
The Metabolic Control of Myeloid Cells in the Tumor Microenvironment

 , and
, and 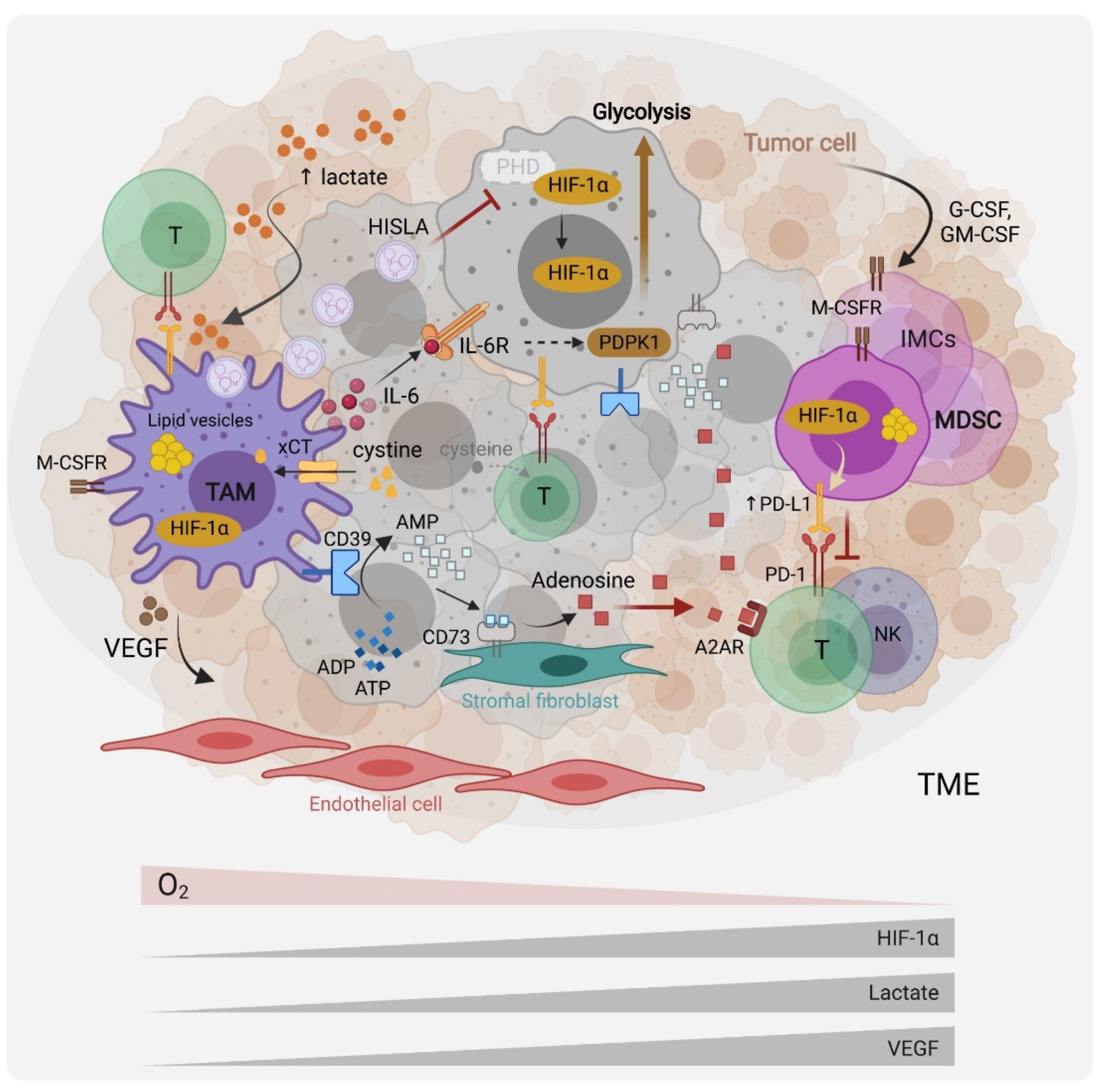{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Role of Hypoxia, Aerobic Glycolysis and Lactate in the Recruitment and Functions of TAMs and MDSCs
3. Amino Acids, Metabolism Controls, Myeloid Cell Phenotypes and Functions in the TME
3.1. Glutamine
3.2. Tryptophan
3.3. Arginine
3.4. Cystine, Glutamate and Oxidative Stress
4. Lipids and Cholesterol Metabolism Shape Immunosuppressive Myeloid Cells in the TME
5. Myelosuppressive Effects of Adenosine
6. Wnt Ligands Potentiate Myeloid Cells—Cancer Cells Metabolic Crosstalk
7. Clinical Trials Targeting Myeloid Cells, including via Their Metabolic Pathways
8. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chakarov, S.; Lim, H.Y.; Tan, L.; Lim, S.Y.; See, P.; Lum, J.; Zhang, X.-M.; Foo, S.; Nakamizo, S.; Duan, K.; et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 2019, 363, eaau0964. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef] [PubMed]
- Lavin, Y.; Mortha, A.; Rahman, A.; Merad, M. Regulation of macrophage development and function in peripheral tissues. Nat. Rev. Immunol. 2015, 15, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-resident macrophages in pancreatic ductal Adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 2017, 47, 323–338. [Google Scholar] [CrossRef]
- Sharma, A.; Seow, J.J.W.; Dutertre, C.-A.; Pai, R.; Blériot, C.; Mishra, A.; Wong, R.M.M.; Singh, G.S.N.; Sudhagar, S.; Khalilnezhad, S.; et al. Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell 2020, 183, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Casanova-Acebes, M.; Dalla, E.; Leader, A.M.; LeBerichel, J.; Nikolic, J.; Morales, B.M.; Brown, M.; Chang, C.; Troncoso, L.; Chen, S.T.; et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nat. Cell Biol. 2021, 595, 578–584. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O′Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Werno, C.; Menrad, H.; Weigert, A.; Dehne, N.; Goerdt, S.; Schledzewski, K.; Kzhyshkowska, J.; Brüne, B. Knockout of HIF-1 in tumor-associated macrophages enhances M2 polarization and attenuates their pro-angiogenic responses. Carcinogenesis 2010, 31, 1863–1872. [Google Scholar] [CrossRef] [Green Version]
- Imtiyaz, H.Z.; Williams, E.P.; Hickey, M.M.; Patel, S.A.; Durham, A.C.; Yuan, L.-J.; Hammond, R.; Gimotty, P.A.; Keith, B.; Simon, M.C. Hypoxia-inducible factor 2α regulates macrophage function in mouse models of acute and tumor inflammation. J. Clin. Investig. 2010, 120, 2699–2714. [Google Scholar] [CrossRef] [Green Version]
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 2013, 24, 695–709. [Google Scholar] [CrossRef] [Green Version]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.R.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.M.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.W.; et al. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, R.K.; Liou, Y.-H.; Knudsen, N.H.; Starost, K.A.; Xu, C.; Hyde, A.L.; Liu, S.; Jacobi, D.; Liao, N.-S.; Lee, C.-H. Bmal1 integrates mitochondrial metabolism and macrophage activation. eLife 2020, 9, 54090. [Google Scholar] [CrossRef] [PubMed]
- De-Brito, N.M.; Duncan-Moretti, J.; Da-Costa, H.C.; Saldanha-Gama, R.; Paula-Neto, H.A.; Dorighello, G.; Simões, R.L.; Barja-Fidalgo, C. Aerobic glycolysis is a metabolic requirement to maintain the M2-like polarization of tumor-associated macrophages. Biochim. Biophys. Acta BBA Bioenerg. 2020, 1867, 118604. [Google Scholar] [CrossRef]
- Wu, J.-Y.; Huang, T.-W.; Hsieh, Y.-T.; Wang, Y.-F.; Yen, C.-C.; Lee, G.-L.; Yeh, C.-C.; Peng, Y.-J.; Kuo, Y.-Y.; Wen, H.-T.; et al. Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol. Cell 2020, 77, 213–227. [Google Scholar] [CrossRef]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.-I.; Cheng, P.; Cho, H.-I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Linke, M.; Fritsch, S.D.; Sukhbaatar, N.; Hengstschläger, M.; Weichhart, T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett. 2017, 591, 3089–3103. [Google Scholar] [CrossRef] [PubMed]
- Welte, T.; Kim, I.S.; Tian, L.; Gao, X.; Wang, H.; Li, J.; Holdman, X.B.; Herschkowitz, J.I.; Pond, A.; Xie, G.; et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat. Cell Biol. 2016, 18, 632–644. [Google Scholar] [CrossRef] [Green Version]
- Katholnig, K.; Schütz, B.; Fritsch, S.D.; Schörghofer, D.; Linke, M.; Sukhbaatar, N.; Matschinger, J.M.; Unterleuthner, D.; Hirtl, M.; Lang, M.; et al. Inactivation of mTORC2 in macrophages is a signature of colorectal cancer that promotes tumorigenesis. JCI Insight 2019, 4, 24. [Google Scholar] [CrossRef]
- Ding, C.; Sun, X.; Wu, C.; Hu, X.; Zhang, H.-G.; Yan, J. Tumor microenvironment modulates immunological outcomes of myeloid cells with mTORC1 disruption. J. Immunol. 2019, 202, 1623–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, S.L.; Oh, M.-H.; Sun, I.-H.; Chan-Li, Y.; Zhao, L.; Powell, J.D.; Horton, M.R. mTORC1 signaling regulates proinflammatory macrophage function and metabolism. J. Immunol. 2021, 207, 913–922. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Aksoylar, H.I.; Yu, J.; Snyder, N.W.; Worth, A.J.; Iyer, S.S.; Wang, J.; Ben-Sahra, I.; Byles, V.; Polynne-Stapornkul, T.; et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife 2016, 5, e11612. [Google Scholar] [CrossRef]
- Linke, M.; Pham, H.T.T.; Katholnig, K.; Schnöller, T.; Miller, A.; Demel, F.; Schütz, B.; Rosner, M.; Kovacic, B.; Sukhbaatar, N.; et al. Chronic signaling via the metabolic checkpoint kinase mTORC1 induces macrophage granuloma formation and marks sarcoidosis progression. Nat. Immunol. 2017, 18, 293–302. [Google Scholar] [CrossRef]
- Wilson, J.L.; Nagele, T.; Linke, M.; Demel, F.; Fritsch, S.D.; Mayr, H.K.; Cai, Z.; Katholnig, K.; Sun, X.; Fragner, L.; et al. Inverse data-driven modeling and multiomics analysis reveals phgdh as a metabolic checkpoint of macrophage polarization and proliferation. Cell Rep. 2020, 30, 1542–1552. [Google Scholar] [CrossRef]
- Byles, V.; Covarrubias, A.J.; Ben-Sahra, I.; Lamming, D.W.; Sabatini, D.M.; Manning, B.D.; Horng, T. The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 2013, 4, 2834. [Google Scholar] [CrossRef] [Green Version]
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver′s seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef]
- Carmona-Fontaine, C.; Deforet, M.; Akkari, L.; Thompson, C.B.; Joyce, J.A.; Xavier, J.B. Metabolic origins of spatial organization in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2017, 114, 2934–2939. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Luo, J.; Kuang, D.; Xu, S.; Duan, Y.; Xia, Y.; Wei, Z.; Xie, X.; Yin, B.; Chen, F.; et al. Lactate inhibits ATP6V0d2 expression in tumor-associated macrophages to promote HIF-2α–mediated tumor progression. J. Clin. Investig. 2019, 129, 631–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nat. Cell Biol. 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Bohn, T.; Rapp, S.; Luther, N.; Klein, M.; Bruehl, T.-J.; Kojima, N.; Lopez, P.A.; Hahlbrock, J.; Muth, S.; Endo, S.; et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 2018, 19, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef] [Green Version]
- Sankowski, R.; Böttcher, C.; Masuda, T.; Geirsdottir, L.; Sindram, E.; Seredenina, T.; Muhs, A.; Scheiwe, C.; Shah, M.J.; Heiland, D.H.; et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 2019, 22, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xu, J.; Ma, Q.; Zhang, X.; Yang, Q.; Wang, L.; Cao, Y.; Xu, Z.; Tawfik, A.; Sun, Y.; et al. Glycolysis links reciprocal activation of myeloid cells and endothelial cells in the retinal angiogenic niche. Sci. Transl. Med. 2020, 12, eaay1371. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, G.; Chu, H.; Wang, X.; Xiong, L.; Cai, G.; Liu, R.; Gao, H.; Tao, B.; Li, W.; et al. Macrophage-associated PGK1 phosphorylation promotes aerobic glycolysis and tumorigenesis. Mol. Cell 2018, 71, 201–215. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Chen, J.; Yang, L.; Liu, J.; Zhang, X.; Zhang, Y.; Tu, Q.; Yin, D.; Lin, D.; Wong, P.P.; et al. Extracellular vesicle-packaged HIF-1α-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat. Cell Biol. 2019, 21, 498–510. [Google Scholar] [CrossRef]
- Palmieri, E.M.; Menga, A.; Martín-Pérez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquière, B.; McVicar, D.W.; et al. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1-like phenotype and inhibits tumor metastasis. Cell Rep. 2017, 20, 1654–1666. [Google Scholar] [CrossRef] [Green Version]
- Menga, A.; Serra, M.; Todisco, S.; Riera-Domingo, C.; Ammarah, U.; Ehling, M.; Palmieri, E.M.; Di Noia, M.A.; Gissi, R.; Favia, M.; et al. Glufosinate constrains synchronous and metachronous metastasis by promoting anti-tumor macrophages. EMBO Mol. Med. 2020, 12, 27. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-T.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.-C.; Chou, C.-H.; Vavakova, M.; et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef]
- Wu, W.-C.; Sun, H.-W.; Chen, J.; Ouyang, H.-Y.; Yu, X.-J.; Chen, H.-T.; Shuang, Z.-Y.; Shi, M.; Wang, Z.; Zheng, L. Immunosuppressive immature myeloid cell generation is controlled by glutamine metabolism in human cancer. Cancer Immunol. Res. 2019, 7, 1605–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.-W.; Wu, W.-C.; Chen, H.-T.; Xu, Y.-T.; Yang, Y.-Y.; Chen, J.; Yu, X.-J.; Wang, Z.; Shuang, Z.-Y.; Zheng, L. Glutamine deprivation promotes the generation and mobilization of MDSCs by enhancing expression of G-CSF and GM-CSF. Front. Immunol. 2021, 11, 616367. [Google Scholar] [CrossRef]
- Oh, M.-H.; Sun, I.-H.; Zhao, L.; Leone, R.D.; Sun, I.M.; Xu, W.; Collins, S.L.; Tam, A.J.; Blosser, R.L.; Patel, C.H.; et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J. Clin. Investig. 2020, 130, 3865–3884. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Kuang, D.-M.; Wu, Y.; Xiao, X.; Li, X.-F.; Li, T.-J.; Zheng, L. Activated CD69+ T cells foster immune privilege by regulating IDO expression in tumor-associated macrophages. J. Immunol. 2012, 188, 1117–1124. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Zhao, J.; Xing, Y.; Zhang, X.; Liu, J.; Ouyang, Q.; Chen, J.; Su, F.; Liu, Q.; Song, E. Immune checkpoint inhibition overcomes ADCP-induced immunosuppression by macrophages. Cell 2018, 175, 442–457.e23. [Google Scholar] [CrossRef] [Green Version]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.-C.; Gutiérrez-Vázquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A.; et al. Author correction: Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 2019, 22, 1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campesato, L.F.; Budhu, S.; Tchaicha, J.; Weng, C.-H.; Gigoux, M.; Cohen, I.J.; Redmond, D.; Mangarin, L.; Pourpe, S.; Liu, C.; et al. Blockade of the AHR restricts a Treg-macrophage suppressive axis induced by L-Kynurenine. Nat. Commun. 2020, 11, 4011. [Google Scholar] [CrossRef]
- Palmieri, E.M.; Gonzalez-Cotto, M.; Baseler, W.A.; Davies, L.C.; Ghesquière, B.; Maio, N.; Rice, C.M.; Rouault, T.A.; Cassel, T.; Higashi, R.M.; et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat. Commun. 2020, 11, 698. [Google Scholar] [CrossRef] [PubMed]
- Brentville, V.A.; Metheringham, R.L.; Daniels, I.; Atabani, S.; Symonds, P.; Cook, K.W.; Vankemmelbeke, M.; Choudhury, R.; Vaghela, P.; Gijon, M.; et al. Combination vaccine based on citrullinated vimentin and enolase peptides induces potent CD4-mediated anti-tumor responses. J. Immunother. Cancer 2020, 8, e000560. [Google Scholar] [CrossRef]
- Mills, C.D.; Shearer, J.; Evans, R.; Caldwell, M.D. Macrophage arginine metabolism and the inhibition or stimulation of cancer. J. Immunol. 1992, 149, 2709–2714. [Google Scholar]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, H.; Li, S.; Li, X.; Wang, W.; Bian, Y.; Wei, S.; Grove, S.; Wang, W.; Vatan, L.; Liu, J.R.; et al. Autophagic adaptation to oxidative stress alters peritoneal residential macrophage survival and ovarian cancer metastasis. JCI Insight 2020, 5, 141115. [Google Scholar] [CrossRef]
- Badeaux, M.D.; Rolig, A.S.; Agnello, G.; Enzler, D.; Kasiewicz, M.J.; Priddy, L.; Wiggins, J.F.; Muir, A.; Sullivan, M.R.; Van Cleef, J.; et al. Arginase therapy combines effectively with immune checkpoint blockade or agonist anti-OX40 immunotherapy to control tumor growth. Cancer Immunol. Res. 2021, 9, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.; Varma, M.; Bryant, C.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 2017, 167, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.; Ma, H.-Y.; Zhong, Z.; Dhar, D.; Liu, X.; Xu, J.; Koyama, Y.; Nishio, T.; Karin, D.; Karin, G.; et al. NADPH oxidase 1 in liver macrophages promotes inflammation and tumor development in mice. Gastroenterology 2019, 156, 1156–1172. [Google Scholar] [CrossRef] [Green Version]
- Roux, C.; Jafari, S.M.; Shinde, R.; Duncan, G.; Cescon, D.W.; Silvester, J.; Chu, M.F.; Hodgson, K.; Berger, T.; Wakeham, A.; et al. Reactive oxygen species modulate macrophage immunosuppressive phenotype through the up-regulation of PD-L1. Proc. Natl. Acad. Sci. USA 2019, 116, 4326–4335. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.; Alcivar, A.L.; Ma, J.; Foo, T.K.; Zywea, S.; Mahdi, A.; Huo, Y.; Kensler, T.; Gatza, M.; Xia, B. NRF2 induction supporting breast cancer cell survival is enabled by oxidative stress–induced DPP3–KEAP1 interaction. Cancer Res. 2017, 77, 2881–2892. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Ye, P.; Mimura, J.; Okada, T.; Sato, H.; Liu, T.; Maruyama, A.; Ohyama, C.; Itoh, K. Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol. Cell. Biol. 2014, 34, 3421–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, P.; Mimura, J.; Okada, T.; Sato, H.; Liu, T.; Maruyama, A.; Ohyama, C.; Itoh, K. Correction for Ye et al., Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol. Cell. Biol. 2015, 35, 2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiramoto, K.; Satoh, H.; Suzuki, T.; Moriguchi, T.; Pi, J.; Shimosegawa, T.; Yamamoto, M. Myeloid lineage—Specific deletion of antioxidant system enhances tumor metastasis. Cancer Prev. Res. 2014, 7, 835–844. [Google Scholar] [CrossRef] [Green Version]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.C.-C.; Everts, B.; Ivanova, Y.; O′Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Yang, Q.; Shi, M.; Zhong, L.; Wu, C.; Meng, T.; Yin, H.; Zhou, J. Polyunsaturated fatty acids promote the expansion of myeloid-derived suppressor cells by activating the JAK/STAT3 pathway. Eur. J. Immunol. 2013, 43, 2943–2955. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Han, Y.; Sillke, Y.R.; Deng, H.; Siddiqui, S.; Treese, C.; Schmidt, F.; Friedrich, M.; Keye, J.; Wan, J.; et al. Lipid droplet-dependent fatty acid metabolism controls the immune suppressive phenotype of tumor-associated macrophages. EMBO Mol. Med. 2019, 11, e10698. [Google Scholar] [CrossRef]
- Su, P.; Wang, Q.; Bi, E.; Ma, X.; Liu, L.; Yang, M.; Qian, J.; Yi, Q. Enhanced lipid accumulation and metabolism are required for the differentiation and activation of tumor-associated macrophages. Cancer Res. 2020, 80, 1438–1450. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Shi, R.; Kang, X.; Zhang, X.; Chen, P.; Zhang, L.; Hou, A.; Wang, R.; Zhao, Y.; Zhao, K.; et al. Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression. Nat. Commun. 2018, 9, 2574. [Google Scholar] [CrossRef] [Green Version]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Del Valle, L.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divakaruni, A.S.; Hsieh, W.Y.; Minarrieta, L.; Duong, T.N.; Kim, K.K.; Desousa, B.R.; Andreyev, A.Y.; Bowman, C.E.; Caradonna, K.; Dranka, B.P.; et al. Etomoxir inhibits macrophage polarization by disrupting CoA homeostasis. Cell Metab. 2018, 28, 490–503. [Google Scholar] [CrossRef] [Green Version]
- Veglia, F.; Tyurin, V.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nat. Cell Biol. 2019, 569, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Prima, V.; Kaliberova, L.N.; Kaliberov, S.; Curiel, D.T.; Kusmartsev, S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc. Natl. Acad. Sci. USA 2017, 114, 1117–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goossens, P.; Rodriguez-Vita, J.; Etzerodt, A.; Masse, M.; Rastoin, O.; Gouirand, V.; Ulas, T.; Papantonopoulou, O.; Van Eck, M.; Auphan-Anezin, N.; et al. Membrane cholesterol efflux drives tumor-associated macrophage reprogramming and tumor progression. Cell Metab. 2019, 29, 1376–1389.e4. [Google Scholar] [CrossRef] [Green Version]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [Green Version]
- Jin, D.; Fan, J.; Wang, L.; Thompson, L.F.; Liu, A.; Daniel, B.J.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 on tumor cells impairs antitumor T-cell responses: A novel mechanism of tumor-induced immune suppression. Cancer Res. 2010, 70, 2245–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cekic, C.; Day, Y.-J.; Sag, D.; Linden, J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. 2014, 74, 7250–7259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Barrio, I.M.; Penski, C.; Schlahsa, L.; Stein, R.G.; Diessner, J.; Wöckel, A.; Dietl, J.; Lutz, M.B.; Mittelbronn, M.; Wischhusen, J.; et al. Adenosine-generating ovarian cancer cells attract myeloid cells which differentiate into adenosine-generating tumor associated macrophages—A self-amplifying, CD39- and CD73-dependent mechanism for tumor immune escape. J. Immunother. Cancer 2016, 4, 49. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, Y.; Chu, Y.; Li, Z.; Yu, X.; Huang, Z.; Xu, J.; Zheng, L. Tumor-derived adenosine promotes macrophage proliferation in human hepatocellular carcinoma. J. Hepatol. 2021, 74, 627–637. [Google Scholar] [CrossRef]
- Bugter, J.M.; Fenderico, N.; Maurice, M.M. Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nat. Rev. Cancer 2021, 21, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Cosin-Roger, J.; Ortiz-Masià, M.D.; Barrachina, M.D. Macrophages as an emerging source of Wnt ligands: Relevance in mucosal integrity. Front. Immunol. 2019, 10, 2297. [Google Scholar] [CrossRef]
- Yang, Y.; Ye, Y.C.; Chen, Y.; Zhao, J.L.; Gao, C.C.; Han, H.; Liu, W.C.; Qin, H.Y. Crosstalk between hepatic tumor cells and macrophages via Wnt/beta-catenin signaling promotes M2-like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis. 2018, 9, 793. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Wu, Y.; Yang, Y.; Wang, J.; Niu, M.; Gao, S.; Qin, T.; Bao, D. Long noncoding RNA LINC00662 promotes M2 macrophage polarization and hepatocellular carcinoma progression via activating Wnt/beta-catenin signaling. Mol. Oncol. 2020, 14, 462–483. [Google Scholar] [CrossRef] [Green Version]
- Douglass, S.M.; Fane, M.E.; Sanseviero, E.; Ecker, B.L.; Kugel, C.H., III; Behera, R.; Kumar, V.; Tcyganov, E.N.; Yin, X.; Liu, Q.; et al. Myeloid-derived suppressor cells are a major source of Wnt5A in the melanoma microenvironment and depend on Wnt5A for full suppressive activity. Cancer Res. 2021, 81, 658–670. [Google Scholar] [CrossRef]
- DeVito, N.C.; Sturdivant, M.; Thievanthiran, B.; Xiao, C.; Plebanek, M.P.; Salama, A.K.; Beasley, G.M.; Holtzhausen, A.; Novotny-Diermayr, V.; Strickler, J.H.; et al. Pharmacological Wnt ligand inhibition overcomes key tumor-mediated resistance pathways to anti-PD-1 immunotherapy. Cell Rep. 2021, 35, 109071. [Google Scholar] [CrossRef]
- Jahchan, N.S.; Mujal, A.M.; Pollack, J.L.; Binnewies, M.; Sriram, V.; Reyno, L.; Krummel, M.F. Tuning the tumor myeloid microenvironment to fight cancer. Front. Immunol. 2019, 10, 1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Yrigoyen, M.; Cassetta, L.; Pollard, J.W. Macrophage targeting in cancer. Ann. N. Y. Acad. Sci. 2021, 1499, 18–41. [Google Scholar] [CrossRef] [PubMed]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation early phase clinical trials consortium phase II study. Neuro Oncol. 2015, 18, 557–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuemmel, S.; Campone, M.; Loirat, D.; López, R.L.; Beck, J.T.; De Laurentiis, M.; Im, S.-A.; Kim, S.-B.; Kwong, A.; Steger, G.G.; et al. A randomized phase II study of anti-CSF-1 monoclonal antibody lacnotuzumab (MCS110) combined with gemcitabine and carboplatin in advanced triple negative breast cancer. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Donthireddy, L.; Marvel, D.; Condamine, T.; Wang, F.; Lavilla-Alonso, S.; Hashimoto, A.; Vonteddu, P.; Behera, R.; Goins, M.A.; et al. Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors. Cancer Cell 2017, 32, 654–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, K.P.; Gluck, L.; Martin, L.P.; Olszanski, A.; Tolcher, A.W.; Ngarmchamnanrith, G.; Rasmussen, E.; Amore, B.M.; Nagorsen, D.; Hill, J.S.; et al. First-in-human study of AMG 820, a monoclonal anti-colony-stimulating factor 1 receptor antibody, in patients with advanced solid tumors. Clin. Cancer Res. 2017, 23, 5703–5710. [Google Scholar] [CrossRef] [Green Version]
- Muller, A.J.; Manfredi, M.G.; Zakharia, Y.; Prendergast, G.C. Inhibiting IDO pathways to treat cancer: Lessons from the ECHO-301 trial and beyond. Semin. Immunopathol. 2019, 41, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Emens, L.A. Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer 2018, 6, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramel, E.; Lillo, S.; Daher, B.; Fioleau, M.; Daubon, T.; Saleh, M. The Metabolic Control of Myeloid Cells in the Tumor Microenvironment. Cells 2021, 10, 2960. https://doi.org/10.3390/cells10112960
Ramel E, Lillo S, Daher B, Fioleau M, Daubon T, Saleh M. The Metabolic Control of Myeloid Cells in the Tumor Microenvironment. Cells. 2021; 10(11):2960. https://doi.org/10.3390/cells10112960
Chicago/Turabian StyleRamel, Eloise, Sebastian Lillo, Boutaina Daher, Marina Fioleau, Thomas Daubon, and Maya Saleh. 2021. "The Metabolic Control of Myeloid Cells in the Tumor Microenvironment" Cells 10, no. 11: 2960. https://doi.org/10.3390/cells10112960