
Metagenomic Profiling of Fecal-Derived Bacterial Membrane Vesicles in Crohn’s Disease Patients

 ,
,
Abstract
:1. Introduction
2. Materials and Method
2.1. Study Population
2.2. Ethical Statement
2.3. Vesicle Isolation from Fecal Samples
2.4. Visualizing MVs by Cryo-Transmission Electron MICROSCOPY (Cryo-TEM)
2.5. Fecal and Bacterial DNA Isolation
2.6. Next Generation Sequencing by Illumina MiSeq
2.7. Sequencing Analysis
2.8. Richness, Diversity, and Taxonomy
2.9. Statistical Analysis
3. Results
3.1. Study Population
3.2. MVs Characterizations
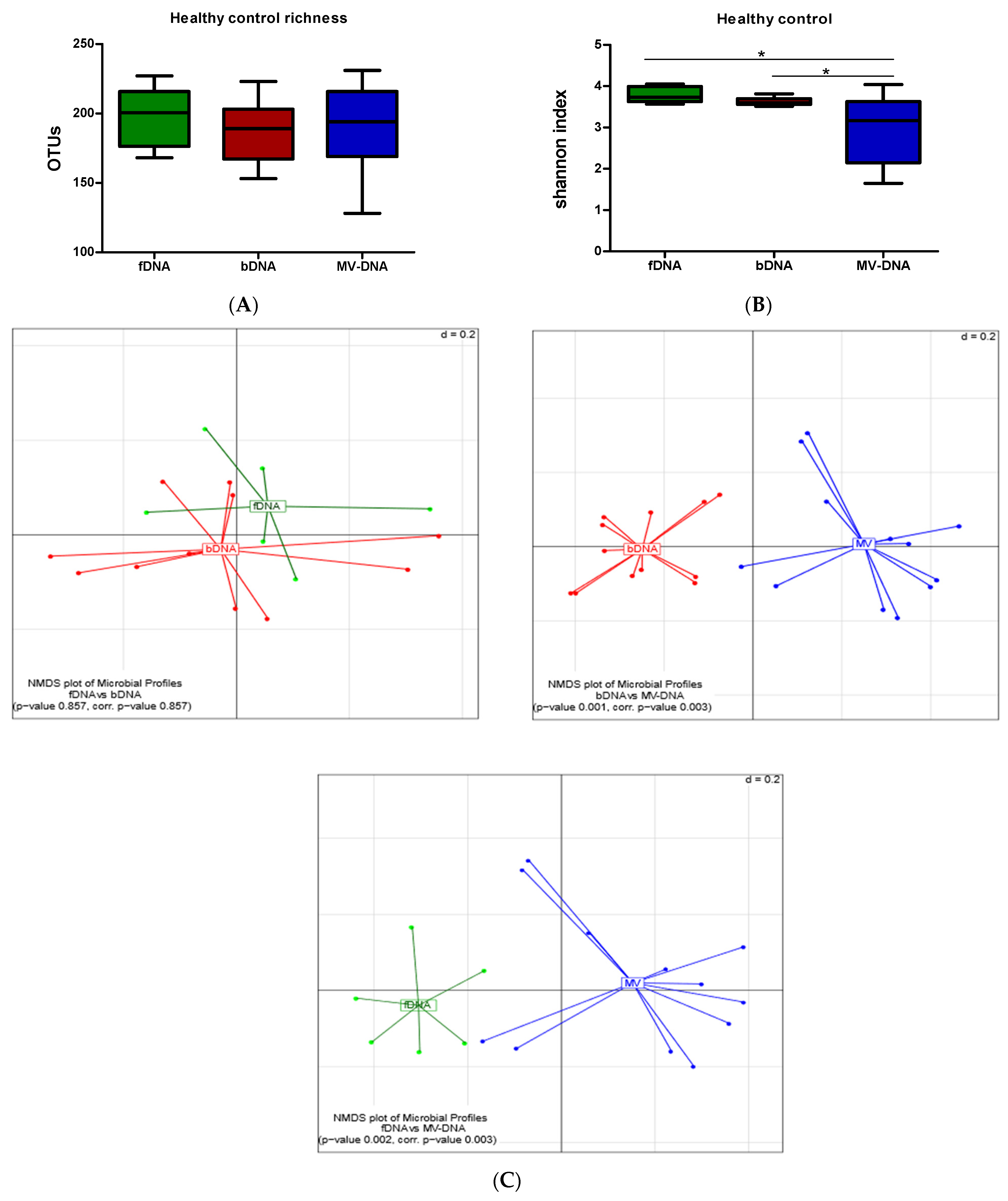
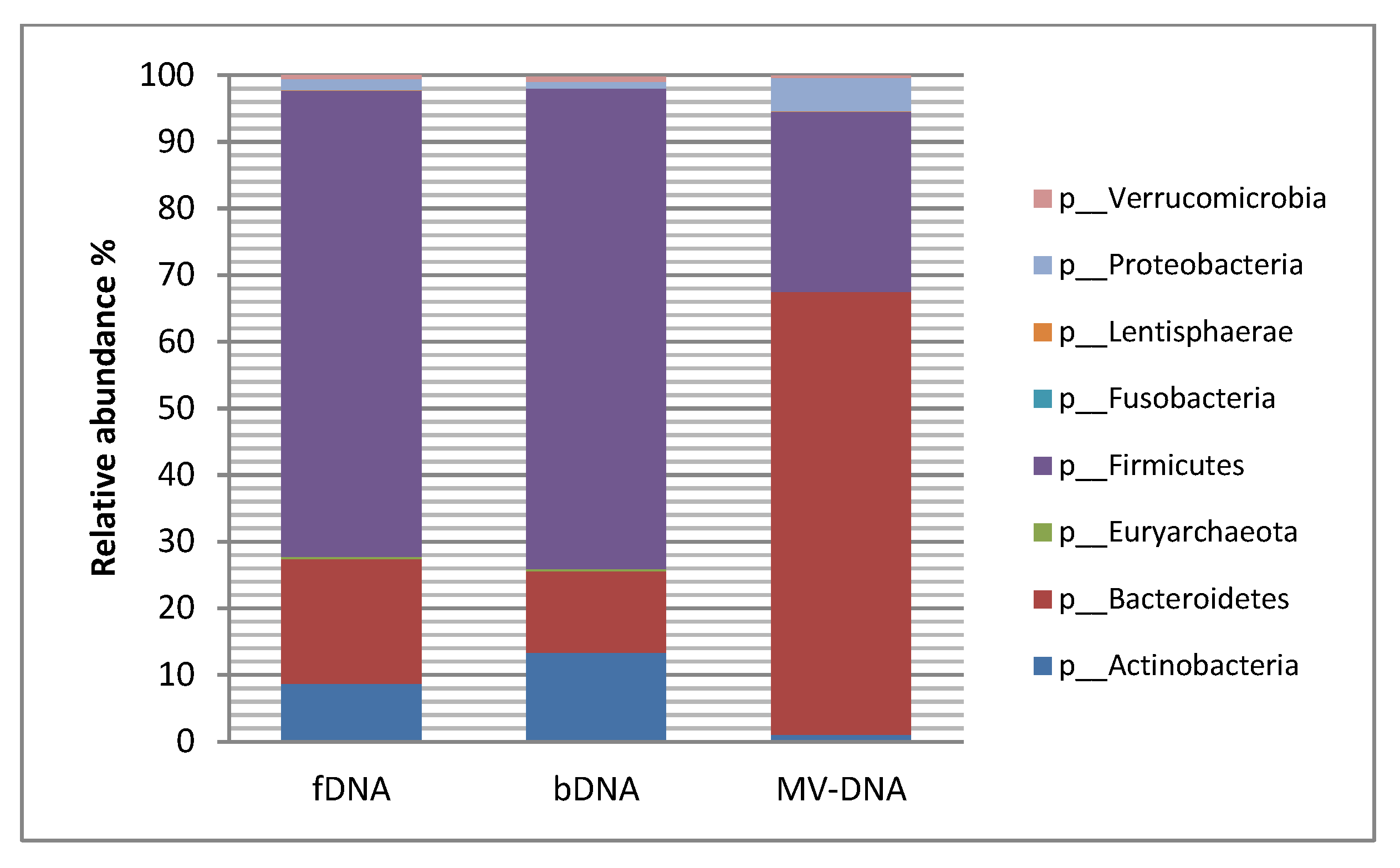
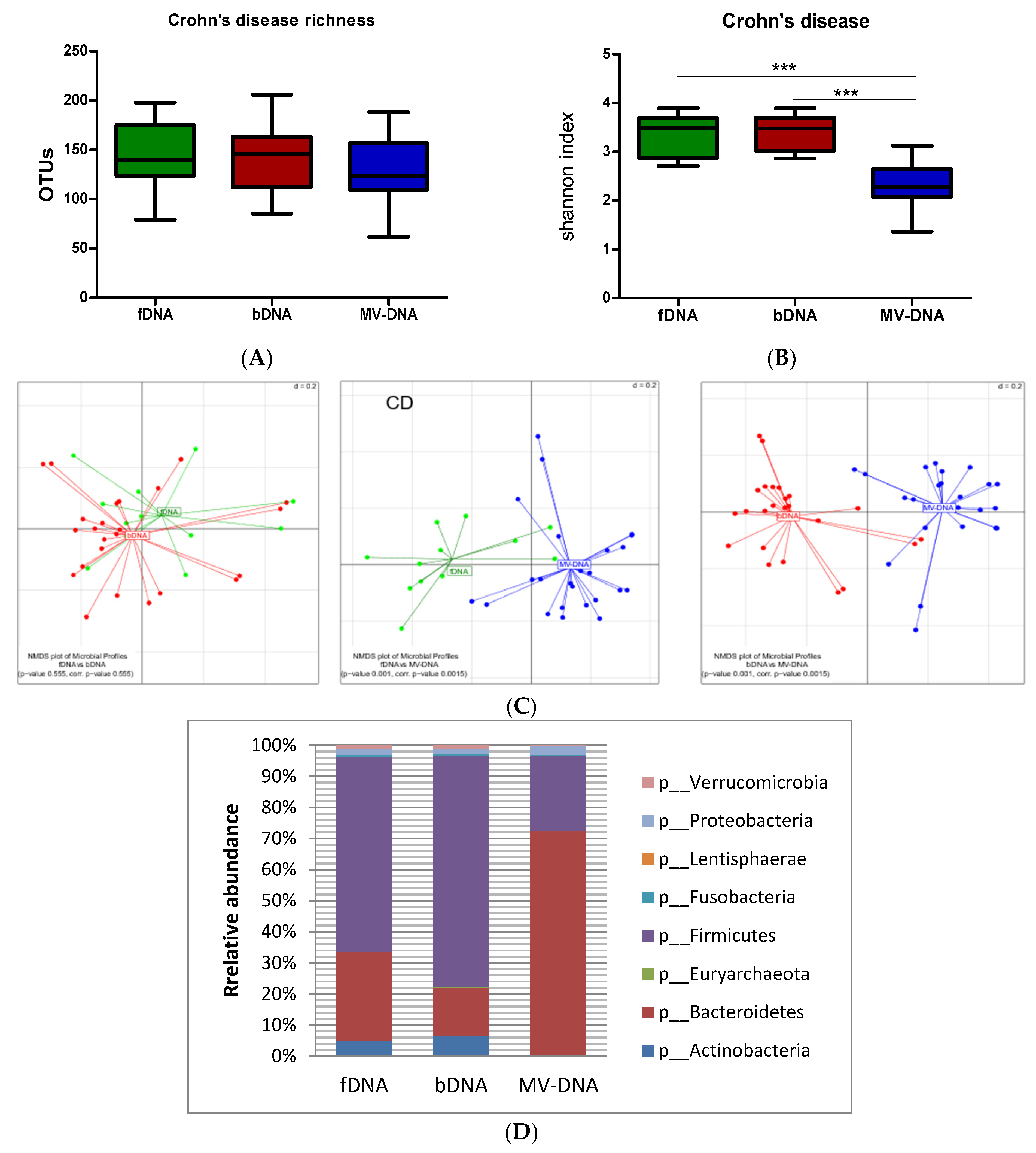
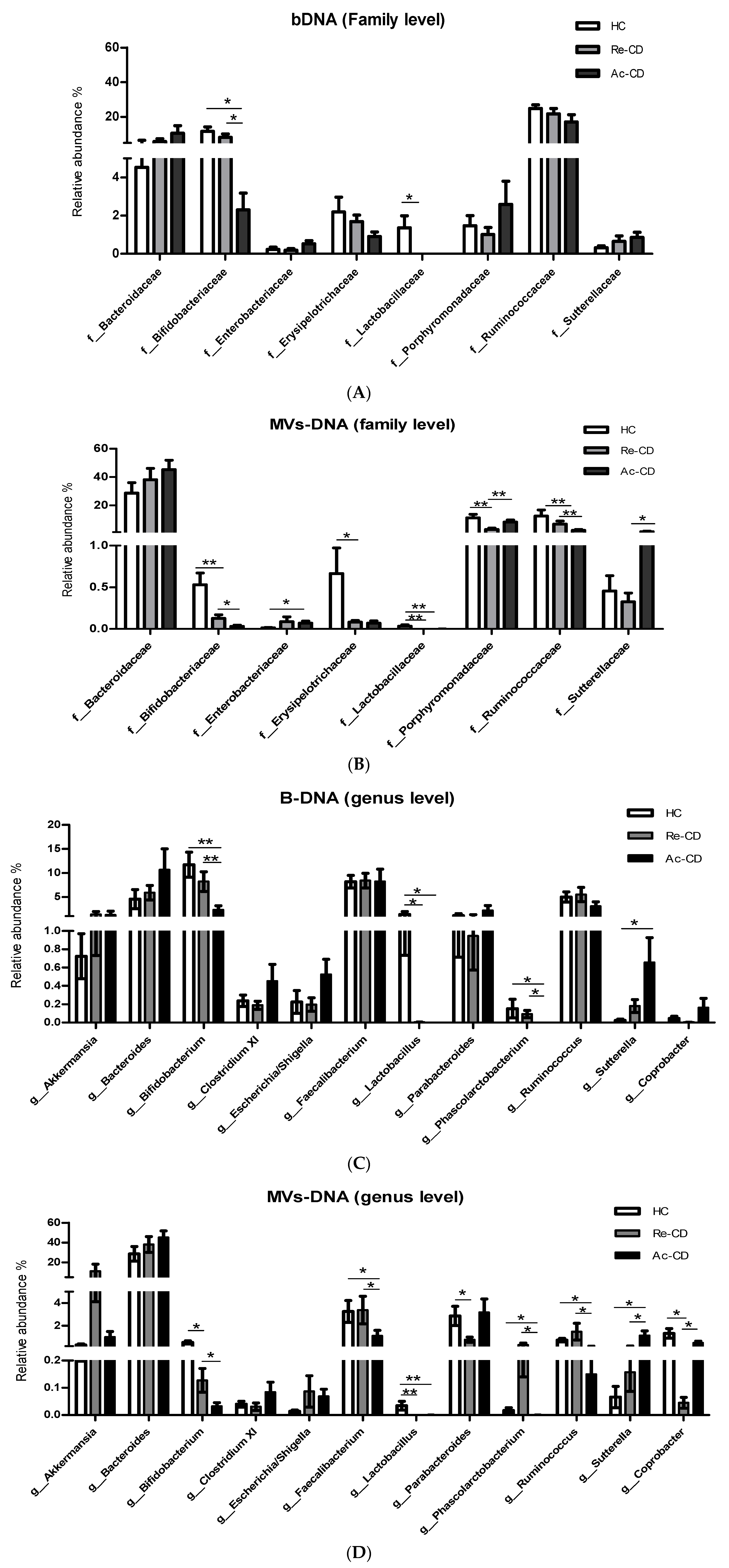
3.3. Microbial Composition and Diversity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, H.D.; Cheon, J.H. Pathogenesis of Inflammatory Bowel Disease and Recent Advances in Biologic Therapies. Immune Netw. 2017, 171, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Sartor, R.B. The intestinal microbiota in inflammatory bowel diseases. Nestle Nutr. Inst. Workshop Ser. 2014, 79, 29–39. [Google Scholar]
- Vindigni, M.S.; Zisman, T.L.; Suskind, D.L.; Damman, C.J. The intestinal microbiome, barrier function, and immune system in inflammatory bowel disease: A tripartite pathophysiological circuit with implications for new therapeutic directions. Ther. Adv. Gastroenterol. 2016, 9, 606–625. [Google Scholar] [CrossRef] [Green Version]
- Torres, J.; Mehandru, S.; Colombel, J.F.; Peyrin-Biroulet, L. Crohn’s disease. Lancet 2017, 389, 1741–1755. [Google Scholar] [CrossRef]
- Schwechheimer, C.; Kuehn, M.J. Outer-membrane vesicles from Gram-negative bacteria: Biogenesis and functions. Nat. Rev. Micro. 2015, 13, 605–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathirana, D.R.; Kaparakis-Liaskos, M. Bacterial membrane vesicles: Biogenesis, immune regulation and pathogenesis. Cell. Microbiol. 2016, 18, 1518–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitto, J.N.; Chapman, R.; Pidot, S.; Costin, A.; Lo, C.; Choi, J.; D’Cruze, T.; Reynolds, E.C.; Dashper, S.G.; Turnbull, L.; et al. Bacterial membrane vesicles transport their DNA cargo into host cells. Sci. Rep. 2017, 7, 7072. [Google Scholar] [CrossRef] [PubMed]
- Nagakubo, T.; Nomura, N.; Toyofuku, M. Cracking Open Bacterial Membrane Vesicles. Front. Microbiol. 2019, 10, 3026. [Google Scholar] [CrossRef] [Green Version]
- Perez-Cruz, C.; Delgado, L.; Lopez-Iglesias, C.; Mercade, E. Outer-inner membrane vesicles naturally secreted by gram-negative pathogenic bacteria. PLoS ONE 2015, 10, e0116896. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.E.; Choi, D.S.; Kim, K.P.; Gho, Y.S. Proteomics in gram-negative bacterial outer membrane vesicles. Mass. Spectrom. Rev. 2008, 27, 535–555. [Google Scholar] [CrossRef]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vazquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current Understanding of Dysbiosis in Disease in Human and Animal Models. Inflamm. Bowel Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedin, C.R.; McCarthy, N.E.; Louis, P.; Farquharson, F.M.; McCartney, S.; Taylor, K.; Prescott, N.J.; Murrells, T.; Stagg, A.J.; Whelan, K.; et al. Altered intestinal microbiota and blood T cell phenotype are shared by patients with Crohn’s disease and their unaffected siblings. Gut 2014, 63, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Butcher, J.; Mack, D.; Stintzi, A. Functional Impacts of the Intestinal Microbiome in the Pathogenesis of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Seksik, P.; Rigottier–Gois, L.; Gramet, G.; Sutren, M.; Pochart, P.; Marteau, P.; Jian, R.; Dore, J. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut 2003, 52, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2014, 63, 1275–1283. [Google Scholar] [CrossRef]
- Geirnaert, A.; Calatayud, M.; Grootaert, C.; Laukens, D.; Devriese, S.; Smagghe, G.; De Vos, M.; Boon, N.; Van De Wiele, T. Butyrate-producing bacteria supplemented in vitro to Crohn’s disease patient microbiota increased butyrate production and enhanced intestinal epithelial barrier integrity. Sci. Rep. 2017, 7, 11450. [Google Scholar] [CrossRef] [Green Version]
- Vargoorani, M.E.; Modarressi, M.H.; Vaziri, F.; Motevaseli, E.; Siadat, S.D. Stimulatory effects of Lactobacillus casei derived extracellular vesicles on toll-like receptor 9 gene expression and cytokine profile in human intestinal epithelial cells. J. Diabetes Metab. Disord. 2020, 19, 223–231. [Google Scholar] [CrossRef]
- Patten, D.A.; Hussein, E.; Davies, S.P.; Humphreys, P.; Collett, A. Commensal-derived OMVs elicit a mild proinflammatory response in intestinal epithelial cells. Microbiology 2017, 163, 702–711. [Google Scholar] [CrossRef]
- Vanaja, S.K.; Russo, A.J.; Behl, B.; Banerjee, I.; Yankova, M.; Deshmukh, S.D.; Rathinam, V.A. Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell 2016, 165, 1106–1119. [Google Scholar] [CrossRef] [Green Version]
- Fabrega, M.J.; Rodríguez-Nogales, A.; Mesa, J.G.; Algieri, F.; Badía, J.; Giménez, R.; Galvez, J.; Baldomà, L. Intestinal Anti-inflammatory Effects of Outer Membrane Vesicles from Escherichia coli Nissle 1917 in DSS-Experimental Colitis in Mice. Front. Microbiol. 2017, 8, 1274. [Google Scholar] [CrossRef]
- van den Heuvel, T.R.; Jonkers, D.M.; Jeuring, S.F.; Romberg-Camps, M.J.; E Oostenbrug, L.; Zeegers, M.P.; Masclee, A.A.; Pierik, M.J. Cohort Profile: The Inflammatory Bowel Disease South Limburg Cohort (IBDSL). Int. J. Epidemiol. 2017, 46, e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Assche, G.; Dignass, A.; Panes, J.; Beaugerie, L.; Karagiannis, J.; Allez, M.; Ochsenkuhn, T.; Orchard, T.; Rogler, G.; Louis, E.; et al. The second European evidence-based Consensus on the diagnosis and management of Crohn’s disease: Definitions and diagnosis. J. Crohns Colitis 2010, 4, 7–27. [Google Scholar] [CrossRef] [Green Version]
- Daperno, M.; D’Haens, G.; Van Assche, G.; Baert, F.; Bulois, P.; Maunoury, V.; Sostegni, R.; Rocca, R.; Pera, A.; Gevers, A.; et al. Development and validation of a new, simplified endoscopic activity score for Crohn’s disease: The SES-CD. Gastrointest. Endosc. 2004, 60, 505–512. [Google Scholar] [CrossRef]
- Benedikter, B.J.; Bouwman, F.G.; Vajen, T.; Heinzmann, A.C.A.; Grauls, G.; Mariman, E.C.; Wouters, E.F.M.; Savelkoul, P.H.; Lopez-Iglesias, C.; Koenen, R.R.; et al. Ultrafiltration combined with size exclusion chromatography efficiently isolates extracellular vesicles from cell culture media for compositional and functional studies. Sci. Rep. 2017, 7, 15297. [Google Scholar] [CrossRef] [PubMed]
- Kameli, N.; Borman, R.; López-Iglesias, C.; Savelkoul, P.; Stassen, F.R. Characterization of Feces-Derived Bacterial Membrane Vesicles and the Impact of Their Origin on the Inflammatory Response. Front. Cell. Infect. Microbiol. 2021, 11, 667987. [Google Scholar]
- Schaar, V.; Nordstrom, T.; Morgelin, M.; Riesbeck, K. Moraxella catarrhalis outer membrane vesicles carry beta-lactamase and promote survival of Streptococcus pneumoniae and Haemophilus influenzae by inactivating amoxicillin. Antimicrob. Agents Chemother. 2011, 55, 3845–3853. [Google Scholar] [CrossRef] [Green Version]
- Salonen, A.; Nikkilä, J.; Jalanka-Tuovinen, J.; Immonen, O.; Rajilic-Stojanovic, M.; Kekkonen, R.A.; Palva, A.; de Vos, W.M. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: Effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J. Microbiol. Methods 2010, 81, 127–134. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; A Walters, W.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A. FastTree 2–Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Lagkouvardos, I.; Fischer, S.; Kumar, N.; Clavel, T. Rhea: A transparent and modular R pipeline for microbial profiling based on 16S rRNA gene amplicons. PeerJ 2017, 5, e2836. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Genet. 2021, 19, 55–71. [Google Scholar] [CrossRef]
- Baumgart, D.C.; Sandborn, W.J. Crohn’s disease. Lancet 2012, 380, 1590–1605. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.W.; Li, D.Y.; Hazen, S.L. Dietary metabolism, the gut microbiome, and heart failure. Nat. Rev. Cardiol. 2019, 16, 137–154. [Google Scholar] [CrossRef]
- Khanna, S.; Raffals, L.E. The Microbiome in Crohn’s Disease: Role in Pathogenesis and Role of Microbiome Replacement Therapies. Gastroenterol. Clin. North. Am. 2017, 46, 481–492. [Google Scholar] [CrossRef]
- Liu, Y.; Defourny, K.A.Y.; Smid, E.J.; Abee, T. Gram-Positive Bacterial Extracellular Vesicles and Their Impact on Health and Disease. Front. Microbiol. 2018, 9, 1502. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.J.; Yang, J.; Seo, H.; Lee, W.H.; Lee, D.H.; Kym, S.; Park, Y.S.; Kim, J.G.; Jang, I.-J.; Kim, Y.-K.; et al. Colorectal cancer diagnostic model utilizing metagenomic and metabolomic data of stool microbial extracellular vesicles. Sci. Rep. 2020, 10, 2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; Choi, J.-P.; Yang, J.; Won, H.-K.; Park, C.S.; Song, W.-J.; Kwon, H.-S.; Kim, T.-B.; Kim, Y.-K.; Park, H.-S.; et al. Metagenome analysis using serum extracellular vesicles identified distinct microbiota in asthmatics. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Yang, J.; McDowell, A.; Kim, E.K.; Seo, H.; Yum, K.; Lee, W.H.; Jee, Y.K.; Kim, Y.K. Consumption of a Leuconostoc holzapfelii-enriched synbiotic beverage alters the composition of the microbiota and microbial extracellular vesicles. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costea, P.I.; Zeller, G.; Sunagawa, S.; Pelletier, E.; Alberti, A.; Levenez, F.; Tramontano, M.; Driessen, M.; Hercog, R.; Jung, F.-E.; et al. Towards standards for human fecal sample processing in metagenomic studies. Nat. Biotechnol. 2017, 35, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Lopetuso, L.R.; Petito, V.; Graziani, C.; Schiavoni, E.; Paroni Sterbini, F.; Poscia, A.; Gaetani, E.; Franceschi, F.; Cammarota, G.; Sanguinetti, M.; et al. Gut Microbiota in Health, Diverticular Disease, Irritable Bowel Syndrome, and Inflammatory Bowel Diseases: Time for Microbial Marker of Gastrointestinal Disorders? Dig. Dis. 2018, 36, 56–65. [Google Scholar] [CrossRef]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, X.; Wang, S.-L.; Zhao, S.-B.; Shi, Y.-H.; Pan, P.; Gu, L.; Yao, J.; Li, Z.-S.; Bai, Y. Extracellular Vesicles with Possible Roles in Gut Intestinal Tract Homeostasis and IBD. Mediat. Inflamm. 2020, 2020, 1945832. [Google Scholar] [CrossRef]
- Cecil, J.D.; Sirisaengtaksin, N.; O’Brien-Simpson, N.; Krachler, A.M. Outer Membrane Vesicle-Host Cell Interactions. Microbiol. Spectr. 2019, 7, 201–214. [Google Scholar] [CrossRef] [Green Version]
- Galazzo, G.; I Tedjo, D.; Wintjens, D.S.J.; Savelkoul, P.H.M.; Masclee, A.A.M.; Bodelier, A.G.L.; Pierik, M.J.; A E Jonkers, D.M.; Penders, J. Faecal Microbiota Dynamics and their Relation to Disease Course in Crohn’s Disease. J. Crohn’s Coliti 2019, 13, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.; Roy, B.C.; Khan, S.A.; Septer, S.; Umar, S. Microbiome, Metabolome and Inflammatory Bowel Disease. Microorganisms 2016, 4, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedjo, D.I.; Smolinska, A.B.; Savelkoul, P.H.; Masclee, A.A.; Van Schooten, F.J.; Pierik, M.J.; Penders, J.; Jonkers, D.M.A.E. The fecal microbiota as a biomarker for disease activity in Crohn’s disease. Sci. Rep. 2016, 6, 35216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef]
- Elian, S.; Souza, E.; Vieira, A.; Teixeira, M.; Arantes, R.; Nicoli, J.; Martins, F. Bifidobacterium longum subsp. infantis BB-02 attenuates acute murine experimental model of inflammatory bowel disease. Benef. Microbes 2015, 6, 277–286. [Google Scholar] [CrossRef]
- Bajer, L.; Kverka, M.; Kostovcik, M.; Macinga, P.; Dvorak, J.; Stehlikova, Z.; Brezina, J.; Wohl, P.; Spicak, J.; Drastich, P. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J. Gastroenterol. 2017, 23, 4548–4558. [Google Scholar] [CrossRef]
- von Schillde, M.-A.; Hörmannsperger, G.; Weiher, M.; Alpert, C.-A.; Hahne, H.; Bäuerl, C.; van Huynegem, K.; Steidler, L.; Hrncir, T.; Martinez, G.P.; et al. Lactocepin Secreted By Lactobacillus Exerts Anti-Inflammatory Effects By Selectively Degrading Proinflammatory Chemokines. Cell Host Microbe 2012, 11, 387–396. [Google Scholar] [CrossRef] [Green Version]
- Kaakoush, N.O. Sutterella Species, IgA-degrading Bacteria in Ulcerative Colitis. Trends Microbiol. 2020, 28, 519–522. [Google Scholar] [CrossRef]
- Santoru, M.L.; Piras, C.; Murgia, A.; Palmas, V.; Camboni, T.; Liggi, S.; Ibba, I.; Lai, M.A.; Orrù, S.; Blois, S.; et al. Cross sectional evaluation of the gut-microbiome metabolome axis in an Italian cohort of IBD patients. Sci. Rep. 2017, 7, 9523. [Google Scholar] [CrossRef]
- Rai, T.; Wu, X.; Shen, B. Frequency and risk factors of low immunoglobulin levels in patients with inflammatory bowel disease. Gastroenterol. Rep. 2015, 3, 115–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Humaran, L.G.B.; Gratadoux, J.-J.; Blugeon, S.; Bridonneau, C.; Furet, J.-P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Denman, S.E.; Morrison, M.; Yu, Z.T.; Dore, J.; Leclerc, M.; McSweeney, C.S. Dysbiosis of Fecal Microbiota in Crohn’s Disease Patients as Revealed by a Custom Phylogenetic Microarray. Inflamm. Bowel Dis. 2010, 16, 2034–2042. [Google Scholar] [CrossRef]
- Kaakoush, N.O. Insights into the Role of Erysipelotrichaceae in the Human Host. Front. Cell. Infect. Microbiol. 2015, 5, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, N.; Soergel, D.A.W.; Repo, S.; E Brenner, S. Association of gut microbiota with post-operative clinical course in Crohn’s disease. BMC Gastroenterol. 2013, 13, 131. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phylum | MAV of HC % | p-Value of fDNA/bDNA | p-Value of fDNA/MV-DNA | p-Value of bDNA/MV-DNA | ||
|---|---|---|---|---|---|---|
| fDNA | bDNA | MV-DNA | ||||
| p__Actinobacteria | 8.665 | 13.35 | 1 | 0.3254 | 0.0009 | 0.0001 |
| p__Bacteroidetes | 18.69 | 12.22 | 66.5 | 0.08 | 0.004 | 0.0001 |
| p__Firmicutes | 70 | 72.1 | 27 | 0.5 | 0.001 | 0.0001 |
| p__Proteobacteria | 1.707 | 1.013 | 5 | 0.2 | 0.9 | 0.3 |
| p__Euryarchaeota | 0.3482 | 0.3087 | 0.006617 | 0.8 | 0.22 | 0.04 |
| p__Fusobacteria | 0.003017 | 0.000227 | 0.001063 | 0.1 | 0.1 | 0.17 |
| p__Lentisphaerae | 0.01331 | 0.000988 | 0.08437 | 0.9 | 0.3 | 0.1 |
| p__Verrucomicrobia | 0.8032 | 0.7239 | 0.3012 | 0.8 | 0.22 | 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kameli, N.; Becker, H.E.F.; Welbers, T.; Jonkers, D.M.A.E.; Penders, J.; Savelkoul, P.; Stassen, F.R. Metagenomic Profiling of Fecal-Derived Bacterial Membrane Vesicles in Crohn’s Disease Patients. Cells 2021, 10, 2795. https://doi.org/10.3390/cells10102795
Kameli N, Becker HEF, Welbers T, Jonkers DMAE, Penders J, Savelkoul P, Stassen FR. Metagenomic Profiling of Fecal-Derived Bacterial Membrane Vesicles in Crohn’s Disease Patients. Cells. 2021; 10(10):2795. https://doi.org/10.3390/cells10102795
Chicago/Turabian StyleKameli, Nader, Heike E. F. Becker, Tessa Welbers, Daisy M. A. E. Jonkers, John Penders, Paul Savelkoul, and Frank R. Stassen. 2021. "Metagenomic Profiling of Fecal-Derived Bacterial Membrane Vesicles in Crohn’s Disease Patients" Cells 10, no. 10: 2795. https://doi.org/10.3390/cells10102795