, J.-F.; Zhang, Y.-G.; Hou, W.-k.; Lu, S.-M. Reduced apoptosis correlates with enhanced autophagy in synovial tissues of rheumatoid arthritis. Inflamm. Res. 2013, 62, 229–237. [Google Scholar] [CrossRef]
- Kato, M.; Ospelt, C.; Gay, R.E.; Gay, S.; Klein, K. Dual role of autophagy in stress-induced cell death in rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol. 2014, 66, 40–48. [Google Scholar] [CrossRef]
- Hubbard, V.M.; Valdor, R.; Patel, B.; Singh, R.; Cuervo, A.M.; Macian, F. Macroautophagy regulates energy metabolism during effector T cell activation. J. Immunol. 2010, 185, 7349–7357. [Google Scholar] [CrossRef] [Green Version]
- Arsov, I.; Adebayo, A.; Kucerova-Levisohn, M.; Haye, J.; MacNeil, M.; Papavasiliou, F.N.; Yue, Z.; Ortiz, B.D. A role for autophagic protein beclin 1 early in lymphocyte development. J. Immunol. 2011, 186, 2201–2209. [Google Scholar] [CrossRef] [Green Version]
- Cenci, S. Autophagy, a new determinant of plasma cell differentiation and antibody responses. Mol. Immunol. 2014, 62, 289–295. [Google Scholar] [CrossRef]
- Conway, K.L.; Kuballa, P.; Khor, B.; Zhang, M.; Shi, H.N.; Virgin, H.W.; Xavier, R.J. ATG5 regulates plasma cell differentiation. Autophagy 2013, 9, 528–537. [Google Scholar] [CrossRef] [Green Version]
- van Loosdregt, J.; Rossetti, M.; Spreafico, R.; Moshref, M.; Olmer, M.; Williams, G.W.; Kumar, P.; Copeland, D.; Pischel, K.; Lotz, M. Increased autophagy in CD4+ T cells of rheumatoid arthritis patients results in T-cell hyperactivation and apoptosis resistance. Eur. J. Immunol. 2016, 46, 2862–2870. [Google Scholar] [CrossRef] [Green Version]
- Ireland, J.M.; Unanue, E.R. Autophagy in antigen-presenting cells results in presentation of citrullinated peptides to CD4 T cells. J. Exp. Med. 2011, 208, 2625–2632. [Google Scholar] [CrossRef]
- Sorice, M.; Iannuccelli, C.; Manganelli, V.; Capozzi, A.; Alessandri, C.; Lococo, E.; Garofalo, T.; Di Franco, M.; Bombardieri, M.; Nerviani, A.; et al. Autophagy generates citrullinated peptides in human synoviocytes: A possible trigger for anti-citrullinated peptide antibodies. Rheumatology 2016, 55, 1374–1385. [Google Scholar] [CrossRef] [Green Version]
- Ireland, J.M.; Unanue, E.R. Processing of proteins in autophagy vesicles of antigen-presenting cells generates citrullinated peptides recognized by the immune system. Autophagy 2012, 8, 429–430. [Google Scholar] [CrossRef] [Green Version]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Kundu, S.; Ghosh, P.; Datta, S.; Ghosh, A.; Chattopadhyay, S.; Chatterjee, M. Oxidative stress as a potential biomarker for determining disease activity in patients with rheumatoid arthritis. Free Radic. Res. 2012, 46, 1482–1489. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Holmdahl, R.; Sareila, O.; Olsson, L.M.; Bäckdahl, L.; Wing, K. Ncf1 polymorphism reveals oxidative regulation of autoimmune chronic inflammation. Immunol. Rev. 2016, 269, 228–247. [Google Scholar] [CrossRef]
- Olofsson, P.; Holmberg, J.; Tordsson, J.; Lu, S.; Åkerström, B.; Holmdahl, R. Positional identification of Ncf1 as a gene that regulates arthritis severity in rats. Nat. Genet. 2003, 33, 25–32. [Google Scholar] [CrossRef]
- Hultqvist, M.; Sareila, O.; Vilhardt, F.; Norin, U.; Olsson, L.M.; Olofsson, P.; Hellman, U.; Holmdahl, R. Positioning of a polymorphic quantitative trait nucleotide in the Ncf1 gene controlling oxidative burst response and arthritis severity in rats. Antioxid. Redox Signal. 2011, 14, 2373–2383. [Google Scholar] [CrossRef]
- Kelkka, T.; Kienhöfer, D.; Hoffmann, M.; Linja, M.; Wing, K.; Sareila, O.; Hultqvist, M.; Laajala, E.; Chen, Z.; Vasconcelos, J. Reactive oxygen species deficiency induces autoimmunity with type 1 interferon signature. Antioxid. Redox Signal. 2014, 21, 2231–2245. [Google Scholar] [CrossRef] [Green Version]
- Becanovic, K.; Jagodic, M.; Sheng, J.R.; Dahlman, I.; Aboul-Enein, F.; Wallstrom, E.; Olofsson, P.; Holmdahl, R.; Lassmann, H.; Olsson, T. Advanced intercross line mapping of Eae5 reveals Ncf-1 and CLDN4 as candidate genes for experimental autoimmune encephalomyelitis. J. Immunol. 2006, 176, 6055–6064. [Google Scholar] [CrossRef] [Green Version]
- Fraenkel, L.; Bathon, J.M.; England, B.R.; St Clair, E.W.; Arayssi, T.; Carandang, K.; Deane, K.D.; Genovese, M.; Huston, K.K.; Kerr, G.; et al. 2021 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Rheumatol. 2021, 73, 1108–1123. [Google Scholar] [CrossRef]
- Paglia, M.D.G.; Silva, M.T.; Lopes, L.C.; Barberato-Filho, S.; Mazzei, L.G.; Abe, F.C.; de Cássia Bergamaschi, C. Use of corticoids and non-steroidal anti-inflammatories in the treatment of rheumatoid arthritis: Systematic review and network meta-analysis. PLoS ONE 2021, 16, e0248866. [Google Scholar] [CrossRef]
- Xie, P.; Xue, W.; Qi, W.; Li, Y.; Yang, L.; Yang, Z.; Shi, A. Safety, Tolerability, and Pharmacokinetics of Ibuprofenamine Hydrochloride Spray (NSAIDs), a New Drug for Rheumatoid Arthritis and Osteoarthritis, in Healthy Chinese Subjects. Drug Des. Dev. Ther. 2021, 15, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Bullock, J.; Rizvi, S.A.A.; Saleh, A.M.; Ahmed, S.S.; Do, D.P.; Ansari, R.A.; Ahmed, J. Rheumatoid Arthritis: A Brief Overview of the Treatment. Med. Princ. Pract. Int. J. Kuwait Univ. Health Sci. Cent. 2018, 27, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.A.; Saag, K.G.; Bridges, S.L., Jr.; Akl, E.A.; Bannuru, R.R.; Sullivan, M.C.; Vaysbrot, E.; McNaughton, C.; Osani, M.; Shmerling, R.H.; et al. 2015 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Czock, D.; Keller, F.; Rasche, F.M.; Häussler, U. Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin. Pharmacokinet. 2005, 44, 61–98. [Google Scholar] [CrossRef] [PubMed]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandewalle, J.; Luypaert, A.; De Bosscher, K.; Libert, C. Therapeutic Mechanisms of Glucocorticoids. Trends Endocrinol. Metab. TEM 2018, 29, 42–54. [Google Scholar] [CrossRef]
- Stacy, J.M.; Greenmyer, J.R.; Beal, J.R.; Sahmoun, A.E.; Diri, E. The efficacy of low dose short-term prednisone therapy for remission induction in newly diagnosed rheumatoid arthritis patients. Adv. Rheumatol. 2021, 61, 50. [Google Scholar] [CrossRef]
- Kirwan, J.R.; Hickey, S.H.; Hällgren, R.; Mielants, H.; Björck, E.; Persson, T.; Wollheim, F.A. The effect of therapeutic glucocorticoids on the adrenal response in a randomized controlled trial in patients with rheumatoid arthritis. Arthritis Rheum. 2006, 54, 1415–1421. [Google Scholar] [CrossRef] [Green Version]
- Cronstein, B.N. Low-dose methotrexate: A mainstay in the treatment of rheumatoid arthritis. Pharmacol. Rev. 2005, 57, 163–172. [Google Scholar] [CrossRef] [Green Version]
- Mavrikakis, I.; Sfikakis, P.P.; Mavrikakis, E.; Rougas, K.; Nikolaou, A.; Kostopoulos, C.; Mavrikakis, M. The incidence of irreversible retinal toxicity in patients treated with hydroxychloroquine: A reappraisal. Ophthalmology 2003, 110, 1321–1326. [Google Scholar] [CrossRef]
- Villa-Hermosilla, M.C.; Fernández-Carballido, A.; Hurtado, C.; Barcia, E.; Montejo, C.; Alonso, M.; Negro, S. Sulfasalazine Microparticles Targeting Macrophages for the Treatment of Inflammatory Diseases Affecting the Synovial Cavity. Pharmaceutics 2021, 13, 951. [Google Scholar] [CrossRef]
- Weinblatt, M.E.; Kremer, J.M.; Coblyn, J.S.; Maier, A.L.; Helfgott, S.M.; Morrell, M.; Byrne, V.M.; Kaymakcian, M.V.; Strand, V. Pharmacokinetics, safety, and efficacy of combination treatment with methotrexate and leflunomide in patients with active rheumatoid arthritis. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 1999, 42, 1322–1328. [Google Scholar] [CrossRef]
- Mohammadi, O.; Kassim, T.A. Azathioprine. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2021. [Google Scholar]
- Catrina, A.I.; Trollmo, C.; Klint, E.; Engstrom, M.; Lampa, J.; Hermansson, Y.; Klareskog, L.; Ulfgren, A.K. Evidence that anti–tumor necrosis factor therapy with both etanercept and infliximab induces apoptosis in macrophages, but not lymphocytes, in rheumatoid arthritis joints. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2005, 52, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Keystone, E.C.; Schiff, M.H.; Kremer, J.M.; Kafka, S.; Lovy, M.; DeVries, T.; Burge, D.J. Once-weekly administration of 50 mg etanercept in patients with active rheumatoid arthritis: Results of a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2004, 50, 353–363. [Google Scholar] [CrossRef]
- Lipsky, P.E.; van der Heijde, D.M.; St Clair, E.W.; Furst, D.E.; Breedveld, F.C.; Kalden, J.R.; Smolen, J.S.; Weisman, M.; Emery, P.; Feldmann, M.; et al. Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N. Engl. J. Med. 2000, 343, 1594–1602. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, Y.; Nakano, K.; Nakayamada, S.; Kubo, S.; Iwata, S.; Hanami, K.; Fukuyo, S.; Miyagawa, I.; Yamaguchi, A.; Kawabe, A.; et al. Serum TNFα levels at 24 h after certolizumab pegol predict effectiveness at week 12 in patients with rheumatoid arthritis from TSUBAME study. Arthritis Res. 2021, 23, 154. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewé, R.; Bijlsma, J.; Burmester, G.; Chatzidionysiou, K.; Dougados, M.; Nam, J.; Ramiro, S.; Voshaar, M.; van Vollenhoven, R.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann. Rheum. Dis. 2017, 76, 960–977. [Google Scholar] [CrossRef]
- Subedi, S.; Gong, Y.; Chen, Y.; Shi, Y. Infliximab and biosimilar infliximab in psoriasis: Efficacy, loss of efficacy, and adverse events. Drug Des. Dev. Ther. 2019, 13, 2491–2502. [Google Scholar] [CrossRef] [Green Version]
- Voulgari, P.V.; Drosos, A.A. Adalimumab for rheumatoid arthritis. Expert Opin. Biol. Ther. 2006, 6, 1349–1360. [Google Scholar] [CrossRef]
- Mou, X.Y.; Jin, D.; Zhang, Q.; Guan, J.T.; Jin, Y. JKAP correlates with lower disease risk and inflammation, and its increment during etanercept treatment associates with commendable treatment efficiency in rheumatoid arthritis patients. Eur. Rev. Med Pharmacol. Sci. 2021, 25, 2654–2661. [Google Scholar] [CrossRef]
- Ngoufack, C.; Semerano, L.; Podglajen, I.; Bruneval, P.; Meune, C.; Valeyre, D.; Dhote, R.; Boissier, M.C.; Saidenberg-Kermanac’h, N. Mitral valve granulomatosis: A paradoxical reaction complicating etanercept treatment in rheumatoid arthritis. A case report. Jt. Bone Spine 2021, 88, 105183. [Google Scholar] [CrossRef]
- Roongta, R.; Mondal, S.; Ghosh, A. Etanercept or methotrexate withdrawal in rheumatoid arthritis patients receiving combination therapy: Comment on the article by Curtis et al. Arthritis Rheumatol. 2021, 73, 1771. [Google Scholar] [CrossRef]
- Strusberg, I.; Mysler, E.; Citera, G.; Siri, D.; de Los Ángeles Correa, M.; Lazaro, M.A.; Pardo Hidalgo, R.; Spindler, A.; Tate, P.; Venarotti, H.; et al. Efficacy, Safety, and Immunogenicity of Biosimilar Etanercept (Enerceptan) Versus Its Original Form in Combination with Methotrexate in Patients with Rheumatoid Arthritis: A Randomized, Multicenter, Evaluator-Blinded, Noninferiority Study. J. Clin. Rheumatol. Pract. Rep. Rheum. Musculoskelet. Dis. 2021, 27, S173–S179. [Google Scholar] [CrossRef]
- Aboobacker, S.; Kurn, H.; Al Aboud, A.M. Secukinumab. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2021. [Google Scholar]
- Cohen, S.; Hurd, E.; Cush, J.; Schiff, M.; Weinblatt, M.E.; Moreland, L.W.; Kremer, J.; Bear, M.B.; Rich, W.J.; McCabe, D. Treatment of rheumatoid arthritis with anakinra, a recombinant human interleukin-1 receptor antagonist, in combination with methotrexate: Results of a twenty-four–week, multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002, 46, 614–624. [Google Scholar] [CrossRef]
- Fleischmann, R.; Genovese, M.C.; Maslova, K.; Leher, H.; Praestgaard, A.; Burmester, G.R. Long-term safety and efficacy of sarilumab over 5 years in patients with rheumatoid arthritis refractory to TNF inhibitors. Rheumatology 2021, 2021, keab355. [Google Scholar] [CrossRef]
- Maini, R.N.; Taylor, P.C.; Szechinski, J.; Pavelka, K.; Bröll, J.; Balint, G.; Emery, P.; Raemen, F.; Petersen, J.; Smolen, J.; et al. Double-blind randomized controlled clinical trial of the interleukin-6 receptor antagonist, tocilizumab, in European patients with rheumatoid arthritis who had an incomplete response to methotrexate. Arthritis Rheum. 2006, 54, 2817–2829. [Google Scholar] [CrossRef]
- McGeachy, M.J.; Cua, D.J.; Gaffen, S.L. The IL-17 Family of Cytokines in Health and Disease. Immunity 2019, 50, 892–906. [Google Scholar] [CrossRef]
- Ramírez, J.; Cañete, J.D. Anakinra for the treatment of rheumatoid arthritis: A safety evaluation. Expert Opin. Drug Saf. 2018, 17, 727–732. [Google Scholar] [CrossRef]
- Ren, H.M.; Lukacher, A.E.; Rahman, Z.S.M.; Olsen, N.J. New developments implicating IL-21 in autoimmune disease. J. Autoimmun. 2021, 122, 102689. [Google Scholar] [CrossRef]
- Elemam, N.M.; Hannawi, S.; Maghazachi, A.A. Role of chemokines and chemokine receptors in rheumatoid arthritis. Immunotargets Ther. 2020, 9, 43. [Google Scholar] [CrossRef] [Green Version]
- Szekanecz, Z.; Koch, A.E. Successes and failures of chemokine-pathway targeting in rheumatoid arthritis. Nat. Rev. Rheumatol. 2016, 12, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, M.; Scheinecker, C. How does abatacept really work in rheumatoid arthritis? Curr. Opin. Rheumatol. 2018, 30, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.M.; Dougados, M.; Emery, P.; Durez, P.; Sibilia, J.; Shergy, W.; Steinfeld, S.; Tindall, E.; Becker, J.C.; Li, T. Treatment of rheumatoid arthritis with the selective costimulation modulator abatacept: Twelve-month results of a phase IIb, double-blind, randomized, placebo-controlled trial. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2005, 52, 2263–2271. [Google Scholar] [CrossRef] [PubMed]
- Rubbert-Roth, A.; Enejosa, J.; Pangan, A.L.; Haraoui, B.; Rischmueller, M.; Khan, N.; Zhang, Y.; Martin, N.; Xavier, R.M. Trial of Upadacitinib or Abatacept in Rheumatoid Arthritis. N. Engl. J. Med. 2020, 383, 1511–1521. [Google Scholar] [CrossRef]
- Schiff, M. Co-stimulation therapy in rheumatoid arthritis: Today and tomorrow. Curr. Treat. Options Rheumatol. 2015, 1, 334–349. [Google Scholar] [CrossRef] [Green Version]
- Clark, E.A.; Ledbetter, J.A. How does B cell depletion therapy work, and how can it be improved? Ann. Rheum. Dis. 2005, 64 (Suppl. 4), 77–80. [Google Scholar] [CrossRef]
- Emery, P.; Rigby, W.; Tak, P.P.; Dörner, T.; Olech, E.; Martin, C.; Millar, L.; Travers, H.; Fisheleva, E. Safety with ocrelizumab in rheumatoid arthritis: Results from the ocrelizumab phase III program. PLoS ONE 2014, 9, e87379. [Google Scholar] [CrossRef]
- Geh, D.; Gordon, C. Epratuzumab for the treatment of systemic lupus erythematosus. Expert Rev. Clin. Immunol. 2018, 14, 245–258. [Google Scholar] [CrossRef]
- Giltiay, N.V.; Shu, G.L.; Shock, A.; Clark, E.A. Targeting CD22 with the monoclonal antibody epratuzumab modulates human B-cell maturation and cytokine production in response to Toll-like receptor 7 (TLR7) and B-cell receptor (BCR) signaling. Arthritis Res. Ther. 2017, 19, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Milani, C.; Castillo, J. Veltuzumab, an anti-CD20 mAb for the treatment of non-Hodgkin’s lymphoma, chronic lymphocytic leukemia and immune thrombocytopenic purpura. Curr. Opin. Mol. 2009, 11, 200–207. [Google Scholar]
- Payandeh, Z.; Rajabibazl, M.; Mortazavi, Y.; Rahimpour, A.; Taromchi, A.H.; Dastmalchi, S. Affinity maturation and characterization of the ofatumumab monoclonal antibody. J. Cell. Biochem. 2019, 120, 940–950. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Wang, T.; Bird, S.; Zou, J.; Dooley, H.; Secombes, C.J. B cell receptor accessory molecule CD79α: Characterisation and expression analysis in a cartilaginous fish, the spiny dogfish (Squalus acanthias). Fish Shellfish Immunol. 2013, 34, 1404–1415. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.-H.; Luo, S.-F.; Ho, L.-J. Targeting the CD40-CD154 signaling pathway for treatment of autoimmune arthritis. Cells 2019, 8, 927. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.-Q.; Pope, R.M. The role of toll-like receptors in rheumatoid arthritis. Curr. Rheumatol. Rep. 2009, 11, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Blair, H.A.; Duggan, S.T. Belimumab: A Review in Systemic Lupus Erythematosus. Drugs 2018, 78, 355–366. [Google Scholar] [CrossRef]
- Liu, Z.; Davidson, A. BAFF inhibition: A new class of drugs for the treatment of autoimmunity. Exp. Cell Res. 2011, 317, 1270–1277. [Google Scholar] [CrossRef] [Green Version]
- Navarra, S.V.; Guzmán, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.; Thomas, M.; Kim, H.-Y.; León, M.G. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: A randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef]
- Kaegi, C.; Steiner, U.C.; Wuest, B.; Crowley, C.; Boyman, O. Systematic review of safety and efficacy of atacicept in treating immune-mediated disorders. Front. Immunol. 2020, 11, 433. [Google Scholar] [CrossRef]
- Xu, S.; Lam, K.-P. Transmembrane Activator and CAML Interactor (TACI): Another Potential Target for Immunotherapy of Multiple Myeloma? Cancers 2020, 12, 1045. [Google Scholar] [CrossRef]
- Kang, Y.; Jiang, X.; Qin, D.; Wang, L.; Yang, J.; Wu, A.; Huang, F.; Ye, Y.; Wu, J. Efficacy and safety of multiple dosages of fostamatinib in adult patients with rheumatoid arthritis: A systematic review and meta-analysis. Front. Pharmacol. 2019, 10, 897. [Google Scholar] [CrossRef] [Green Version]
- Hodge, J.A.; Kawabata, T.T.; Krishnaswami, S.; Clark, J.D.; Telliez, J.B.; Dowty, M.E.; Menon, S.; Lamba, M.; Zwillich, S. The mechanism of action of tofacitinib—An oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin. Exp. Rheumatol. 2016, 34, 318–328. [Google Scholar]
- Zhang, X.; Chua, L.; Ernest, C., 2nd; Macias, W.; Rooney, T.; Tham, L.S. Dose/Exposure-Response Modeling to Support Dosing Recommendation for Phase III Development of Baricitinib in Patients with Rheumatoid Arthritis. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 804–813. [Google Scholar] [CrossRef]
- Pavelka, K. Targeted and biological drugs in the treatment of inflammatory rheumatic diseases. Vnitr. Lek. 2021, 67, 195–200. [Google Scholar] [CrossRef]
- Kim, E.S.; Keam, S.J. Filgotinib in Rheumatoid Arthritis: A Profile of Its Use. Clin. Drug Investig. 2021, 41, 741–749. [Google Scholar] [CrossRef]
- Iwamoto, N.; Sato, S.; Kurushima, S.; Michitsuji, T.; Nishihata, S.; Okamoto, M.; Tsuji, Y.; Endo, Y.; Shimizu, T.; Sumiyoshi, R.; et al. Real-world comparative effectiveness and safety of tofacitinib and baricitinib in patients with rheumatoid arthritis. Arthritis Res. 2021, 23, 197. [Google Scholar] [CrossRef]
- Mohty, M. Mechanisms of action of antithymocyte globulin: T-cell depletion and beyond. Leukemia 2007, 21, 1387–1394. [Google Scholar] [CrossRef] [Green Version]
- Lassoued, S.; Moyano, C.; Beldjerd, M.; Pauly, P.; Lassoued, D.; Billey, T. Bortezomib improved the joint manifestations of rheumatoid arthritis in three patients. Jt. Bone Spine 2019, 86, 381–382. [Google Scholar] [CrossRef]
- Lee, S.W.; Kim, J.H.; Park, Y.B.; Lee, S.K. Bortezomib attenuates murine collagen-induced arthritis. Ann. Rheum. Dis. 2009, 68, 1761–1767. [Google Scholar] [CrossRef] [Green Version]
- Durkin, A.; Vu, H.Y.; Lee, H. The VR23 Antitumor Compound Also Shows Strong Anti-Inflammatory Effects in a Human Rheumatoid Arthritis Cell Model and Acute Lung Inflammation in Mice. J. Immunol. 2020, 204, 788–795. [Google Scholar] [CrossRef]
- Mahévas, T.; Arnulf, B.; Bouaziz, J.-D.; Livideanu, C.B.; Osio, A.; Servy, A.; Cribier, B.; Sassolas, B.; Jachiet, M.; Michel, L. Plasma cell–directed therapies in monoclonal gammopathy–associated scleromyxedema. Blood J. Am. Soc. Hematol. 2020, 135, 1101–1110. [Google Scholar] [CrossRef]
- Woodle, E.S.; Tremblay, S.; Driscoll, J. Targeting Plasma Cells with Proteasome Inhibitors: Principles from Primates. J. Am. Soc. Nephrol. 2017, 28, 1951–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghannam, S.; Pène, J.; Moquet-Torcy, G.; Jorgensen, C.; Yssel, H. Mesenchymal stem cells inhibit human Th17 cell differentiation and function and induce a T regulatory cell phenotype. J. Immunol. 2010, 185, 302–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, A.R.R.; Dahlke, M.H. Immunomodulation by mesenchymal stem cells (MSCs): Mechanisms of action of living, apoptotic, and dead MSCs. Front. Immunol. 2019, 10, 1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.J.; Rim, Y.A.; Nam, Y.; Ju, J.H. Recent Developments in Clinical Applications of Mesenchymal Stem Cells in the Treatment of Rheumatoid Arthritis and Osteoarthritis. Front. Immunol. 2021, 12, 631291. [Google Scholar] [CrossRef]
- Gabay, C.; Riek, M.; Scherer, A.; Finckh, A. Effectiveness of biologic DMARDs in monotherapy versus in combination with synthetic DMARDs in rheumatoid arthritis: Data from the Swiss Clinical Quality Management Registry. Rheumatology 2015, 54, 1664–1672. [Google Scholar] [CrossRef] [Green Version]
- Teitsma, X.M.; Marijnissen, A.K.; Bijlsma, J.W.; Lafeber, F.P.; Jacobs, J.W. Tocilizumab as monotherapy or combination therapy for treating active rheumatoid arthritis: A meta-analysis of efficacy and safety reported in randomized controlled trials. Arthritis Res. 2016, 18, 211. [Google Scholar] [CrossRef] [Green Version]
- Westhovens, R.; Rigby, W.F.C.; van der Heijde, D.; Ching, D.W.T.; Stohl, W.; Kay, J.; Chopra, A.; Bartok, B.; Matzkies, F.; Yin, Z.; et al. Filgotinib in combination with methotrexate or as monotherapy versus methotrexate monotherapy in patients with active rheumatoid arthritis and limited or no prior exposure to methotrexate: The phase 3, randomised controlled FINCH 3 trial. Ann. Rheum. Dis. 2021, 80, 727–738. [Google Scholar] [CrossRef]
- Hammaker, D.; Firestein, G.S. Epigenetics of inflammatory arthritis. Curr. Opin. Rheumatol. 2018, 30, 188–196. [Google Scholar] [CrossRef]
- Khan, N.M.; Haqqi, T.M. Epigenetics in osteoarthritis: Potential of HDAC inhibitors as therapeutics. Pharmacol. Res. 2018, 128, 73–79. [Google Scholar] [CrossRef]
- Lohman, R.J.; Iyer, A.; Fairlie, T.J.; Cotterell, A.; Gupta, P.; Reid, R.C.; Vesey, D.A.; Sweet, M.J.; Fairlie, D.P. Differential Anti-inflammatory Activity of HDAC Inhibitors in Human Macrophages and Rat Arthritis. J. Pharmacol. Exp. Ther. 2016, 356, 387–396. [Google Scholar] [CrossRef]
- Alghadir, A.; Miraj, M.; Ali, S. Efficacy of Curcumin with Iontophoretic Application on Paw Edema and Hematological Responses in Collagen-Induced Arthritis Rat Models. Evid.-Based Complementary Altern. Med. 2020, 2020, 4606520. [Google Scholar] [CrossRef]
- Wang, Q.; Ye, C.; Sun, S.; Li, R.; Shi, X.; Wang, S.; Zeng, X.; Kuang, N.; Liu, Y.; Shi, Q.; et al. Curcumin attenuates collagen-induced rat arthritis via anti-inflammatory and apoptotic effects. Int. Immunopharmacol. 2019, 72, 292–300. [Google Scholar] [CrossRef]
- Yang, M.; Akbar, U.; Mohan, C. Curcumin in Autoimmune and Rheumatic Diseases. Nutrients 2019, 11, 1004. [Google Scholar] [CrossRef] [Green Version]
- Escobedo-Martínez, C.; Guzmán-Gutiérrez, S.L.; Carrillo-López, M.I.; Deveze-Álvarez, M.A.; Trujillo-Valdivia, A.; Meza-Morales, W.; Enríquez, R.G. Diacetylcurcumin: Its Potential Antiarthritic Effect on a Freund’s Complete Adjuvant-Induced Murine Model. Molecules 2019, 24, 2643. [Google Scholar] [CrossRef] [Green Version]
- Sana, E.; Zeeshan, M.; Ain, Q.U.; Khan, A.U.; Hussain, I.; Khan, S.; Lepeltier, E.; Ali, H. Topical delivery of curcumin-loaded transfersomes gel ameliorated rheumatoid arthritis by inhibiting NF-κβ pathway. Nanomedicine 2021, 16, 819–837. [Google Scholar] [CrossRef]
- Javadi, M.; Khadem Haghighian, H.; Goodarzy, S.; Abbasi, M.; Nassiri-Asl, M. Effect of curcumin nanomicelle on the clinical symptoms of patients with rheumatoid arthritis: A randomized, double-blind, controlled trial. Int. J. Rheum. Dis. 2019, 22, 1857–1862. [Google Scholar] [CrossRef]
- Banji, D.; Pinnapureddy, J.; Banji, O.J.; Saidulu, A.; Hayath, M.S. Synergistic activity of curcumin with methotrexate in ameliorating Freund’s Complete Adjuvant induced arthritis with reduced hepatotoxicity in experimental animals. Eur. J. Pharmacol. 2011, 668, 293–298. [Google Scholar] [CrossRef]
- Chandran, B.; Goel, A. A randomized, pilot study to assess the efficacy and safety of curcumin in patients with active rheumatoid arthritis. Phytother. Res. 2012, 26, 1719–1725. [Google Scholar] [CrossRef]
- Hemshekhar, M.; Anaparti, V.; El-Gabalawy, H.; Mookherjee, N. A bioavailable form of curcumin, in combination with vitamin-D- and omega-3-enriched diet, modifies disease onset and outcomes in a murine model of collagen-induced arthritis. Arthritis Res. 2021, 23, 39. [Google Scholar] [CrossRef]
- Poonia, N.; Lather, V.; Kaur, B.; Kirthanashri, S.V.; Pandita, D. Optimization and Development of Methotrexate- and Resveratrol-Loaded Nanoemulsion Formulation Using Box-Behnken Design for Rheumatoid Arthritis. Assay Drug Dev. Technol. 2020, 18, 356–368. [Google Scholar] [CrossRef]
- Sankrityayan, H.; Majumdar, A.S. Curcumin and folic acid abrogated methotrexate induced vascular endothelial dysfunction. Can. J. Physiol. Pharmacol. 2016, 94, 89–96. [Google Scholar] [CrossRef]
- Singh, P.; Dabre, S. Controlled Release Gel Encompassing Curcumin Microspheres and Diclofenac Diethylamine for Feat Against Arthritis Inflammation. Curr. Rheumatol. Rev. 2020, 16, 110–119. [Google Scholar] [CrossRef]
- Yan, F.; Li, H.; Zhong, Z.; Zhou, M.; Lin, Y.; Tang, C.; Li, C. Co-Delivery of Prednisolone and Curcumin in Human Serum Albumin Nanoparticles for Effective Treatment of Rheumatoid Arthritis. Int. J. Nanomed. 2019, 14, 9113–9125. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Wang, T.; Jiao, Y.; Zhang, X. Immunometabolic Pathways and Its Therapeutic Implication in Autoimmune Diseases. Clin. Rev. Allergy Immunol. 2021, 60, 55–67. [Google Scholar] [CrossRef]
- Xiu, Y.; Xu, H.; Zhao, C.; Li, J.; Morita, Y.; Yao, Z.; Xing, L.; Boyce, B.F. Chloroquine reduces osteoclastogenesis in murine osteoporosis by preventing TRAF3 degradation. J. Clin. Investig. 2014, 124, 297–310. [Google Scholar] [CrossRef]
- Goel, P.; Gerriets, V. Chloroquine. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2021. [Google Scholar]
- Dai, S.; Wang, B.; Li, W.; Wang, L.; Song, X.; Guo, C.; Li, Y.; Liu, F.; Zhu, F.; Wang, Q. Systemic application of 3-methyladenine markedly inhibited atherosclerotic lesion in ApoE−/− mice by modulating autophagy, foam cell formation and immune-negative molecules. Cell Death Dis. 2016, 7, e2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laha, D.; Deb, M.; Das, H. KLF2 (kruppel-like factor 2 [lung]) regulates osteoclastogenesis by modulating autophagy. Autophagy 2019, 15, 2063–2075. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Alasti, F.; Smolen, J.S. Optimisation of a treat-to-target approach in rheumatoid arthritis: Strategies for the 3-month time point. Ann. Rheum. Dis. 2016, 75, 1479–1485. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.H.; Losina, E.; Lu, B.; Zak, A.; Corrigan, C.; Lee, S.B.; Agosti, J.; Bitton, A.; Harrold, L.R.; Pincus, T. Implementation of treat-to-target in rheumatoid arthritis through a learning collaborative: Results of a randomized controlled trial. Arthritis Rheumatol. 2017, 69, 1374–1380. [Google Scholar] [CrossRef] [PubMed]
- Di Giovine, F.S.; Nuki, G.; Duff, G.W. Tumour necrosis factor in synovial exudates. Ann. Rheum. Dis. 1988, 47, 768–772. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.C.; Lee, Y.H. Comparative efficacy and safety of biosimilar-infliximab and originator-infliximab in combination with methotrexate in patients with active rheumatoid arthritis: A meta-analysis of randomized controlled trials. Int. J. Rheum. Dis. 2018, 21, 922–929. [Google Scholar] [CrossRef]
- Radner, H.; Aletaha, D. Anti-TNF in rheumatoid arthritis: An overview. Wien. Med. Wochenschr. 2015, 165, 3–9. [Google Scholar] [CrossRef]
- Atiqi, S.; Hooijberg, F.; Loeff, F.C.; Rispens, T.; Wolbink, G.J. Immunogenicity of TNF-Inhibitors. Front. Immunol. 2020, 11, 312. [Google Scholar] [CrossRef]
- Mitoma, H.; Horiuchi, T.; Tsukamoto, H.; Ueda, N. Molecular mechanisms of action of anti-TNF-α agents—Comparison among therapeutic TNF-α antagonists. Cytokine 2018, 101, 56–63. [Google Scholar] [CrossRef]
- Moots, R.J.; Xavier, R.M.; Mok, C.C.; Rahman, M.U.; Tsai, W.C.; Al-Maini, M.H.; Pavelka, K.; Mahgoub, E.; Kotak, S.; Korth-Bradley, J.; et al. The impact of anti-drug antibodies on drug concentrations and clinical outcomes in rheumatoid arthritis patients treated with adalimumab, etanercept, or infliximab: Results from a multinational, real-world clinical practice, non-interventional study. PLoS ONE 2017, 12, e0175207. [Google Scholar] [CrossRef] [Green Version]
- Schett, G.; Tohidast-Akrad, M.; Smolen, J.S.; Schmid, B.J.; Steiner, C.W.; Bitzan, P.; Zenz, P.; Redlich, K.; Xu, Q.; Steiner, G. Activation, differential localization, and regulation of the stress-activated protein kinases, extracellular signal–regulated kinase, c-Jun N-terminal kinase, and p38 mitogen-activated protein kinase, in synovial tissue and cells in rheumatoid arthritis. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2000, 43, 2501–2512. [Google Scholar] [CrossRef]
- Abramson, S.B.; Amin, A. Blocking the effects of IL-1 in rheumatoid arthritis protects bone and cartilage. Rheumatology 2002, 41, 972–980. [Google Scholar] [CrossRef] [Green Version]
- Mejbri, M.; Theodoropoulou, K.; Hofer, M.; Cimaz, R. Interleukin-1 Blockade in Systemic Juvenile Idiopathic Arthritis. Paediatr. Drugs 2020, 22, 251–262. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.R.; Singh, J.A. Use of biologics in rheumatoid arthritis: Current and emerging paradigms of care. Clin. Ther. 2011, 33, 679–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimondo, M.G.; Biggioggero, M.; Crotti, C.; Becciolini, A.; Favalli, E.G. Profile of sarilumab and its potential in the treatment of rheumatoid arthritis. Drug Des. Dev. Ther. 2017, 11, 1593–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheppard, M.; Laskou, F.; Stapleton, P.P.; Hadavi, S.; Dasgupta, B. Tocilizumab (Actemra). Hum. Vaccines Immunother. 2017, 13, 1972–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackie, S.L.; Vital, E.M.; Ponchel, F.; Emery, P. Co-stimulatory blockade as therapy for rheumatoid arthritis. Curr. Rheumatol. Rep. 2005, 7, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A.; Deeks, E.D. Abatacept: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1221–1233. [Google Scholar] [CrossRef] [PubMed]
- Pombo-Suarez, M.; Gomez-Reino, J.J. Abatacept for the treatment of rheumatoid arthritis. Expert Rev. Clin. Immunol. 2019, 15, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef] [PubMed]
- Marston, B.; Palanichamy, A.; Anolik, J.H. B cells in the pathogenesis and treatment of rheumatoid arthritis. Curr. Opin. Rheumatol. 2010, 22, 307–315. [Google Scholar] [CrossRef]
- Payandeh, Z.; Bahrami, A.A.; Hoseinpoor, R.; Mortazavi, Y.; Rajabibazl, M.; Rahimpour, A.; Taromchi, A.H.; Khalil, S. The applications of anti-CD20 antibodies to treat various B cells disorders. Biomed. Pharmacother. 2019, 109, 2415–2426. [Google Scholar] [CrossRef]
- Polson, A.G.; Zheng, B.; Elkins, K.; Chang, W.; Du, C.; Dowd, P.; Yen, L.; Tan, C.; Hongo, J.A.; Koeppen, H.; et al. Expression pattern of the human FcRH/IRTA receptors in normal tissue and in B-chronic lymphocytic leukemia. Int. Immunol. 2006, 18, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Vincent, F.B.; Saulep-Easton, D.; Figgett, W.A.; Fairfax, K.A.; Mackay, F. The BAFF/APRIL system: Emerging functions beyond B cell biology and autoimmunity. Cytokine Growth Factor Rev. 2013, 24, 203–215. [Google Scholar] [CrossRef]
- Davidson, A. The rationale for BAFF inhibition in systemic lupus erythematosus. Curr. Rheumatol. Rep. 2012, 14, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Pine, P.R.; Chang, B.; Schoettler, N.; Banquerigo, M.L.; Wang, S.; Lau, A.; Zhao, F.; Grossbard, E.B.; Payan, D.G.; Brahn, E. Inflammation and bone erosion are suppressed in models of rheumatoid arthritis following treatment with a novel Syk inhibitor. Clin. Immunol. 2007, 124, 244–257. [Google Scholar] [CrossRef]
- Barnas, J.L.; Looney, R.J.; Anolik, J.H. B cell targeted therapies in autoimmune disease. Curr. Opin. Immunol. 2019, 61, 92–99. [Google Scholar] [CrossRef]
- Fragoulis, G.E.; McInnes, I.B.; Siebert, S. JAK-inhibitors. New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology 2019, 58, i43–i54. [Google Scholar] [CrossRef] [Green Version]
- Morinobu, A. JAK inhibitors for the treatment of rheumatoid arthritis. Immunol. Med. 2020, 43, 148–155. [Google Scholar] [CrossRef]
- Malemud, C.J. The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 2018, 10, 117–127. [Google Scholar] [CrossRef]
- Lopez-Santalla, M.; Fernandez-Perez, R.; Garin, M.I. Mesenchymal Stem/Stromal Cells for Rheumatoid Arthritis Treatment: An Update on Clinical Applications. Cells 2020, 9, 1852. [Google Scholar] [CrossRef]
- Luque-Campos, N.; Contreras-López, R.A.; Jose Paredes-Martínez, M.; Torres, M.J.; Bahraoui, S.; Wei, M.; Espinoza, F.; Djouad, F.; Elizondo-Vega, R.J.; Luz-Crawford, P. Mesenchymal Stem Cells Improve Rheumatoid Arthritis Progression by Controlling Memory T Cell Response. Front. Immunol. 2019, 10, 798. [Google Scholar] [CrossRef]
- Liu, H.; Li, R.; Liu, T.; Yang, L.; Yin, G.; Xie, Q. Immunomodulatory Effects of Mesenchymal Stem Cells and Mesenchymal Stem Cell-Derived Extracellular Vesicles in Rheumatoid Arthritis. Front. Immunol. 2020, 11, 1912. [Google Scholar] [CrossRef]
- Gowhari Shabgah, A.; Shariati-Sarabi, Z.; Tavakkol-Afshari, J.; Ghasemi, A.; Ghoryani, M.; Mohammadi, M. A significant decrease of BAFF, APRIL, and BAFF receptors following mesenchymal stem cell transplantation in patients with refractory rheumatoid arthritis. Gene 2020, 732, 144336. [Google Scholar] [CrossRef]
- Hofmann, K.; Clauder, A.-K.; Manz, R.A. Targeting B cells and plasma cells in autoimmune diseases. Front. Immunol. 2018, 9, 835. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Deng, Y.; Feng, Y.; Long, D.; Ma, K.; Wang, X.; Zhao, M.; Lu, L.; Lu, Q. Epigenetic regulation in B-cell maturation and its dysregulation in autoimmunity. Cell. Mol. Immunol. 2018, 15, 676–684. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewé, R.B.M.; Bijlsma, J.W.J.; Burmester, G.R.; Dougados, M.; Kerschbaumer, A.; McInnes, I.B.; Sepriano, A.; van Vollenhoven, R.F.; de Wit, M.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann. Rheum. Dis. 2020, 79, 685–699. [Google Scholar] [CrossRef] [Green Version]
- Hiepe, F.; Alexander, T.; Voll, R.E. [Plasma cells]. Z. Rheumatol. 2015, 74, 20–25. [Google Scholar] [CrossRef]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Cao, X. Epigenetic regulation of the innate immune response to infection. Nat. Rev. Immunol. 2019, 19, 417–432. [Google Scholar] [CrossRef]
- Araki, Y.; Wang, Z.; Zang, C.; Wood, W.H., 3rd; Schones, D.; Cui, K.; Roh, T.Y.; Lhotsky, B.; Wersto, R.P.; Peng, W.; et al. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity 2009, 30, 912–925. [Google Scholar] [CrossRef] [Green Version]
- de la Calle-Fabregat, C.; Niemantsverdriet, E.; Cañete, J.D.; Li, T.; van der Helm-van Mil, A.H.M.; Rodríguez-Ubreva, J.; Ballestar, E. The DNA methylation Profile of Undifferentiated Arthritis Patients Anticipates their Subsequent Differentiation to Rheumatoid Arthritis. Arthritis Rheumatol. 2021. [Google Scholar] [CrossRef]
- Nair, N.; Barton, A.; Wilson, A.G. Cell-specific epigenetic drivers of pathogenesis in rheumatoid arthritis. Epigenomics 2021, 13, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.Y.; Hsieh, S.C.; Liu, C.W.; Lu, C.H.; Liao, H.T.; Chen, M.H.; Li, K.J.; Wu, C.H.; Shen, C.Y.; Kuo, Y.M.; et al. The Expression of Non-Coding RNAs and Their Target Molecules in Rheumatoid Arthritis: A Molecular Basis for Rheumatoid Pathogenesis and Its Potential Clinical Applications. Int. J. Mol. Sci. 2021, 22, 5689. [Google Scholar] [CrossRef]
- Zhang, L.L.; Wu, X.X.; Wang, X.F.; Di, D.S.; Huang, Q.; Liu, R.S.; Shuai, Z.W.; Ye, D.Q.; Leng, R.X. Genetic variant in microRNA-146a gene is associated with risk of rheumatoid arthritis. Ann. Med. 2021, 53, 824–829. [Google Scholar] [CrossRef]
- Ai, R.; Laragione, T.; Hammaker, D.; Boyle, D.L.; Wildberg, A.; Maeshima, K.; Palescandolo, E.; Krishna, V.; Pocalyko, D.; Whitaker, J.W. Comprehensive epigenetic landscape of rheumatoid arthritis fibroblast-like synoviocytes. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Brondello, J.-M.; Djouad, F.; Jorgensen, C. Where to Stand with Stromal Cells and Chronic Synovitis in Rheumatoid Arthritis? Cells 2019, 8, 1257. [Google Scholar] [CrossRef] [Green Version]
- Buhrmann, C.; Mobasheri, A.; Busch, F.; Aldinger, C.; Stahlmann, R.; Montaseri, A.; Shakibaei, M. Curcumin modulates nuclear factor kappaB (NF-kappaB)-mediated inflammation in human tenocytes in vitro: Role of the phosphatidylinositol 3-kinase/Akt pathway. J. Biol. Chem. 2011, 286, 28556–28566. [Google Scholar] [CrossRef] [Green Version]
- Buhrmann, C.; Mobasheri, A.; Matis, U.; Shakibaei, M. Curcumin mediated suppression of nuclear factor-κB promotes chondrogenic differentiation of mesenchymal stem cells in a high-density co-culture microenvironment. Arthritis Res. 2010, 12, R127. [Google Scholar] [CrossRef] [Green Version]
- Buhrmann, C.; Shayan, P.; Aggarwal, B.B.; Shakibaei, M. Evidence that TNF-β (lymphotoxin α) can activate the inflammatory environment in human chondrocytes. Arthritis Res. 2013, 15, R202. [Google Scholar] [CrossRef] [Green Version]
- Csaki, C.; Mobasheri, A.; Shakibaei, M. Synergistic chondroprotective effects of curcumin and resveratrol in human articular chondrocytes: Inhibition of IL-1beta-induced NF-kappaB-mediated inflammation and apoptosis. Arthritis Res. 2009, 11, R165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, C.; Park, S.-h.; Lee, A.; Yuan, R.; Ivashkiv, L.B.; Kalliolias, G.D. TNF-induced inflammatory genes escape repression in fibroblast-like synoviocytes: Transcriptomic and epigenomic analysis. Ann. Rheum. Dis. 2019, 78, 1205–1214. [Google Scholar] [CrossRef]
- Mobasheri, A.; Henrotin, Y.; Biesalski, H.K.; Shakibaei, M. Scientific evidence and rationale for the development of curcumin and resveratrol as nutraceutricals for joint health. Int. J. Mol. Sci. 2012, 13, 4202–4232. [Google Scholar] [CrossRef]
- Shakibaei, M.; Mobasheri, A.; Lueders, C.; Busch, F.; Shayan, P.; Goel, A. Curcumin enhances the effect of chemotherapy against colorectal cancer cells by inhibition of NF-κB and Src protein kinase signaling pathways. PLoS ONE 2013, 8, e57218. [Google Scholar] [CrossRef] [Green Version]
- Diomede, F.; Fonticoli, L.; Guarnieri, S.; Della Rocca, Y.; Rajan, T.S.; Fontana, A.; Trubiani, O.; Marconi, G.D.; Pizzicannella, J. The Effect of Liposomal Curcumin as an Anti-Inflammatory Strategy on Lipopolysaccharide e from Porphyromonas gingivalis Treated Endothelial Committed Neural Crest Derived Stem Cells: Morphological and Molecular Mechanisms. Int. J. Mol. Sci. 2021, 22, 7534. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.; Pradhan, B.; Nayak, R.; Behera, C.; Das, S.; Patra, S.K.; Efferth, T.; Jena, M.; Bhutia, S.K. Dietary polyphenols in chemoprevention and synergistic effect in cancer: Clinical evidences and molecular mechanisms of action. Phytomed. Int. J. Phytother. Phytopharm. 2021, 90, 153554. [Google Scholar] [CrossRef]
- Zhang, S.; Li, L.; Chen, W.; Xu, S.; Feng, X.; Zhang, L. Natural products: The role and mechanism in low-density lipoprotein oxidation and atherosclerosis. Phytother. Res. 2021, 35, 2945–2967. [Google Scholar] [CrossRef]
- Kunnumakkara, A.B.; Bordoloi, D.; Padmavathi, G.; Monisha, J.; Roy, N.K.; Prasad, S.; Aggarwal, B.B. Curcumin, the golden nutraceutical: Multitargeting for multiple chronic diseases. Br. J. Pharmacol. 2017, 174, 1325–1348. [Google Scholar] [CrossRef] [Green Version]
- Buhrmann, C.; Brockmueller, A.; Harsha, C.; Kunnumakkara, A.B.; Kubatka, P.; Aggarwal, B.B.; Shakibaei, M. Evidence That Tumor Microenvironment Initiates Epithelial-To-Mesenchymal Transition and Calebin A can Suppress it in Colorectal Cancer Cells. Front. Pharm. 2021, 12, 699842. [Google Scholar] [CrossRef]
- Buhrmann, C.; Kraehe, P.; Lueders, C.; Shayan, P.; Goel, A.; Shakibaei, M. Curcumin suppresses crosstalk between colon cancer stem cells and stromal fibroblasts in the tumor microenvironment: Potential role of EMT. PLoS ONE 2014, 9, e107514. [Google Scholar] [CrossRef] [Green Version]
- Buhrmann, C.; Shayan, P.; Banik, K.; Kunnumakkara, A.B.; Kubatka, P.; Koklesova, L.; Shakibaei, M. Targeting NF-κB Signaling by Calebin A, a Compound of Turmeric, in Multicellular Tumor Microenvironment: Potential Role of Apoptosis Induction in CRC Cells. Biomedicines 2020, 8, 236. [Google Scholar] [CrossRef]
- Satoskar, R.; Shah, S.; Shenoy, S. Evaluation of anti-inflammatory property of curcumin (diferuloyl methane) in patients with postoperative inflammation. Int. J. Clin. Pharmacol. Ther. Toxicol. 1986, 24, 651–654. [Google Scholar]
- Shakibaei, M.; Buhrmann, C.; Kraehe, P.; Shayan, P.; Lueders, C.; Goel, A. Curcumin chemosensitizes 5-fluorouracil resistant MMR-deficient human colon cancer cells in high density cultures. PLoS ONE 2014, 9, e85397. [Google Scholar] [CrossRef] [Green Version]
- Toden, S.; Okugawa, Y.; Buhrmann, C.; Nattamai, D.; Anguiano, E.; Baldwin, N.; Shakibaei, M.; Boland, C.R.; Goel, A. Novel Evidence for Curcumin and Boswellic Acid-Induced Chemoprevention through Regulation of miR-34a and miR-27a in Colorectal Cancer. Cancer Prev. Res. 2015, 8, 431–443. [Google Scholar] [CrossRef] [Green Version]
- Toden, S.; Okugawa, Y.; Jascur, T.; Wodarz, D.; Komarova, N.L.; Buhrmann, C.; Shakibaei, M.; Boland, C.R.; Goel, A. Curcumin mediates chemosensitization to 5-fluorouracil through miRNA-induced suppression of epithelial-to-mesenchymal transition in chemoresistant colorectal cancer. Carcinogenesis 2015, 36, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Zhai, K.; Brockmüller, A.; Kubatka, P.; Shakibaei, M.; Büsselberg, D. Curcumin’s Beneficial Effects on Neuroblastoma: Mechanisms, Challenges, and Potential Solutions. Biomolecules 2020, 10, 1469. [Google Scholar] [CrossRef]
- Buhrmann, C.; Brockmueller, A.; Mueller, A.L.; Shayan, P.; Shakibaei, M. Curcumin Attenuates Environment-Derived Osteoarthritis by Sox9/NF-kB Signaling Axis. Int. J. Mol. Sci. 2021, 22, 7645. [Google Scholar] [CrossRef]
- Buhrmann, C.; Honarvar, A.; Setayeshmehr, M.; Karbasi, S.; Shakibaei, M.; Valiani, A. Herbal Remedies as Potential in Cartilage Tissue Engineering: An Overview of New Therapeutic Approaches and Strategies. Molecules 2020, 25, 3075. [Google Scholar] [CrossRef]
- Shakibaei, M.; John, T.; Schulze-Tanzil, G.; Lehmann, I.; Mobasheri, A. Suppression of NF-κB activation by curcumin leads to inhibition of expression of cyclo-oxygenase-2 and matrix metalloproteinase-9 in human articular chondrocytes: Implications for the treatment of osteoarthritis. Biochem. Pharmacol. 2007, 73, 1434–1445. [Google Scholar] [CrossRef]
- Hanai, H.; Iida, T.; Takeuchi, K.; Watanabe, F.; Maruyama, Y.; Andoh, A.; Tsujikawa, T.; Fujiyama, Y.; Mitsuyama, K.; Sata, M. Curcumin maintenance therapy for ulcerative colitis: Randomized, multicenter, double-blind, placebo-controlled trial. Clin. Gastroenterol. Hepatol. 2006, 4, 1502–1506. [Google Scholar] [CrossRef]
- Lal, B.; Kapoor, A.; Agrawal, P.; Asthana, O.; Srimal, R. Role of curcumin in idiopathic inflammatory orbital pseudotumours. Phytother. Res. Int. J. Pharmacol. Toxicol. Eval. Nat. Prod. Deriv. 2000, 14, 443–447. [Google Scholar] [CrossRef]
- Daskalakis, M.; Brocks, D.; Sheng, Y.H.; Islam, M.S.; Ressnerova, A.; Assenov, Y.; Milde, T.; Oehme, I.; Witt, O.; Goyal, A.; et al. Reactivation of endogenous retroviral elements via treatment with DNMT- and HDAC-inhibitors. Cell Cycle 2018, 17, 811–822. [Google Scholar] [CrossRef]
- Nakano, K.; Boyle, D.L.; Firestein, G.S. Regulation of DNA methylation in rheumatoid arthritis synoviocytes. J. Immunol. 2013, 190, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Vojinovic, J.; Damjanov, N. HDAC inhibition in rheumatoid arthritis and juvenile idiopathic arthritis. Mol. Med. 2011, 17, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Aguilera, O.; Depreux, P.; Halby, L.; Arimondo, P.B.; Goossens, L. DNA Methylation Targeting: The DNMT/HMT Crosstalk Challenge. Biomolecules 2017, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Van Veggel, M.; Westerman, E.; Hamberg, P. Clinical Pharmacokinetics and Pharmacodynamics of Panobinostat. Clin. Pharmacokinet. 2018, 57, 21–29. [Google Scholar] [CrossRef]
- Wu, D.J.; Adamopoulos, I.E. Autophagy and autoimmunity. Clin. Immunol. 2017, 176, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Nirk, E.L.; Reggiori, F.; Mauthe, M. Hydroxychloroquine in rheumatic autoimmune disorders and beyond. EMBO Mol. Med. 2020, 12, e12476. [Google Scholar] [CrossRef]
- Suto, T.; Karonitsch, T. The immunobiology of mTOR in autoimmunity. J. Autoimmun. 2020, 110, 102373. [Google Scholar] [CrossRef]
- Abbasi, M.; Mousavi, M.J.; Jamalzehi, S.; Alimohammadi, R.; Bezvan, M.H.; Mohammadi, H.; Aslani, S. Strategies toward rheumatoid arthritis therapy; the old and the new. J. Cell. Physiol. 2019, 234, 10018–10031. [Google Scholar] [CrossRef]
- Burmester, G.R.; Pope, J.E. Novel treatment strategies in rheumatoid arthritis. Lancet 2017, 389, 2338–2348. [Google Scholar] [CrossRef]
- Salomon-Escoto, K.; Kay, J. The “Treat to Target” Approach to Rheumatoid Arthritis. Rheum. Dis. Clin. N. Am. 2019, 45, 487–504. [Google Scholar] [CrossRef]



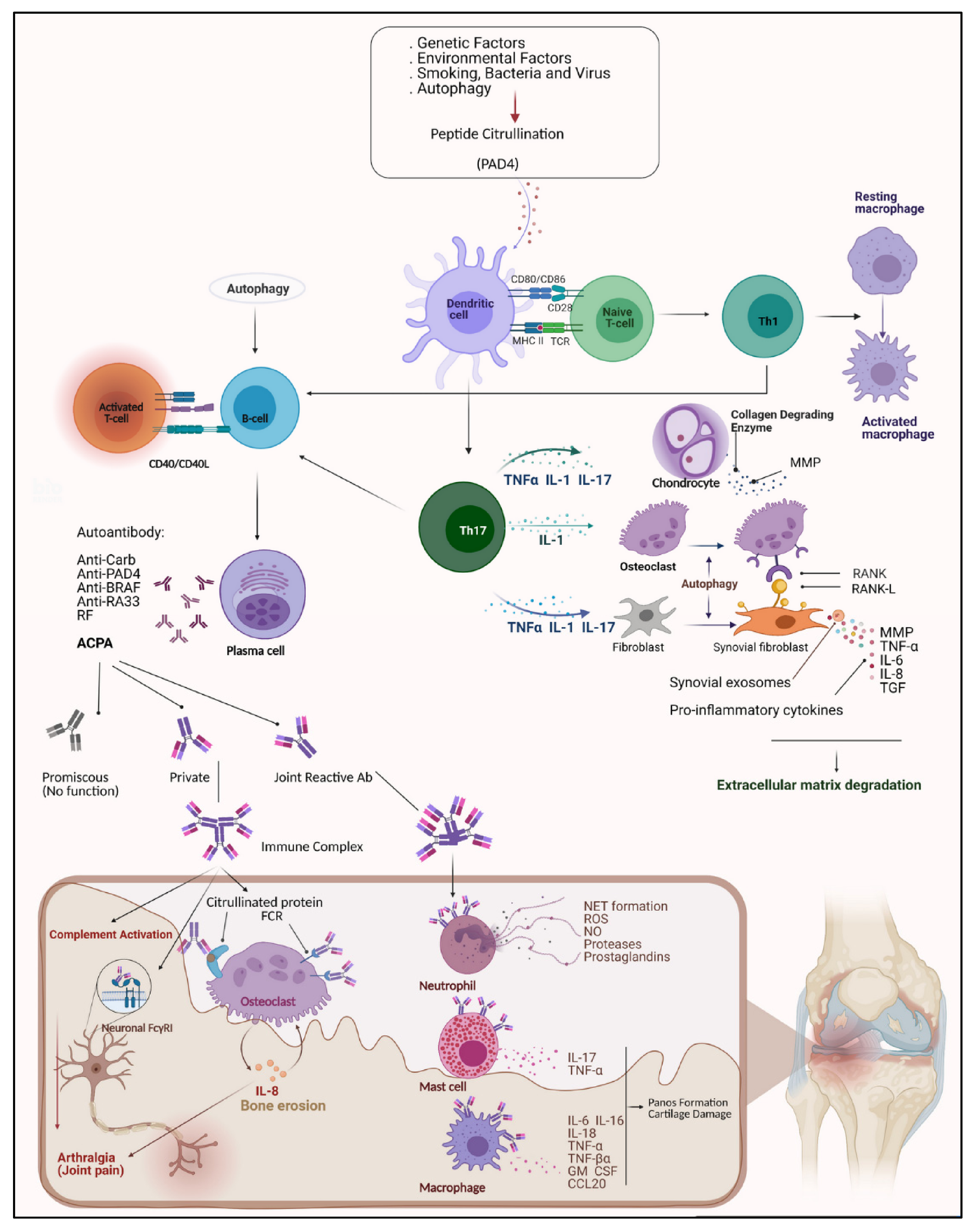{kind=link}
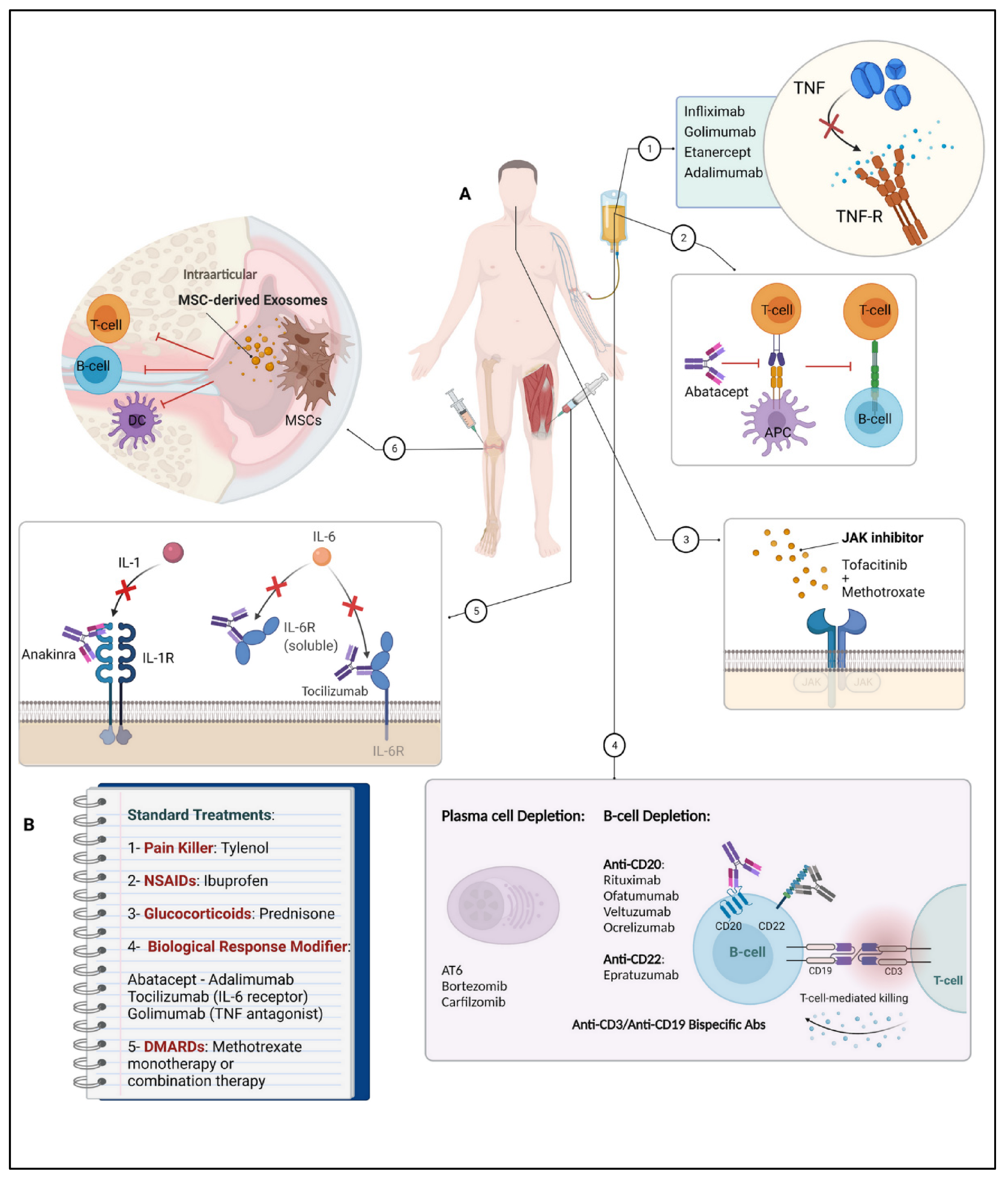{kind=link}
| Standard Treatments | |||
|---|---|---|---|
| First-Line Treatments | Therapy | Mechanisms of Action | Refs. |
| NSAIDs | Inhibition of COX-1 and/or COX-2 enzymes, blocked formation of pro-inflammatory prostaglandins from the arachidonic acid metabolism, thereby interrupting the inflammatory cycle. | [160,161,162,163] | |
| (i.e., Ibuprofen, Naproxen, Diclofenac, Etoricoxib, Celecoxib) | |||
| Glucocorticoids (GCs) | Activation or suppression of protein synthesis, including cytokines, chemokines, inflammatory enzymes, and adhesion molecules by activating the cytosolic glucocorticoid receptor; thus, modification of immune responses and inflammatory mechanisms. | [164,165,166,167,168] | |
| (i.e., Prednisone, Prednisolone, Budesonide) | |||
| DMARDs | Impedes immune cell proliferation. Relieves symptoms such as joint pain and prevents joint damage. | [169] | |
| (i.e., Methotrexate (MTX), | |||
| Hydroxychloroquine (HCQ), | Interferes with deoxyribonucleotides metabolism, impairs the antigen presentation, and lysosomal membrane stabilization is increased. | [170] | |
| Sulfasalazine, | Anti-inflammatory, immunosuppressive effects, which are attributed to its breakdown products sulfapyridine and 5-aminosalicylic acid. | [171] | |
| Leflunomide, | Interferes with pyrimidine synthesis, which is needed for lymphocyte activation. | [172] | |
| Azathioprine) | Blocks purine metabolism. | [173] | |
| New/Future Treatment Strategies | |||
| Biological Agents | Therapy | Mechanisms of Action | Refs. |
| TNF inhibitors (TNFi) | Selective inhibition of TNF-α, one of the major inflammatory cytokines, by specific binding to both its soluble subunit and its transmembrane precursor, leading to prevention of pro-inflammatory cell recruitment. Given the risk of developing Abs to the murine components of the molecule and the decreasing efficacy of monotherapy, RA therapy is only recommended in combination with MTX. | [162,174,175,176,177,178,179,180] | |
| (i.e., Infliximab, Adalimumab, Golimumab, Certolizumab, | |||
| Etanercept) | A biological TNF antagonist inhibiting the interaction of TNF-α and lymphotoxin (TNF-β) with cell-surface receptors, whereby TNF-mediated cellular responses and the activity of other pro-inflammatory cytokines are modulated. | [181,182,183,184] | |
| IL-inhibitors | Acting as IL-antagonists, thereby inhibiting cytokine binding to its receptors. Consequently IL-mediated signaling and its pro-inflammatory effects are suppressed. | [185,186,187,188,189,190,191] | |
| (IL-1: i.e., Anakinra, IL-6: i.e., Tocilizumab, Sarilumab, IL-17: i.e., Secukinumab, IL-21 Abs) | |||
| CXCL chemokine inhibitors | Inhibition of chemokines and chemokine receptors, which are found in inflamed synovia, to reduce the synovial migration of B-cells and the formation of ectopic germinal centers. | [192,193] | |
| (i.e., CXCL-10, -12, -13) | |||
| Co-stimulation blockers | Modulation of T-cell co-stimulation by binding to CD80 and CD86 receptors, thereby suppressing T-cell activation and B-cell stimulation. | [194,195,196,197] | |
| (i.e., Abatacept) | |||
| Anti-B-cell-agents | The monoclonal Abs cause depletion of B-cells through various mechanisms, i.e., apoptosis, complement-dependent cytotoxicity, and mediation of antibody-dependent cellular cytotoxicity. | [198,199,200,201,202,203] | |
| (CD20: i.e., Rituximab, Ofatumumab, Veltuzumab, Ocrelizumab, CD22: i.e., Epratuzumab, | |||
| CD79 Abs, | Targeting CD79 leads to inhibition of BCR signaling pathway, thereby depleting B-cells and ectopic germinal centers. | [204] | |
| CD40/CD154 Abs, | Blockade of complex formation leads to attenuation of CD154-mediated T-cell co-stimulation, inhibition of CD40-mediated B-cell stimulation, and supports CD4+ T-cells’ conversion to Tregs, mediating immunosuppression. | [205] | |
| TLR7/9 Abs | Inhibition of the binding between BCR and TLR (dominantly TLR7/9) leads to inactivation of B-cells. | [206] | |
| BAFF: Belimumab | Selective inhibition of BAFF (B-cell activating factor) prevents B-cell differentiation, thus leading to B-cell depletion. | [207,208,209] | |
| BAFF/APRIL: Atacicept, | Dual inhibitor of BAFF and APRIL (proliferation inducing ligand) by binding soluble BAFF and APRIL, thus interfering the interaction of these cytokines to their related receptors leading to B-cell depletion. | [210,211] | |
| PTK/Syk: i.e., Fostamatinib) | Inhibition of Syk, which is a vital non-receptor-type protein tyrosine kinase (PTK) activating down-stream MAPKs and the PI3K signaling pathway, leads to anti-inflammatory effects. | [212] | |
| Synthetical Agents | JAK-inhibitors | Reducing the production of inflammatory cytokines by suppressing the JAK signaling pathway. Either as monotherapy or in combination with MTX for the treatment of active RA. Ensuring optimal use of JAK inhibitors in the treatment of RA in practice. | [213,214,215,216,217] |
| (i.e., Tofacitinib, Baricitinib, Upadacitinib, Filgotinib) | |||
| Cell Therapy | Plasma cell therapy | The polyclonal Abs with T-cell-depleting properties can modulate the immune response by affecting the function of various immune effectors including B-cells and Tregs. | [218] |
| (i.e., Anti-thymocyte-globuline (ATG), | |||
| Bortezomib, Carfilzomib) | Proteasome inhibitor that suppresses activation of the pro-inflammatory transcription factor nuclear factor kappa B (NF-κB) by blocking degradation of the NF-κB inhibitor, thereby secretion of pro-inflammatory cytokines is reduced. | [219,220,221,222,223] | |
| Mesenchymal stem cell (MSC) therapy | Modulation of the immune response via cell-to-cell communication and MSC-secreted cytokines. For example, MSCs are able to directly inhibit T-cell function and differentiation or shifting them to functional Tregs. | [224,225,226] | |
| Combination Therapy | Combination of biologics and methotrexate | There is much evidence that treatment with a combination of biologics and MTX is significantly more effective than treatment with biologics or MTX alone in some patients. Combination therapy is usually the treatment of choice when monotherapy with DMARDs is not effective. | [227,228,229] |
| (i.e., Tocilizumab and MTX Filgotinib and MTX) | |||
| Epigenetic Therapy | DNA-methyltransferase (DNMT) and Histone-deacetylase (HDAC) inhibitors | Modification of epigenetic marks such as hypomethylation and histone marks as the main treatment goal to restore abnormalities that contribute to the development of RA back to normal levels. Moreover, HDAC inhibitors’ effects are related to immune cell apoptosis. | [230,231,232] |
| (i.e., Givinostat, Vorinostat,Panobinostat, Romidepsin) | |||
| Polyphenols | Therapeutic efficacy in the treatment of arthritis in CIA rats, reduction of inflammatory response by inhibition of NF-κB pathway resulted in significant relief of arthritic symptoms, by epigenetic regulation. | [233,234,235,236,237,238] | |
| Curcumin | |||
| Curcumin and Prednisolone, or MTX or diclofenac, or Vitamin D3 | Combination therapy showed significantly higher therapeutic efficacy than simple treatment alone in patients with active RA and also in rats, by epigenetic regulation. | [239,240,241,242,243,244,245] | |
| Targeting Autophagy | Autophagy inhibitors | Activation of autophagy by inhibiting mTOR. | [246,247] |
| (i.e., Rapamycin, | |||
| Chloroquine (CQ), Hydroxychloroquine (HCQ) | Autophagy suppression by inhibiting the lysosome reduces the activity of T-cells and apoptosis resistance. | [248,249] | |
| 3-Methyladenine (3-MA) | Inhibition of autophagy at an early stage of autophagosome development by blocking P13K signaling. | [250,251] | |
| Treat-to-Target | Monitoring of disease activity and adjusting therapeutic drug management | Establishment of a treatment goal and measuring treatment progress and disease activity regularly, whereby the treatment strategy is adjusted until the goal is reached. The improvement of disease activity achieved in 3 months should be at least 50% by applying new therapeutic strategies, otherwise the treatment should be modified. | [252,253] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mueller, A.-L.; Payandeh, Z.; Mohammadkhani, N.; Mubarak, S.M.H.; Zakeri, A.; Alagheband Bahrami, A.; Brockmueller, A.; Shakibaei, M. Recent Advances in Understanding the Pathogenesis of Rheumatoid Arthritis: New Treatment Strategies. Cells 2021, 10, 3017. https://doi.org/10.3390/cells10113017
Mueller A-L, Payandeh Z, Mohammadkhani N, Mubarak SMH, Zakeri A, Alagheband Bahrami A, Brockmueller A, Shakibaei M. Recent Advances in Understanding the Pathogenesis of Rheumatoid Arthritis: New Treatment Strategies. Cells. 2021; 10(11):3017. https://doi.org/10.3390/cells10113017
Chicago/Turabian StyleMueller, Anna-Lena, Zahra Payandeh, Niloufar Mohammadkhani, Shaden M. H. Mubarak, Alireza Zakeri, Armina Alagheband Bahrami, Aranka Brockmueller, and Mehdi Shakibaei. 2021. "Recent Advances in Understanding the Pathogenesis of Rheumatoid Arthritis: New Treatment Strategies" Cells 10, no. 11: 3017. https://doi.org/10.3390/cells10113017