
CRMP2 Is Involved in Regulation of Mitochondrial Morphology and Motility in Neurons


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Isolation and Purification of Brain Mitochondria
2.3. Cell Culture
2.4. Immunocytochemistry
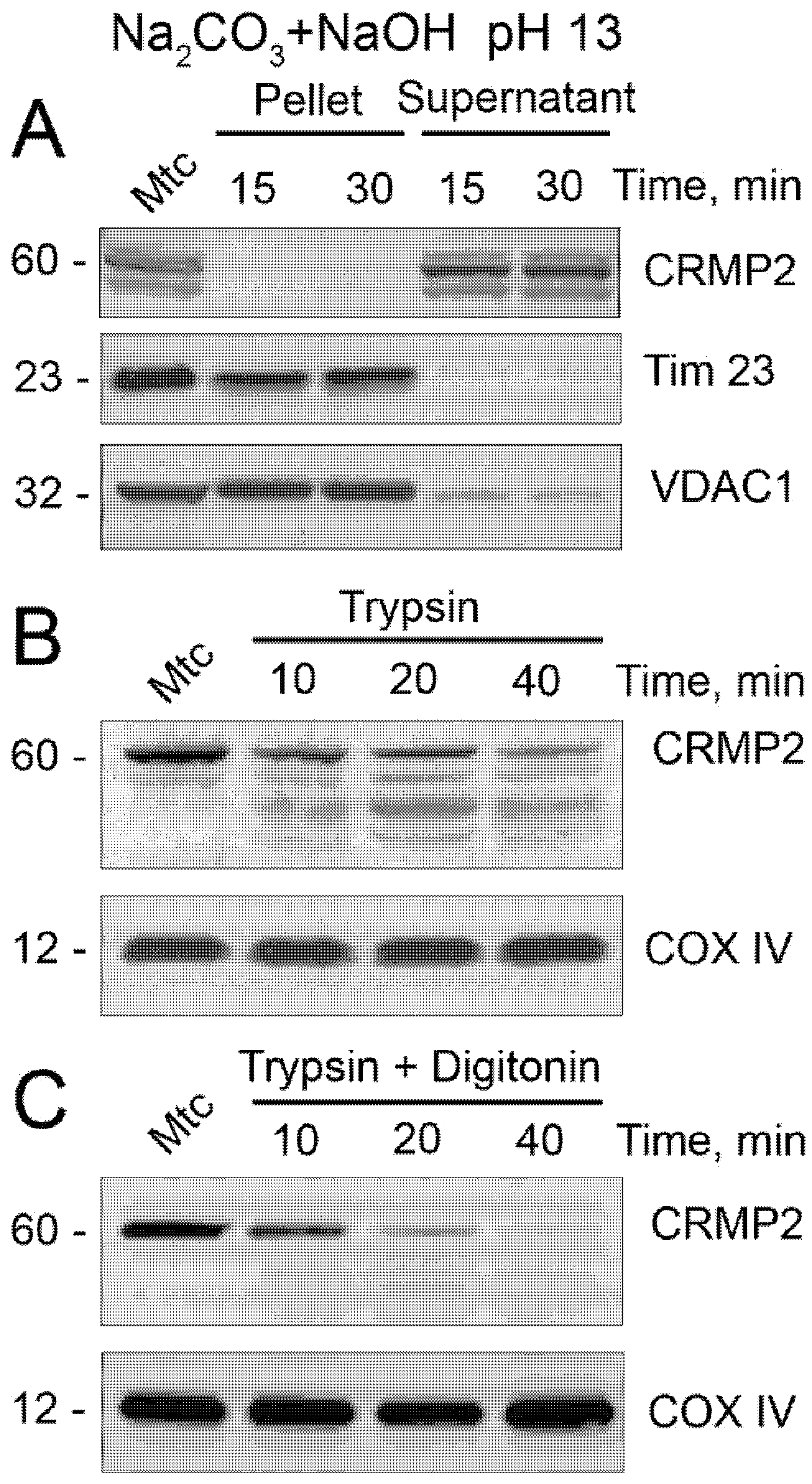
2.5. Alkali and Trypsin Treatment
2.6. Immunoblotting
2.7. Co-Immunoprecipitation
2.8. Neuronal Transfection
2.9. Mitochondrial Morphology
2.10. Mitochondrial Motility
2.11. Statistics
3. Results
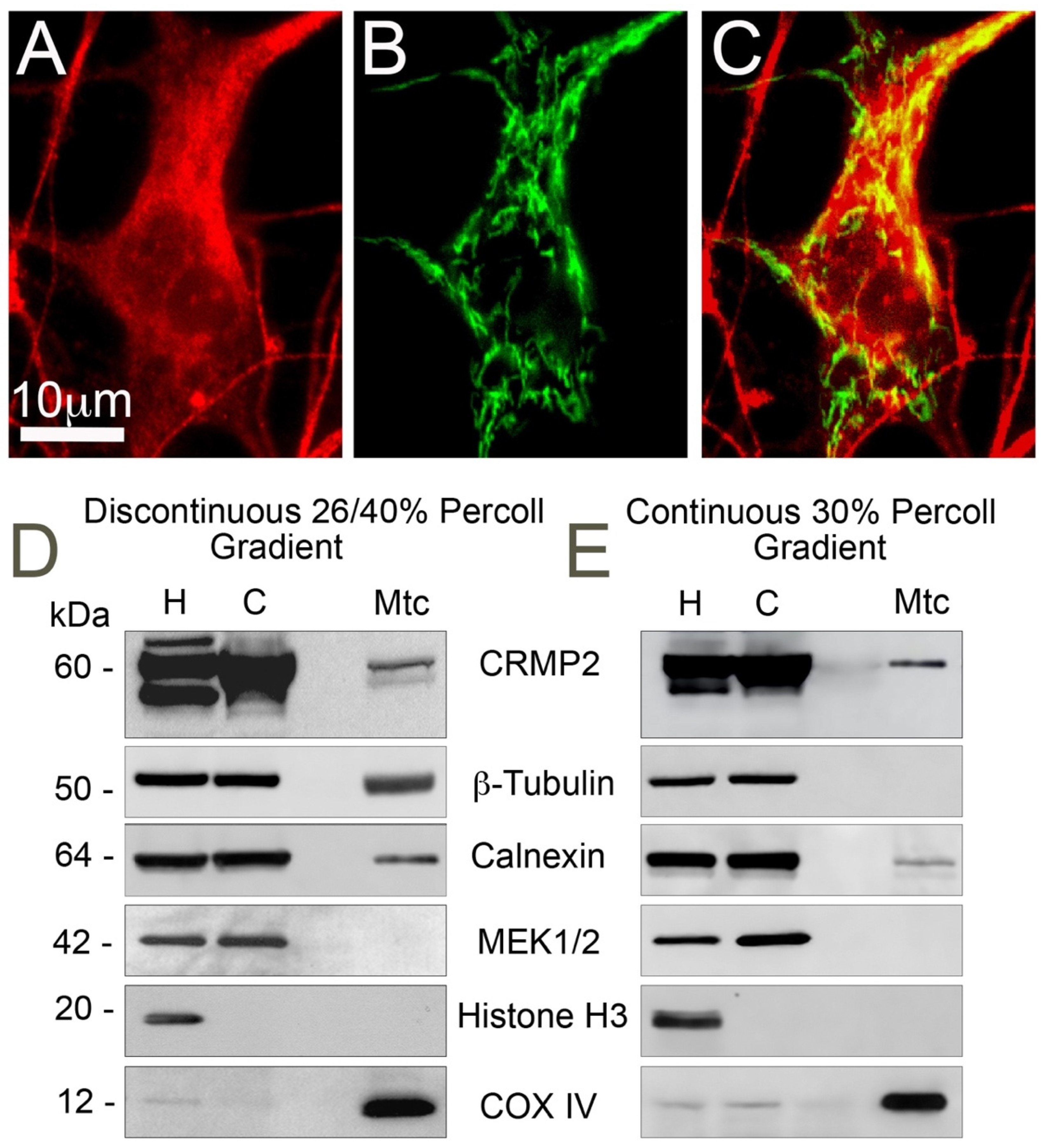
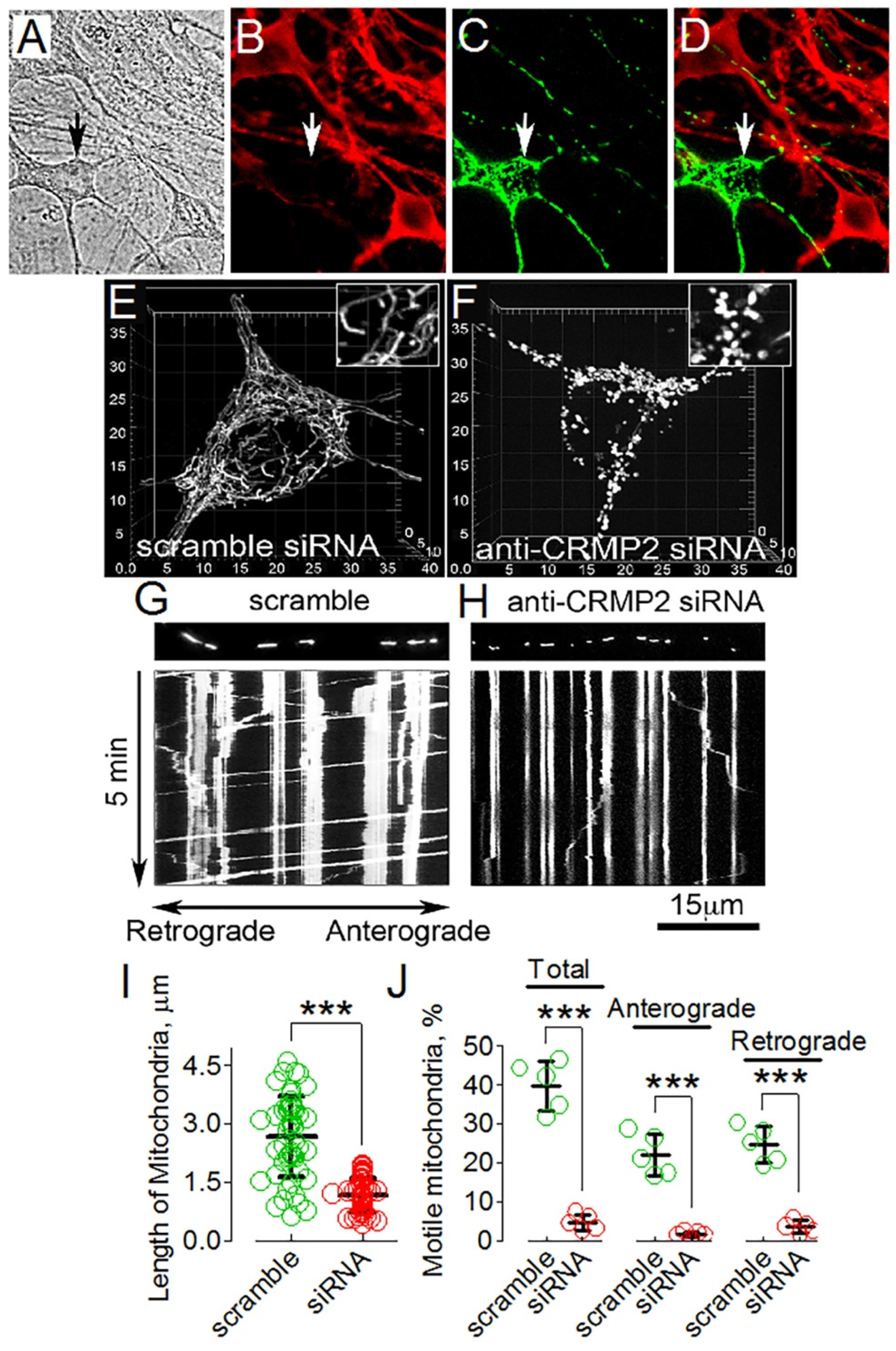
3.1. CRMP2 Localization on Mitochondria
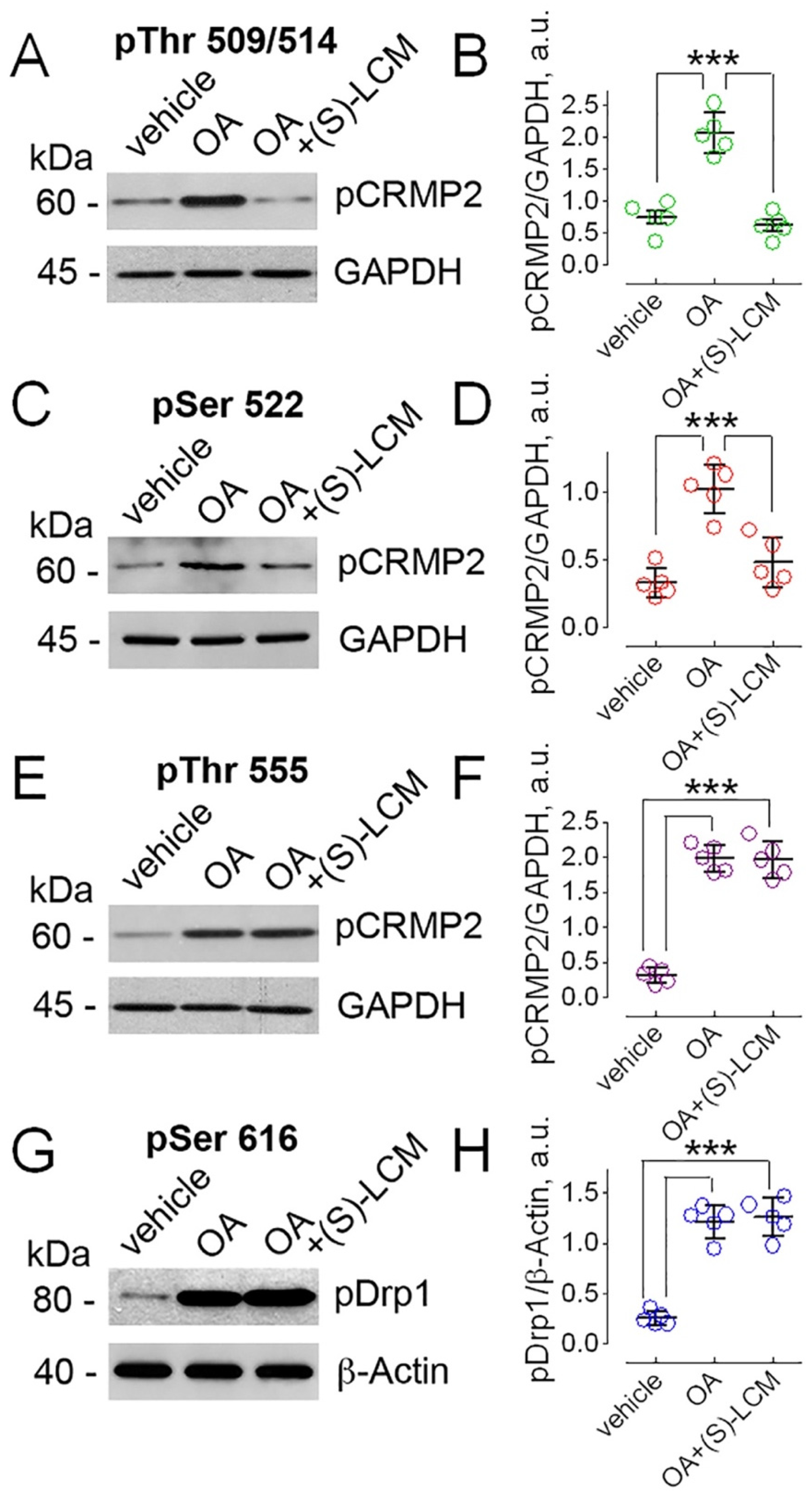
3.2. Manipulations with CRMP2 Phosphorylation
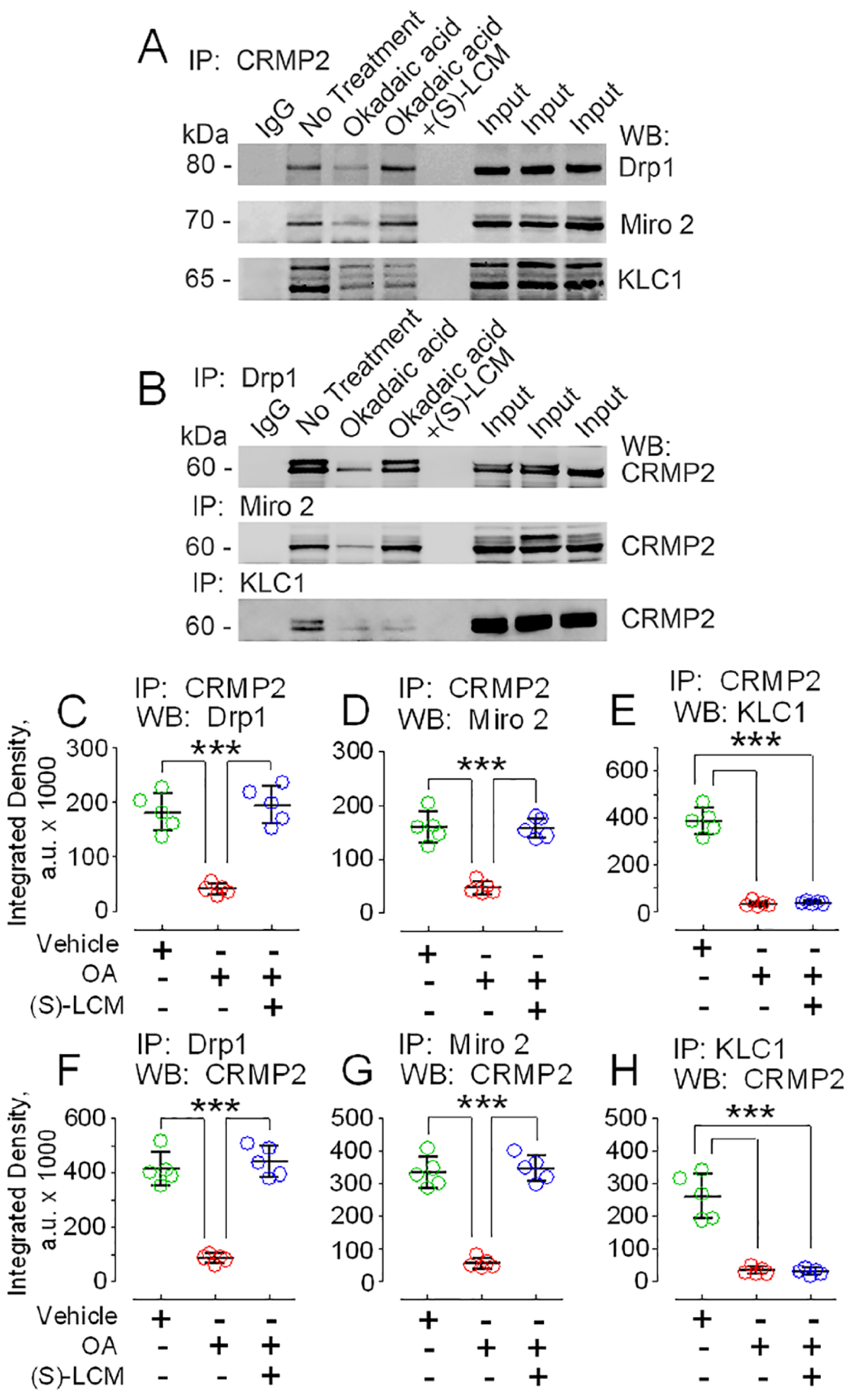
3.3. CRMP2 and Proteins Involved in Regulating Mitochondrial Morphology and Motility
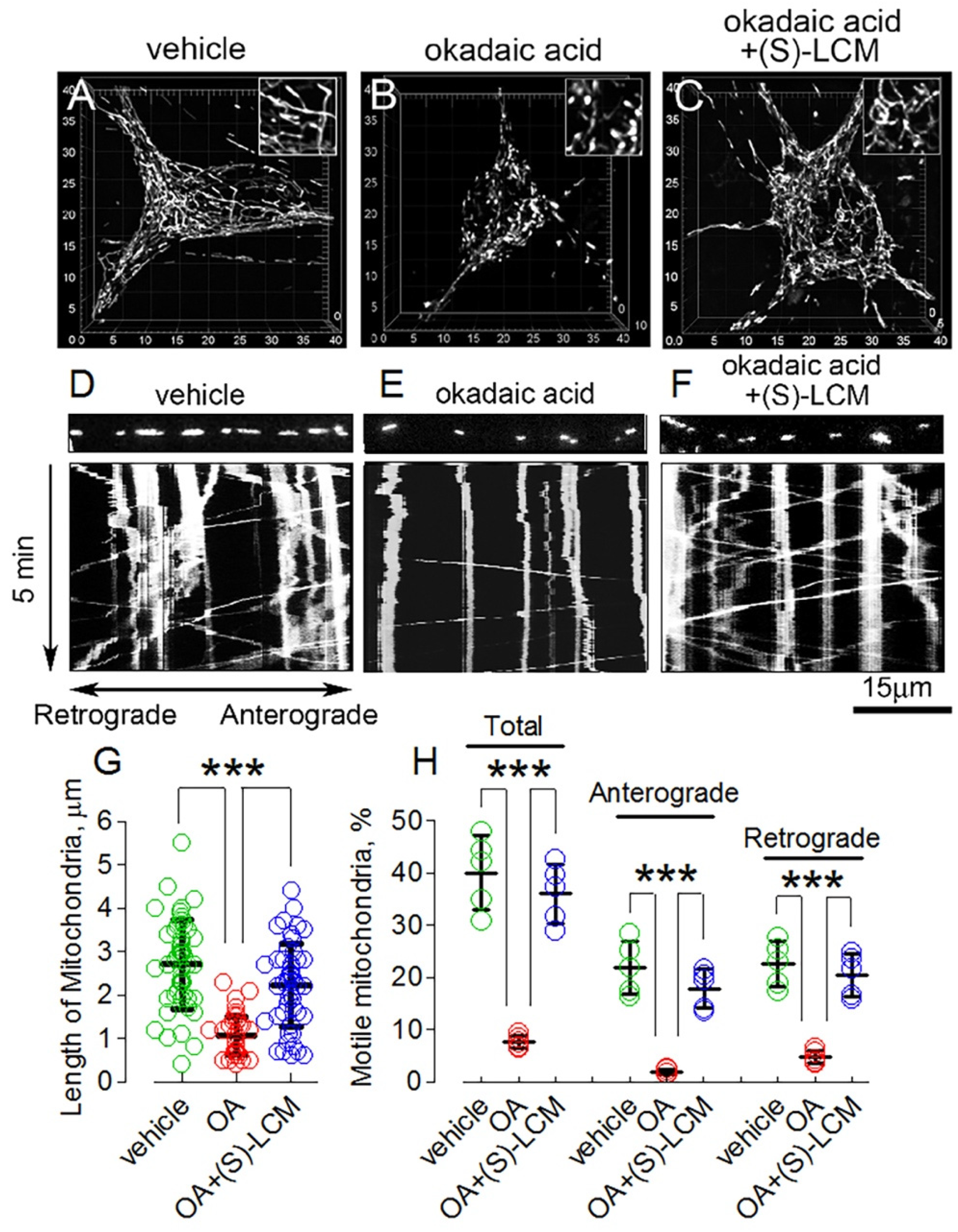
3.4. CRMP2 Phosphorylation State and Alterations in Mitochondrial Morphology and Motility
3.5. CRMP2 Deletion Correlates with Alterations in Mitochondrial Morphology and Motility
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, H.; Chan, D.C. Mitochondrial dynamics--fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef] [PubMed]
- Bossy-Wetzel, E.; Petrilli, A.; Knott, A.B. Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci. 2008, 31, 609–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, D.H.; Nakamura, T.; Lipton, S.A. Mitochondrial dynamics in cell death and neurodegeneration. Cell Mol. Life Sci. 2010, 67, 3435–3447. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Strittmatter, S.M. A family of rat CRMP genes is differentially expressed in the nervous system. J. Neurosci. 1996, 16, 6197–6207. [Google Scholar] [CrossRef] [PubMed]
- Goshima, Y.; Nakamura, F.; Strittmatter, P.; Strittmatter, S.M. Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC. Nature 1995, 376, 509–514. [Google Scholar] [CrossRef]
- Hensley, K.; Venkova, K.; Christov, A.; Gunning, W.; Park, J. Collapsin response mediator protein-2: An emerging pathologic feature and therapeutic target for neurodisease indications. Mol. Neurobiol. 2011, 43, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Khanna, R.; Wilson, S.M.; Brittain, J.M.; Weimer, J.; Sultana, R.; Butterfield, A.; Hensley, K. Opening Pandora’s jar: A primer on the putative roles of CRMP2 in a panoply of neurodegenerative, sensory and motor neuron, and central disorders. Future Neurol. 2012, 7, 749–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charrier, E.; Reibel, S.; Rogemond, V.; Aguera, M.; Thomasset, N.; Honnorat, J. Collapsin response mediator proteins (CRMPs): Involvement in nervous system development and adult neurodegenerative disorders. Mol. Neurobiol. 2003, 28, 51–64. [Google Scholar] [CrossRef]
- Brustovetsky, T.; Pellman, J.J.; Yang, X.F.; Khanna, R.; Brustovetsky, N. Collapsin response mediator protein 2 (CRMP2) interacts with N-methyl-D-aspartate (NMDA) receptor and Na+/Ca2+ exchanger and regulates their functional activity. J. Biol. Chem. 2014, 289, 7470–7482. [Google Scholar] [CrossRef] [Green Version]
- Moutal, A.; White, K.A.; Chefdeville, A.; Laufmann, R.N.; Vitiello, P.F.; Feinstein, D.; Weimer, J.M.; Khanna, R. Dysregulation of CRMP2 Post-Translational Modifications Drive Its Pathological Functions. Mol. Neurobiol. 2019, 56, 6736–6755. [Google Scholar] [CrossRef]
- Kimura, T.; Watanabe, H.; Iwamatsu, A.; Kaibuchi, K. Tubulin and CRMP-2 complex is transported via Kinesin. J. Neurochem. 2005, 93, 1371–1382. [Google Scholar] [CrossRef]
- Arimura, N.; Hattori, A.; Kimura, T.; Nakamuta, S.; Funahashi, Y.; Hirotsune, S.; Furuta, K.; Urano, T.; Toyoshima, Y.Y.; Kaibuchi, K. CRMP-2 directly binds to cytoplasmic dynein and interferes with its activity. J. Neurochem. 2009, 111, 380–390. [Google Scholar] [CrossRef]
- Yoshimura, T.; Kawano, Y.; Arimura, N.; Kawabata, S.; Kikuchi, A.; Kaibuchi, K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 2005, 120, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Cole, A.R.; Noble, W.; van Aalten, L.; Plattner, F.; Meimaridou, R.; Hogan, D.; Taylor, M.; LaFrancois, J.; Gunn-Moore, F.; Verkhratsky, A.; et al. Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer’s disease progression. J. Neurochem. 2007, 103, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Arimura, N.; Inagaki, N.; Chihara, K.; Menager, C.; Nakamura, N.; Amano, M.; Iwamatsu, A.; Goshima, Y.; Kaibuchi, K. Phosphorylation of collapsin response mediator protein-2 by Rho-kinase. Evidence for two separate signaling pathways for growth cone collapse. J. Biol. Chem. 2000, 275, 23973–23980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arimura, N.; Menager, C.; Kawano, Y.; Yoshimura, T.; Kawabata, S.; Hattori, A.; Fukata, Y.; Amano, M.; Goshima, Y.; Inagaki, M.; et al. Phosphorylation by Rho kinase regulates CRMP-2 activity in growth cones. Mol. Cell Biol. 2005, 25, 9973–9984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, K.A.; Pimplikar, S.W. Activation of GSK-3 and phosphorylation of CRMP2 in transgenic mice expressing APP intracellular domain. J. Cell Biol. 2005, 171, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Uchida, Y.; Ohshima, T.; Sasaki, Y.; Suzuki, H.; Yanai, S.; Yamashita, N.; Nakamura, F.; Takei, K.; Ihara, Y.; Mikoshiba, K.; et al. Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: Implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes Cells 2005, 10, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.R.; Causeret, F.; Yadirgi, G.; Hastie, C.J.; McLauchlan, H.; McManus, E.J.; Hernandez, F.; Eickholt, B.J.; Nikolic, M.; Sutherland, C. Distinct priming kinases contribute to differential regulation of collapsin response mediator proteins by glycogen synthase kinase-3 in vivo. J. Biol. Chem. 2006, 281, 16591–16598. [Google Scholar] [CrossRef] [Green Version]
- Astle, M.V.; Ooms, L.M.; Cole, A.R.; Binge, L.C.; Dyson, J.M.; Layton, M.J.; Petratos, S.; Sutherland, C.; Mitchell, C.A. Identification of a proline-rich inositol polyphosphate 5-phosphatase (PIPP)*collapsin response mediator protein 2 (CRMP2) complex that regulates neurite elongation. J. Biol. Chem. 2011, 286, 23407–23418. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.Q.; Zheng, H.Y.; Peng, C.X.; Liu, D.; Li, H.L.; Wang, Q.; Wang, J.Z. Protein phosphatase 2A facilitates axonogenesis by dephosphorylating CRMP. J. Neurosci. 2010, 30, 3839–3848. [Google Scholar] [CrossRef] [Green Version]
- Mokhtar, S.H.; Kim, M.J.; Magee, K.A.; Aui, P.M.; Thomas, S.; Bakhuraysah, M.M.; Alrehaili, A.A.; Lee, J.Y.; Steer, D.L.; Kenny, R.; et al. Amyloid-beta-dependent phosphorylation of collapsin response mediator protein-2 dissociates kinesin in Alzheimer’s disease. Neural Regen. Res. 2018, 13, 1066–1080. [Google Scholar]
- Kim, J.; Moody, J.P.; Edgerly, C.K.; Bordiuk, O.L.; Cormier, K.; Smith, K.; Beal, M.F.; Ferrante, R.J. Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum. Mol. Genet. 2010, 19, 3919–3935. [Google Scholar] [CrossRef]
- Costa, V.; Giacomello, M.; Hudec, R.; Lopreiato, R.; Ermak, G.; Lim, D.; Malorni, W.; Davies, K.J.; Carafoli, E.; Scorrano, L. Mitochondrial fission and cristae disruption increase the response of cell models of Huntington’s disease to apoptotic stimuli. EMBO Mol. Med. 2010, 2, 490–503. [Google Scholar] [CrossRef]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Zhou, Y.; Poquiz, P.; Tjong, J.; Pouladi, M.A.; Hayden, M.R.; et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 2011, 17, 377–382. [Google Scholar] [CrossRef] [Green Version]
- Shirendeb, U.; Reddy, A.P.; Manczak, M.; Calkins, M.J.; Mao, P.; Tagle, D.A.; Reddy, P.H. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: Implications for selective neuronal damage. Hum. Mol. Genet. 2011, 20, 1438–1455. [Google Scholar] [CrossRef] [Green Version]
- Trushina, E.; Dyer, R.B.; Badger, J.D.; Ure, D.; Eide, L.; Tran, D.D.; Vrieze, B.T.; Legendre-Guillemin, V.; McPherson, P.S.; Mandavilli, B.S.; et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell Biol. 2004, 24, 8195–8209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, D.T.; Rintoul, G.L.; Pandipati, S.; Reynolds, I.J. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol. Dis. 2006, 22, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.L.; Li, S.; Wang, C.E.; Li, H.; Wang, J.; Rong, J.; Xu, X.; Mastroberardino, P.G.; Greenamyre, J.T.; Li, X.J. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 2008, 28, 2783–2792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum. Mol. Genet. 2012, 21, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Williamson, R.; van Aalten, L.; Mann, D.M.; Platt, B.; Plattner, F.; Bedford, L.; Mayer, J.; Howlett, D.; Usardi, A.; Sutherland, C.; et al. CRMP2 hyperphosphorylation is characteristic of Alzheimer’s disease and not a feature common to other neurodegenerative diseases. J. Alzheimers. Dis. 2011, 27, 615–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef] [PubMed]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef] [PubMed]
- Rembutsu, M.; Soutar, M.P.; van Aalten, L.; Gourlay, R.; Hastie, C.J.; McLauchlan, H.; Morrice, N.A.; Cole, A.R.; Sutherland, C. Novel procedure to investigate the effect of phosphorylation on protein complex formation in vitro and in cells. Biochemistry 2008, 47, 2153–2161. [Google Scholar] [CrossRef]
- Hamilton, J.; Brustovetsky, T.; Brustovetsky, N. Mutant huntingtin fails to directly impair brain mitochondria. J. Neurochem. 2019, 151, 716–731. [Google Scholar] [CrossRef]
- Lai, J.C.K.; Clark, J.B.; Boulton, A.A.; Baker, G.B.; Butterworth, R.F. (Eds.) Neuromethods; Humana Press: Clifton, NJ, USA, 1989; pp. 43–98. [Google Scholar]
- Noterman, M.F.; Chaubey, K.; Lin-Rahardja, K.; Rajadhyaksha, A.M.; Pieper, A.A.; Taylor, E.B. Dual-process brain mitochondria isolation preserves function and clarifies protein composition. PNAS 2021, 118, 2019046118. [Google Scholar] [CrossRef]
- Dubinsky, J.M. Intracellular calcium levels during the period of delayed excitotoxicity. J. Neurosci. 1993, 13, 623–631. [Google Scholar] [CrossRef] [Green Version]
- Griparic, L.; van der Bliek, A.M. Assay and properties of the mitochondrial dynamin related protein Opa1. Methods Enzymol. 2005, 404, 620–631. [Google Scholar]
- Brustovetsky, N.; Jemmerson, R.; Dubinsky, J.M. Calcium-induced Cytochrome c release from rat brain mitochondria is altered by digitonin. Neurosci. Lett. 2002, 332, 91–94. [Google Scholar] [CrossRef]
- Brustovetsky, T.; Bolshakov, A.; Brustovetsky, N. Calpain activation and Na(+)/Ca(2+) exchanger degradation occur downstream of calcium deregulation in hippocampal neurons exposed to excitotoxic glutamate. J. Neurosci. Res. 2010, 88, 1317–1328. [Google Scholar]
- Brittain, J.M.; Chen, L.; Wilson, S.M.; Brustovetsky, T.; Gao, X.; Ashpole, N.M.; Molosh, A.I.; You, H.; Hudmon, A.; Shekhar, A.; et al. Neuroprotection against traumatic brain injury by a peptide derived from the collapsin response mediator protein 2 (CRMP2). J. Biol. Chem. 2011, 286, 37778–37792. [Google Scholar] [CrossRef] [Green Version]
- Chi, X.X.; Schmutzler, B.S.; Brittain, J.M.; Wang, Y.; Hingtgen, C.M.; Nicol, G.D.; Khanna, R. Regulation of N-type voltage-gated calcium channels (Cav2.2) and transmitter release by collapsin response mediator protein-2 (CRMP-2) in sensory neurons. J. Cell Sci. 2009, 122, 4351–4362. [Google Scholar] [CrossRef] [Green Version]
- Brustovetsky, T.; Li, V.; Brustovetsky, N. Stimulation of glutamate receptors in cultured hippocampal neurons causes Ca2+-dependent mitochondrial contraction. Cell Calcium 2009, 46, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukata, Y.; Itoh, T.J.; Kimura, T.; Menager, C.; Nishimura, T.; Shiromizu, T.; Watanabe, H.; Inagaki, N.; Iwamatsu, A.; Hotani, H.; et al. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat. Cell Biol. 2002, 4, 583–591. [Google Scholar] [CrossRef]
- Pellman, J.J.; Hamilton, J.; Brustovetsky, T.; Brustovetsky, N. Ca(2+) handling in isolated brain mitochondria and cultured neurons derived from the YAC128 mouse model of Huntington’s disease. J. Neurochem. 2015, 134, 652–667. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.; Pellman, J.J.; Brustovetsky, T.; Harris, R.A.; Brustovetsky, N. Oxidative metabolism in YAC128 mouse model of Huntington’s disease. Hum. Mol. Genet. 2015, 24, 4862–4878. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, H.; Martin, B.L.; Brautigan, D.L.; Karaki, H.; Ozaki, H.; Kato, Y.; Fusetani, N.; Watabe, S.; Hashimoto, K.; Uemura, D.; et al. Calyculin A and okadaic acid: Inhibitors of protein phosphatase activity. Biochem. Biophys. Res. Commun. 1989, 159, 871–877. [Google Scholar] [CrossRef]
- Haystead, T.A.; Sim, A.T.; Carling, D.; Honnor, R.C.; Tsukitani, Y.; Cohen, P.; Hardie, D.G. Effects of the tumour promoter okadaic acid on intracellular protein phosphorylation and metabolism. Nature 1989, 337, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.W.; Yoon, S.Y.; Choi, J.E.; Kim, S.M.; Lee, H.S.; Choe, H.; Lee, S.C.; Kim, D.H. Maintained activity of glycogen synthase kinase-3beta despite of its phosphorylation at serine-9 in okadaic acid-induced neurodegenerative model. Biochem. Biophys. Res. Commun. 2010, 395, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Kim, D.H.; Choi, J.E.; Chang, E.J.; Seung, Y. Increased phosphorylation of dynamin-related protein 1 and mitochondrial fission in okadaic acid-treated neurons. Brain Res. 2012, 1454, 100–110. [Google Scholar] [CrossRef]
- Wilson, S.M.; Moutal, A.; Melemedjian, O.K.; Wang, Y.; Ju, W.; Francois-Moutal, L.; Khanna, M.; Khanna, R. The functionalized amino acid (S)-Lacosamide subverts CRMP2-mediated tubulin polymerization to prevent constitutive and activity-dependent increase in neurite outgrowth. Front. Cell Neurosci. 2014, 8, 196. [Google Scholar] [CrossRef] [Green Version]
- Moutal, A.; Francois-Moutal, L.; Perez-Miller, S.; Cottier, K.; Chew, L.A.; Yeon, S.K.; Dai, J.; Park, K.D.; Khanna, M.; Khanna, R. (S)-Lacosamide Binding to Collapsin Response Mediator Protein 2 (CRMP2) Regulates CaV2.2 Activity by Subverting Its Phosphorylation by Cdk5. Mol. Neurobiol. 2016, 53, 1959–1976. [Google Scholar] [CrossRef]
- Brot, S.; Smaoune, H.; Youssef-Issa, M.; Malleval, C.; Benetollo, C.; Besancon, R.; Auger, C.; Moradi-Ameli, M.; Honnorat, J. Collapsin response-mediator protein 5 (CRMP5) phosphorylation at threonine 516 regulates neurite outgrowth inhibition. Eur. J. Neurosci. 2014, 40, 3010–3020. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Taivassalo, T.; Gouspillou, G.; Hepple, R.T. Mitochondria: Isolation, structure and function. J. Physiol 2011, 589, 4413–4421. [Google Scholar] [CrossRef]
- Yamashita, N.; Goshima, Y. Collapsin response mediator proteins regulate neuronal development and plasticity by switching their phosphorylation status. Mol. Neurobiol. 2012, 45, 234–246. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, Z.-H. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J. Cell Biol. 2014, 204, 1087–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glater, E.E.; Megeath, L.J.; Stowers, R.S.; Schwarz, T.L. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol. 2006, 173, 545–557. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.H.; Lin, C.C.; Yang, M.C.; Wei, C.C.; Liao, H.D.; Lin, R.C.; Tu, W.Y.; Kao, T.C.; Hsu, C.M.; Cheng, J.T.; et al. GSK3beta-mediated Drp1 phosphorylation induced elongated mitochondrial morphology against oxidative stress. PLoS ONE 2012, 7, 49112. [Google Scholar] [CrossRef]
- Cho, B.; Cho, H.M.; Kim, H.J.; Jeong, J.; Park, S.K.; Hwang, E.M.; Park, J.Y.; Kim, W.R.; Kim, H.; Sun, W. CDK5-dependent inhibitory phosphorylation of Drp1 during neuronal maturation. Exp. Mol. Med. 2014, 46, 105. [Google Scholar] [CrossRef] [Green Version]
- Jahani-Asl, A.; Huang, E.; Irrcher, I.; Rashidian, J.; Ishihara, N.; Lagace, D.C.; Slack, R.S.; Park, D.S. CDK5 phosphorylates DRP1 and drives mitochondrial defects in NMDA-induced neuronal death. Hum. Mol. Genet. 2015, 24, 4573–4583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, M.H.; Wu, C.C.; Chan, W.H.; Chien, K.Y.; Yu, J.S. GSK-3 mediates the okadaic acid-induced modification of collapsin response mediator protein-2 in human SK-N-SH neuroblastoma cells. J. Cell Biochem. 2008, 103, 1833–1848. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brustovetsky, T.; Khanna, R.; Brustovetsky, N. CRMP2 Is Involved in Regulation of Mitochondrial Morphology and Motility in Neurons. Cells 2021, 10, 2781. https://doi.org/10.3390/cells10102781
Brustovetsky T, Khanna R, Brustovetsky N. CRMP2 Is Involved in Regulation of Mitochondrial Morphology and Motility in Neurons. Cells. 2021; 10(10):2781. https://doi.org/10.3390/cells10102781
Chicago/Turabian StyleBrustovetsky, Tatiana, Rajesh Khanna, and Nickolay Brustovetsky. 2021. "CRMP2 Is Involved in Regulation of Mitochondrial Morphology and Motility in Neurons" Cells 10, no. 10: 2781. https://doi.org/10.3390/cells10102781