
The Pathophysiological Role of Heat Shock Response in Autoimmunity: A Literature Review


Abstract
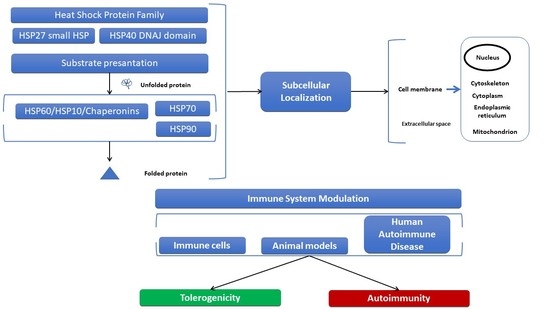
:
1. Introduction
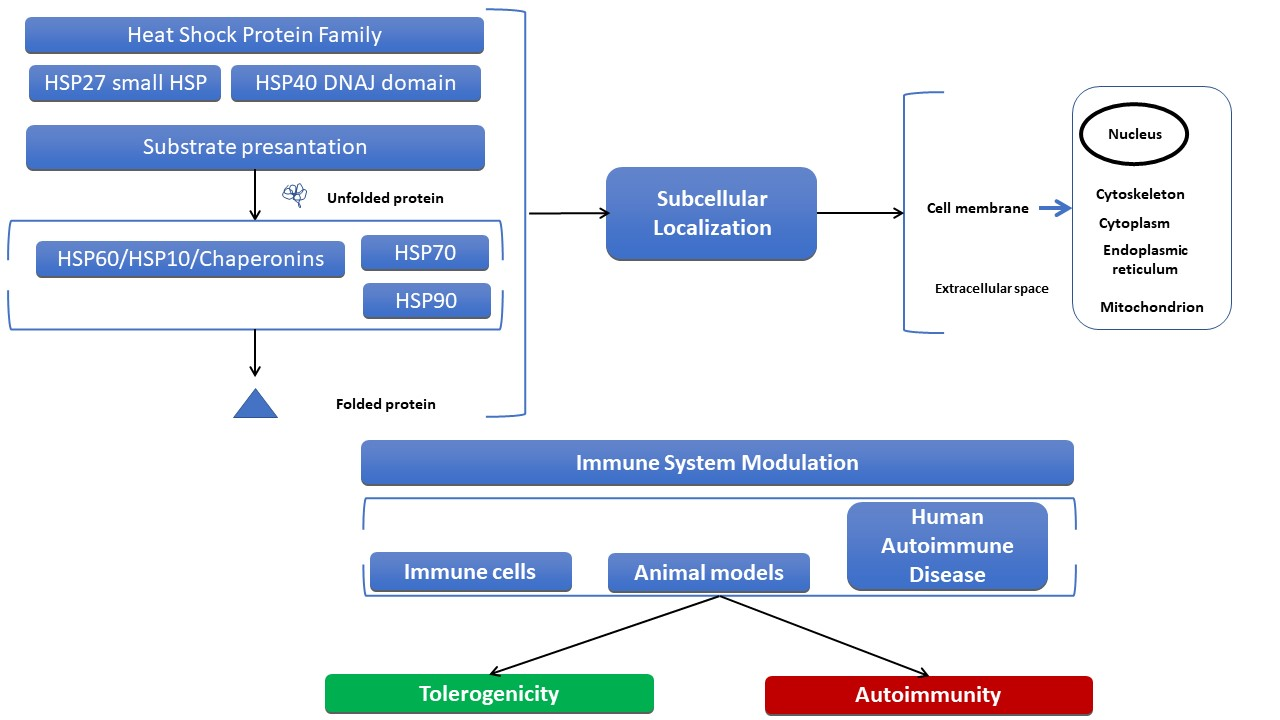
2. Structural Characteristics, Subcellular Localization of HSPs, and Elicited Immune Responses
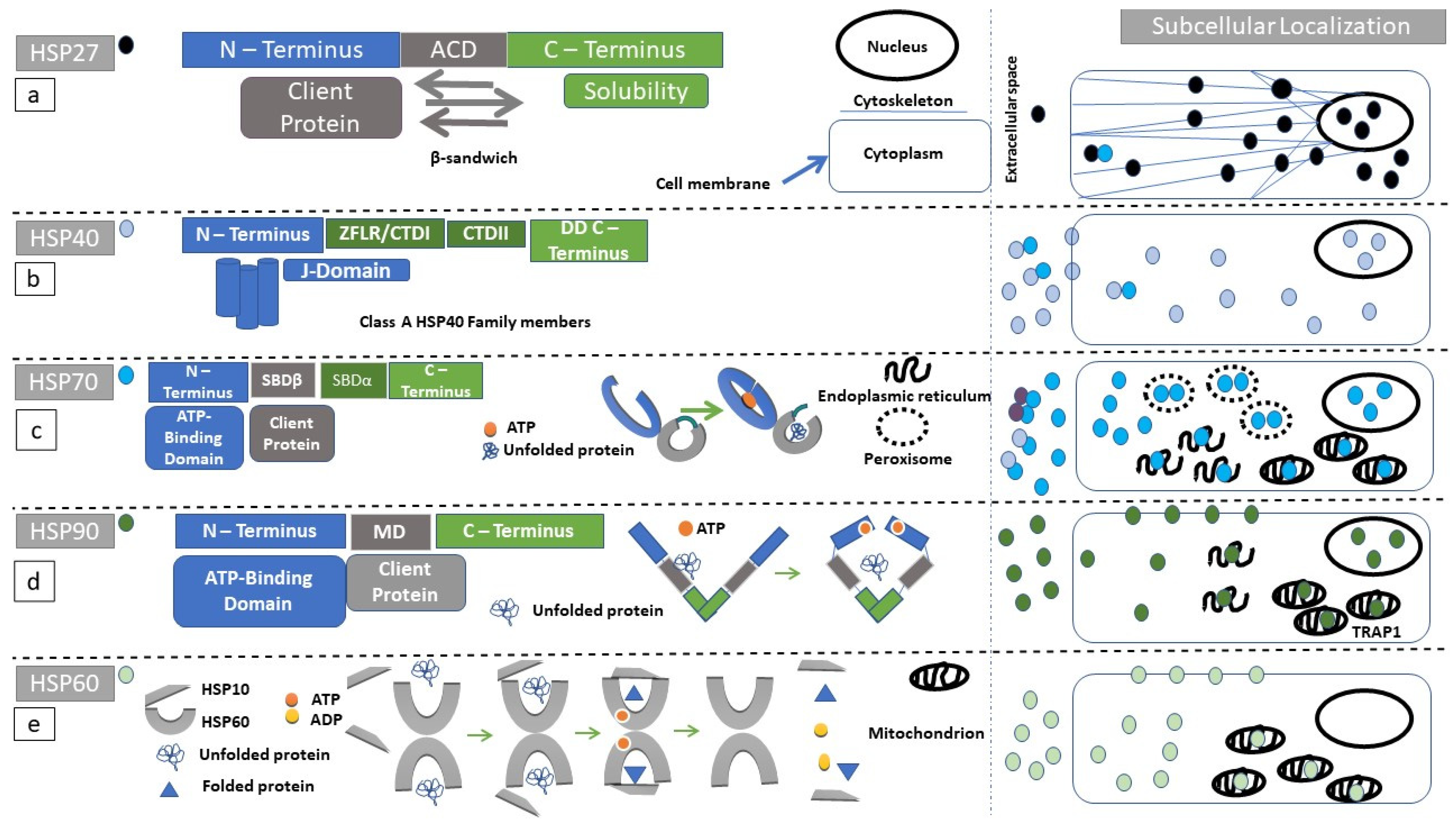
2.1. Structure and Subcellular Localization of the Small HSP Family
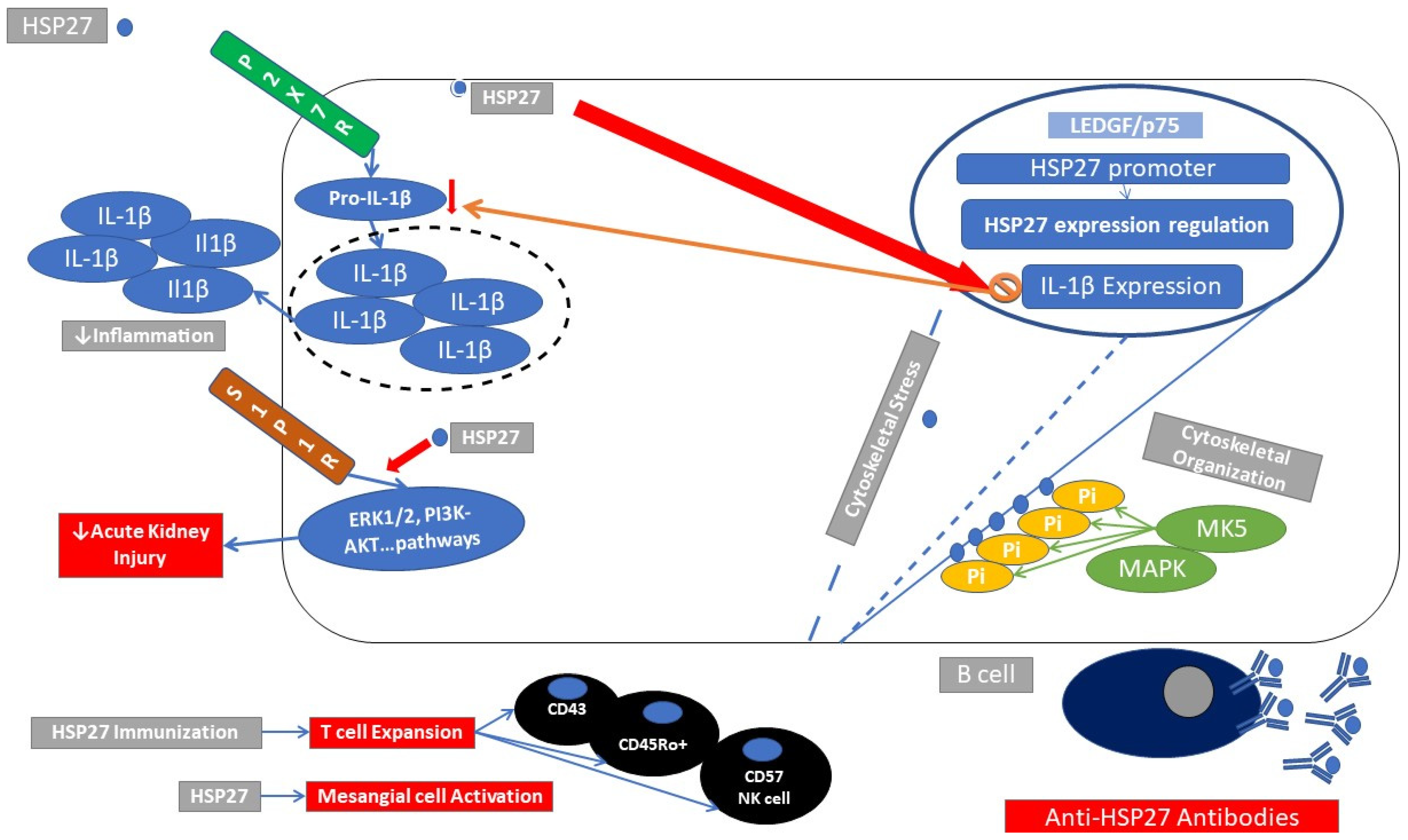
2.2. Immune Response Elicited through HSP27
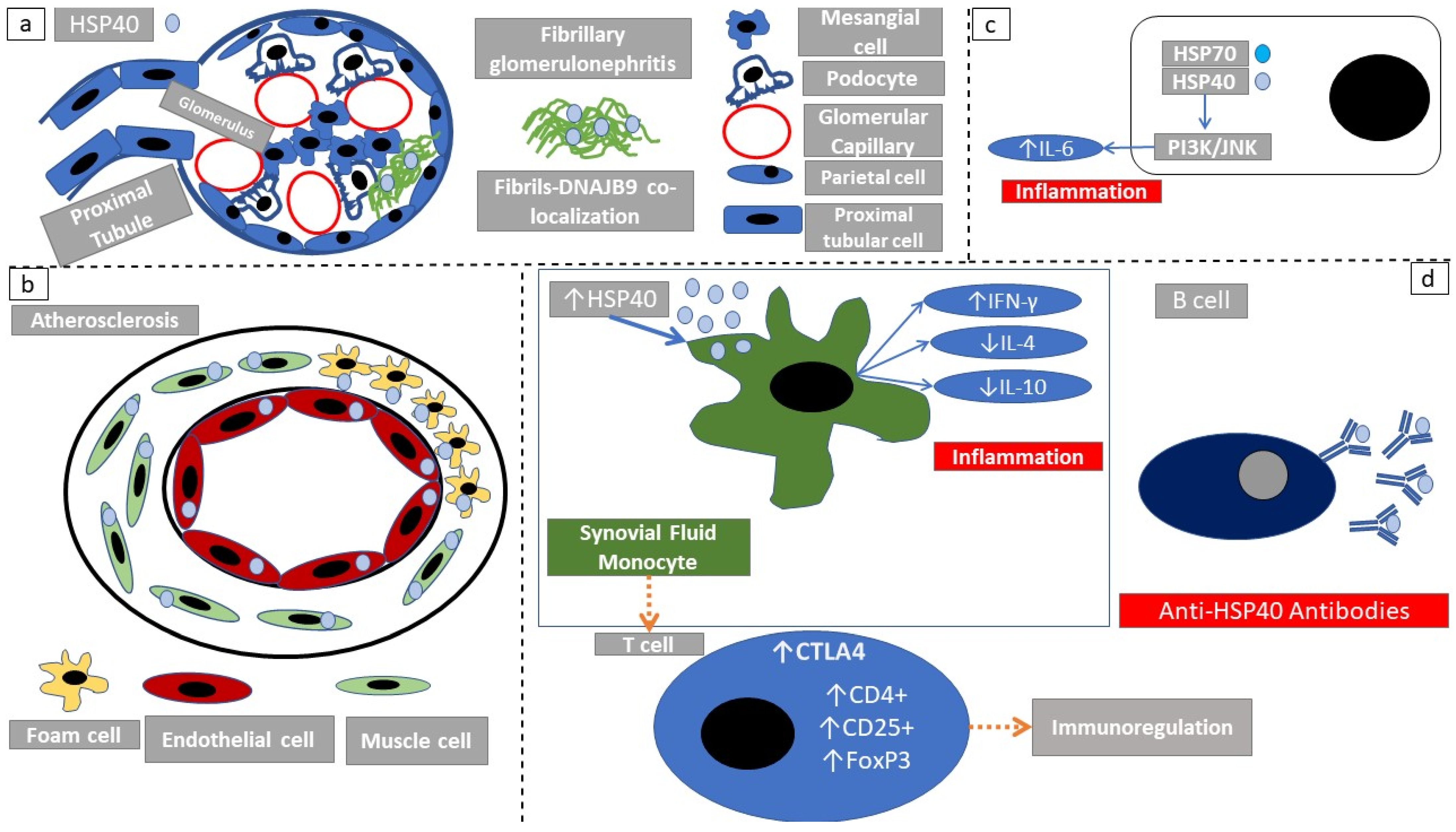
2.3. Structure and Subcellular Localization of HSP40 Family Members
2.4. Immune Response Elicited through HSP40
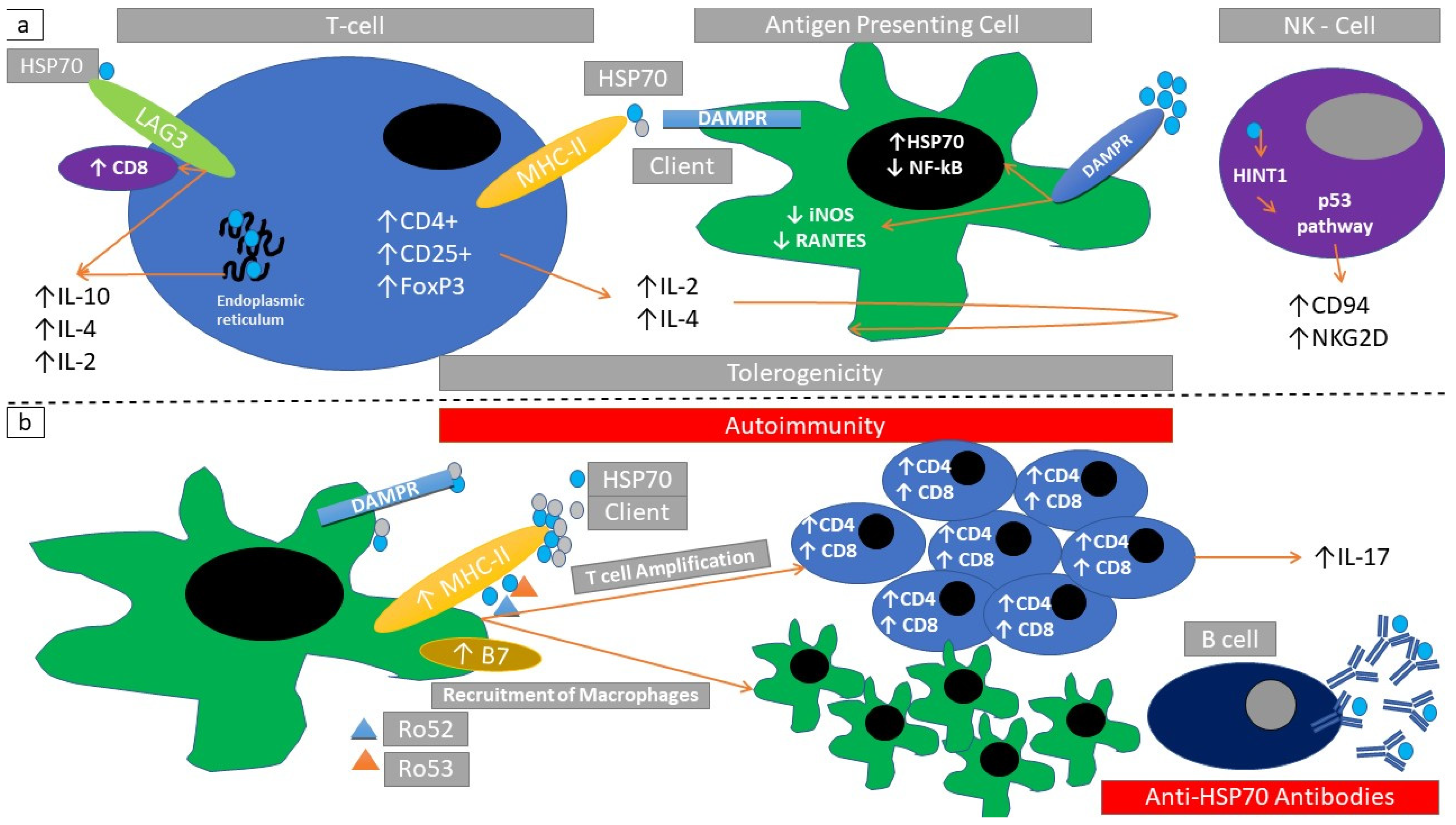
2.5. Structure and Subcellular Localization of HSP70 Superfamily Members
2.6. Immune Response Elicited through HSP70
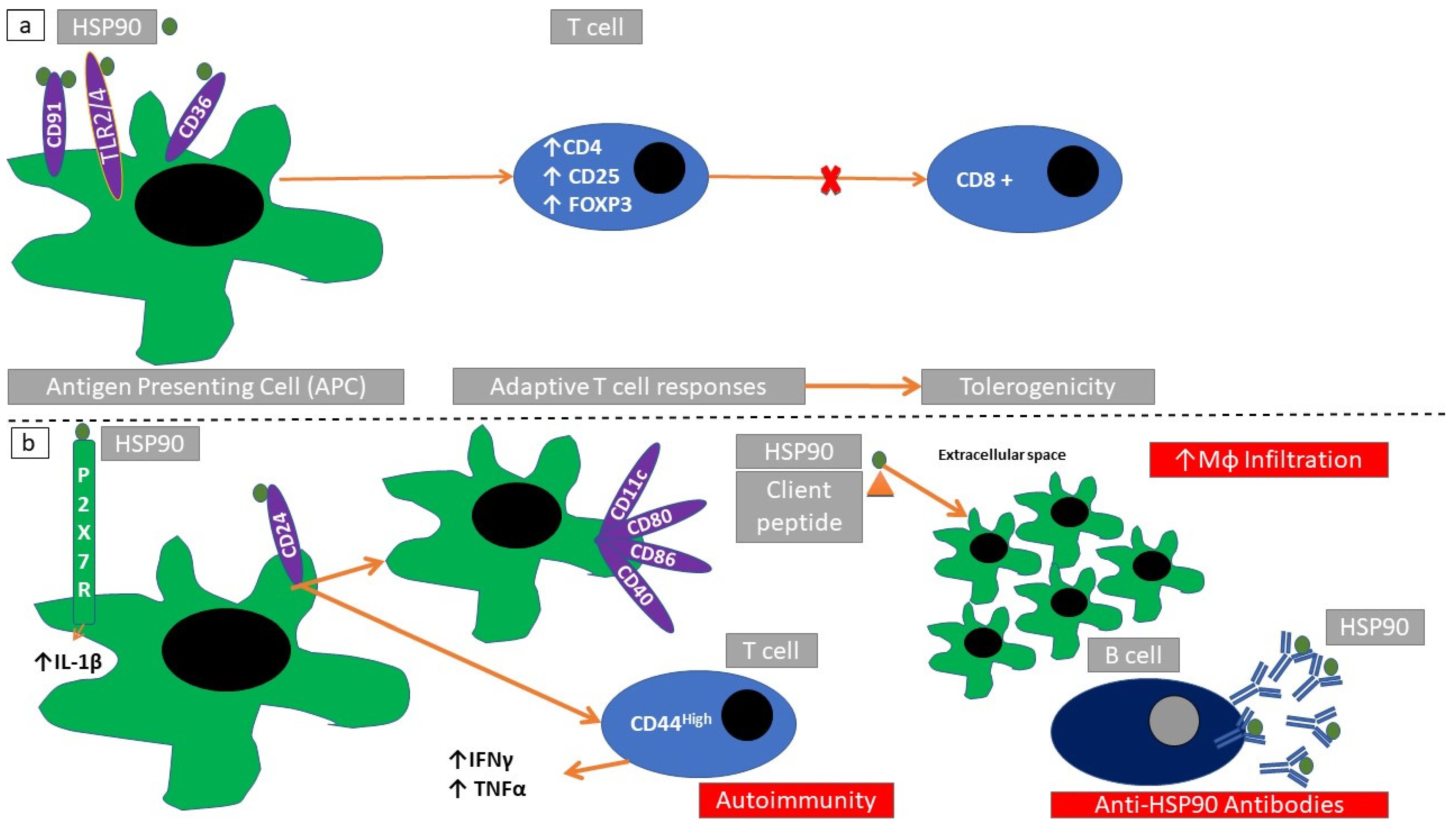
2.7. Structure and Subcellular Localization of HSP90
2.8. Immune Responses Elicited through HSP90
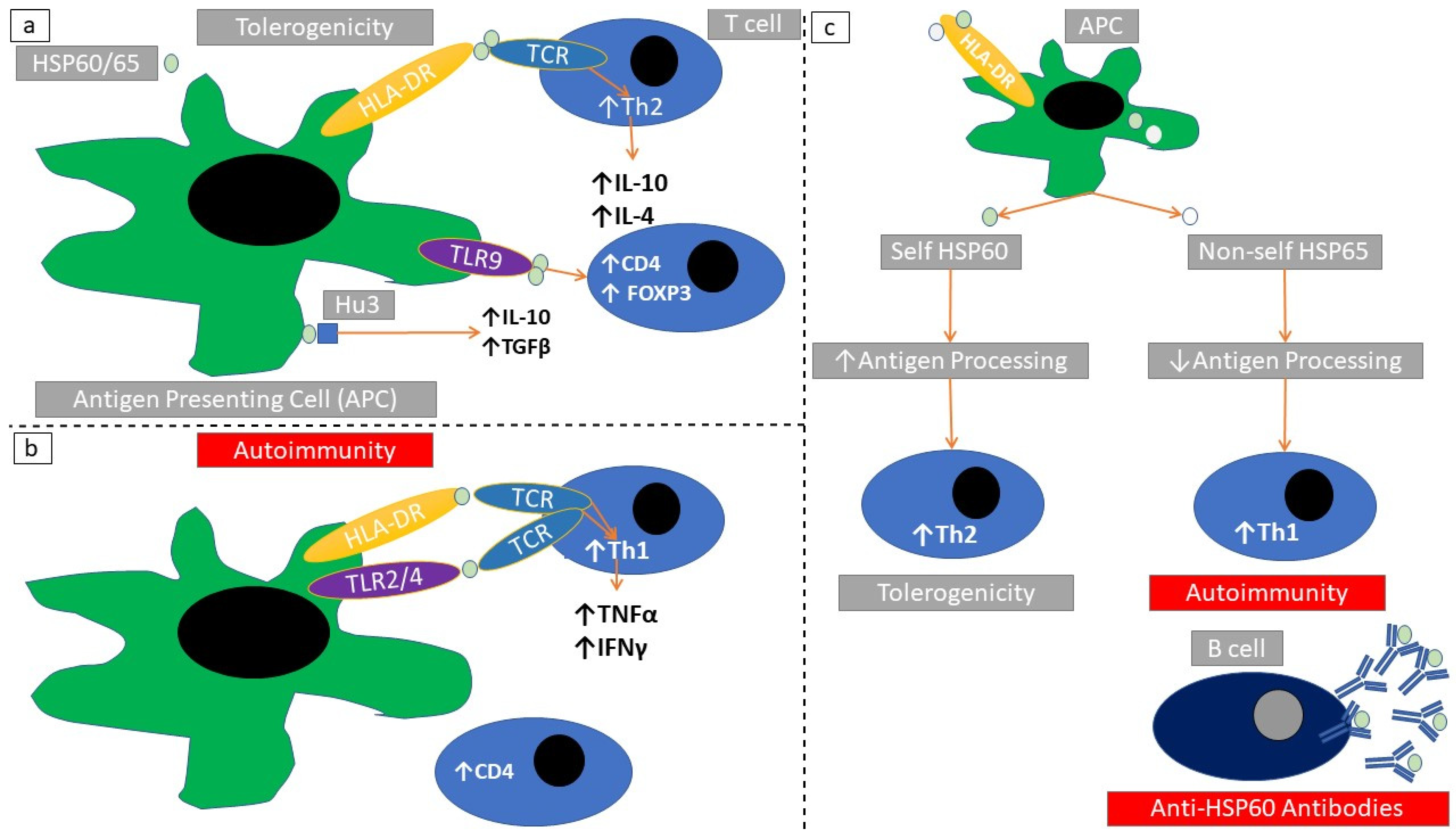
2.9. Structure and Subcellular Localization of Chaperonins
2.10. Immune Responses Elicited through HSP60
3. Therapeutic Implications
4. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Calderwood, S.K.; Repasky, E.A.; Neckers, L.; Hightower, L.E. The IXth CSSI international symposium on heat shock proteins in biology and medicine: Stress responses in health and disease: Alexandria Old Town, Alexandria, Virginia, November 10–13, 2018. Cell Stress Chaperones 2019, 24, 1–6. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Hightower, L.E. Report on the VIIth International Symposium on Heat Shock Proteins in Biology & Medicine. Cell Stress Chaperones 2015, 20, 213–216. [Google Scholar] [CrossRef] [Green Version]
- Krakowiak, J.; Zheng, X.; Patel, N.; Feder, Z.A.; Anandhakumar, J.; Valerius, K.; Gross, D.S.; Khalil, A.S.; Pincus, D. Hsf1 and Hsp70 constitute a two-component feedback loop that regulates the yeast heat shock response. Elife 2018, 7. [Google Scholar] [CrossRef]
- Kmiecik, S.W.; Le Breton, L.; Mayer, M.P. Feedback regulation of heat shock factor 1 (Hsf1) activity by Hsp70-mediated trimer unzipping and dissociation from DNA. EMBO J. 2020, 39, e104096. [Google Scholar] [CrossRef]
- Tukaj, S. Heat Shock Protein 70 as a Double Agent Acting Inside and Outside the Cell: Insights into Autoimmunity. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Stevenson, M.A.; Murshid, A. Heat shock proteins, autoimmunity, and cancer treatment. Autoimmune Dis. 2012, 2012, 486069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaxton, J.E.; Liu, B.; Zheng, P.; Liu, Y.; Li, Z. Deletion of CD24 impairs development of heat shock protein gp96-driven autoimmune disease through expansion of myeloid-derived suppressor cells. J. Immunol. 2014, 192, 5679–5686. [Google Scholar] [CrossRef] [PubMed]
- Winfield, J.B. Stress proteins, arthritis, and autoimmunity. Arthritis Rheum. 1989, 32, 1497–1504. [Google Scholar] [CrossRef] [PubMed]
- Rajaiah, R.; Moudgil, K.D. Heat-shock proteins can promote as well as regulate autoimmunity. Autoimmun. Rev. 2009, 8, 388–393. [Google Scholar] [CrossRef] [Green Version]
- Zuo, D.; Subjeck, J.; Wang, X.Y. Unfolding the Role of Large Heat Shock Proteins: New Insights and Therapeutic Implications. Front. Immunol. 2016, 7, 75. [Google Scholar] [CrossRef] [Green Version]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hino, M.; Kurogi, K.; Okubo, M.A.; Murata-Hori, M.; Hosoya, H. Small heat shock protein 27 (HSP27) associates with tubulin/microtubules in HeLa cells. Biochem. Biophys. Res. Commun. 2000, 271, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.J.; Kanon, B.; Kampinga, H.H. HSPB7 is a SC35 speckle resident small heat shock protein. Biochim. Biophys. Acta 2009, 1793, 1343–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batulan, Z.; Pulakazhi Venu, V.K.; Li, Y.; Koumbadinga, G.; Alvarez-Olmedo, D.G.; Shi, C.; O’Brien, E.R. Extracellular Release and Signaling by Heat Shock Protein 27: Role in Modifying Vascular Inflammation. Front. Immunol. 2016, 7, 285. [Google Scholar] [CrossRef] [Green Version]
- Haslbeck, M.; Weinkauf, S.; Buchner, J. Small heat shock proteins: Simplicity meets complexity. J. Biol. Chem. 2019, 294, 2121–2132. [Google Scholar] [CrossRef] [Green Version]
- Charmpilas, N.; Kyriakakis, E.; Tavernarakis, N. Small heat shock proteins in ageing and age-related diseases. Cell Stress Chaperones 2017, 22, 481–492. [Google Scholar] [CrossRef]
- Reddy, V.S.; Madala, S.K.; Trinath, J.; Reddy, G.B. Extracellular small heat shock proteins: Exosomal biogenesis and function. Cell Stress Chaperones 2018, 23, 441–454. [Google Scholar] [CrossRef]
- Kostenko, S.; Johannessen, M.; Moens, U. PKA-induced F-actin rearrangement requires phosphorylation of Hsp27 by the MAPKAP kinase MK5. Cell. Signal. 2009, 21, 712–718. [Google Scholar] [CrossRef]
- Mainz, A.; Peschek, J.; Stavropoulou, M.; Back, K.C.; Bardiaux, B.; Asami, S.; Prade, E.; Peters, C.; Weinkauf, S.; Buchner, J.; et al. The chaperone alphaB-crystallin uses different interfaces to capture an amorphous and an amyloid client. Nat. Struct. Mol. Biol. 2015, 22, 898–905. [Google Scholar] [CrossRef]
- Ehrnsperger, M.; Hergersberg, C.; Wienhues, U.; Nichtl, A.; Buchner, J. Stabilization of proteins and peptides in diagnostic immunological assays by the molecular chaperone Hsp25. Anal. Biochem. 1998, 259, 218–225. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell. Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Ezemaduka, A.N.; Wang, Z.; Hu, H.; Shi, X.; Liu, C.; Lu, X.; Fu, X.; Chang, Z.; Yin, C.C. A novel mechanism for small heat shock proteins to function as molecular chaperones. Sci. Rep. 2015, 5, 8811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampinga, H.H.; Brunsting, J.F.; Stege, G.J.; Konings, A.W.; Landry, J. Cells overexpressing Hsp27 show accelerated recovery from heat-induced nuclear protein aggregation. Biochem. Biophys. Res. Commun. 1994, 204, 1170–1177. [Google Scholar] [CrossRef]
- Cashikar, A.G.; Duennwald, M.; Lindquist, S.L. A chaperone pathway in protein disaggregation. Hsp26 alters the nature of protein aggregates to facilitate reactivation by Hsp104. J. Biol. Chem. 2005, 280, 23869–23875. [Google Scholar] [CrossRef] [Green Version]
- Berkowitz, P.; Hu, P.; Liu, Z.; Diaz, L.A.; Enghild, J.J.; Chua, M.P.; Rubenstein, D.S. Desmosome signaling. Inhibition of p38MAPK prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J. Biol. Chem. 2005, 280, 23778–23784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadadi, E.; Zhang, B.; Baidzajevas, K.; Yusof, N.; Puan, K.J.; Ong, S.M.; Yeap, W.H.; Rotzschke, O.; Kiss-Toth, E.; Wilson, H.; et al. Differential IL-1beta secretion by monocyte subsets is regulated by Hsp27 through modulating mRNA stability. Sci. Rep. 2016, 6, 39035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajramovic, J.J.; Bsibsi, M.; Geutskens, S.B.; Hassankhan, R.; Verhulst, K.C.; Stege, G.J.; de Groot, C.J.; van Noort, J.M. Differential expression of stress proteins in human adult astrocytes in response to cytokines. J. Neuroimmunol. 2000, 106, 14–22. [Google Scholar] [CrossRef]
- Ben-Ami Shor, D.; Blank, M.; Reuter, S.; Matthias, T.; Beiglass, I.; Volkov, A.; Barshack, I.; Shoenfeld, Y. Anti-ribosomal-P antibodies accelerate lupus glomerulonephritis and induce lupus nephritis in naive mice. J. Autoimmun. 2014, 54, 118–126. [Google Scholar] [CrossRef]
- Thanner, J.; Bekos, C.; Veraar, C.; Janik, S.; Laggner, M.; Boehm, P.M.; Schiefer, A.I.; Mullauer, L.; Klepetko, W.; Ankersmit, H.J.; et al. Heat shock protein 90alpha in thymic epithelial tumors and non-thymomatous myasthenia gravis. Oncoimmunology 2020, 9, 1756130. [Google Scholar] [CrossRef]
- Ciocca, D.R.; Frayssinet, P.; Cuello-Carrion, F.D. A pilot study with a therapeutic vaccine based on hydroxyapatite ceramic particles and self-antigens in cancer patients. Cell Stress Chaperones 2007, 12, 33–43. [Google Scholar] [CrossRef]
- Bartels, K.; Grenz, A.; Eltzschig, H.K. Sphingosine-1-phosphate receptor signaling during acute kidney injury: The tissue is the issue. Kidney Int. 2014, 85, 733–735. [Google Scholar] [CrossRef] [Green Version]
- Ham, A.; Kim, M.; Kim, J.Y.; Brown, K.M.; Fruttiger, M.; D’Agati, V.D.; Lee, H.T. Selective deletion of the endothelial sphingosine-1-phosphate 1 receptor exacerbates kidney ischemia-reperfusion injury. Kidney Int. 2014, 85, 807–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skrzeczynska-Moncznik, J.; Bzowska, M.; Nogiec, A.; Sroka, A.; Zarebski, M.; Vallieres, L.; Guzik, K. Rapid externalization of 27-kDa heat shock protein (HSP27) and atypical cell death in neutrophils treated with the sphingolipid analog drug FTY720. J. Leukoc. Biol. 2015, 98, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verleden, G.M.; Glanville, A.R.; Lease, E.D.; Fisher, A.J.; Calabrese, F.; Corris, P.A.; Ensor, C.R.; Gottlieb, J.; Hachem, R.R.; Lama, V.; et al. Chronic lung allograft dysfunction: Definition, diagnostic criteria, and approaches to treatment-A consensus report from the Pulmonary Council of the ISHLT. J. Heart Lung Transplant. 2019, 38, 493–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, K.L.; Nunley, D.R.; Moffatt-Bruce, S.; Pope-Harman, A.; Huang, Q.; Shamo, E.N.; Phillips, G.S.; Baran, C.; Batra, S.; Marsh, C.B.; et al. The role of heat shock protein 27 in bronchiolitis obliterans syndrome after lung transplantation. J. Heart Lung Transplant. 2010, 29, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Liang, C.; Zhou, L. Structural and functional analysis of the Hsp70/Hsp40 chaperone system. Protein Sci. 2020, 29, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Hattori, H.; Liu, Y.C.; Tohnai, I.; Ueda, M.; Kaneda, T.; Kobayashi, T.; Tanabe, K.; Ohtsuka, K. Intracellular localization and partial amino acid sequence of a stress-inducible 40-kDa protein in HeLa cells. Cell Struct. Funct. 1992, 17, 77–86. [Google Scholar] [CrossRef]
- Ancevska-Taneva, N.; Onoprishvili, I.; Andria, M.L.; Hiller, J.M.; Simon, E.J. A member of the heat shock protein 40 family, hlj1, binds to the carboxyl tail of the human mu opioid receptor. Brain Res. 2006, 1081, 28–33. [Google Scholar] [CrossRef]
- Li, J.; Qian, X.; Sha, B. Heat shock protein 40: Structural studies and their functional implications. Protein Pept. Lett. 2009, 16, 606–612. [Google Scholar] [CrossRef] [Green Version]
- Said, S.M.; Rocha, A.B.; Royal, V.; Valeri, A.M.; Larsen, C.P.; Theis, J.D.; Vrana, J.A.; McPhail, E.D.; Bandi, L.; Safabakhsh, S.; et al. Immunoglobulin-Negative DNAJB9-Associated Fibrillary Glomerulonephritis: A Report of 9 Cases. Am. J. Kidney Dis. 2021, 77, 454–458. [Google Scholar] [CrossRef]
- Taha, E.A.; Ono, K.; Eguchi, T. Roles of Extracellular HSPs as Biomarkers in Immune Surveillance and Immune Evasion. Int. J. Mol. Sci. 2019, 20, 4588. [Google Scholar] [CrossRef] [Green Version]
- Nasr, S.H.; Fogo, A.B. New developments in the diagnosis of fibrillary glomerulonephritis. Kidney Int. 2019, 96, 581–592. [Google Scholar] [CrossRef]
- Baker, L.W.; Khan, M.; Cortese, C.; Aslam, N. Fibrillary glomerulonephritis or complement 3 glomerulopathy: A rare case of diffuse necrotising crescentic glomerulonephritis with C3-dominant glomerular deposition and positive DNAJB9. BMJ Case Rep. 2021, 14. [Google Scholar] [CrossRef]
- Nguyen, T.Q.; Jaramillo, A.; Thompson, R.W.; Dintzis, S.; Oppat, W.F.; Allen, B.T.; Sicard, G.A.; Mohanakumar, T. Increased expression of HDJ-2 (hsp40) in carotid artery atherosclerosis: A novel heat shock protein associated with luminal stenosis and plaque ulceration. J. Vasc. Surg. 2001, 33, 1065–1071. [Google Scholar] [CrossRef] [Green Version]
- Banecka-Majkutewicz, Z.; Grabowski, M.; Kadzinski, L.; Papkov, A.; Wegrzyn, A.; Banecki, B. Increased levels of antibodies against heat shock proteins in stroke patients. Acta Biochim. Pol. 2014, 61, 379–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zininga, T.; Ramatsui, L.; Shonhai, A. Heat Shock Proteins as Immunomodulants. Molecules 2018, 23, 2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.; Ma, C.; Ye, G.; Shi, Y.; Xu, W.; Zhong, L.; Wang, J.; Yin, Y.; Zhang, X.; Wang, H. DnaJ (hsp40) of Streptococcus pneumoniae is involved in bacterial virulence and elicits a strong natural immune reaction via PI3K/JNK. Mol. Immunol. 2017, 83, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Massa, M.; Passalia, M.; Manzoni, S.M.; Campanelli, R.; Ciardelli, L.; Yung, G.P.; Kamphuis, S.; Pistorio, A.; Meli, V.; Sette, A.; et al. Differential recognition of heat-shock protein dnaJ-derived epitopes by effector and Treg cells leads to modulation of inflammation in juvenile idiopathic arthritis. Arthritis Rheum. 2007, 56, 1648–1657. [Google Scholar] [CrossRef] [PubMed]
- Ospelt, C.; Camici, G.G.; Engler, A.; Kolling, C.; Vogetseder, A.; Gay, R.E.; Michel, B.A.; Gay, S. Smoking induces transcription of the heat shock protein system in the joints. Ann. Rheum. Dis. 2014, 73, 1423–1426. [Google Scholar] [CrossRef]
- Kasperkiewicz, M.; Tukaj, S.; Gembicki, A.J.; Sillo, P.; Gorog, A.; Zillikens, D.; Karpati, S. Evidence for a role of autoantibodies to heat shock protein 60, 70, and 90 in patients with dermatitis herpetiformis. Cell Stress Chaperones 2014, 19, 837–843. [Google Scholar] [CrossRef] [Green Version]
- Navasa, N.; Martin-Ruiz, I.; Atondo, E.; Sutherland, J.D.; Angel Pascual-Itoiz, M.; Carreras-Gonzalez, A.; Izadi, H.; Tomas-Cortazar, J.; Ayaz, F.; Martin-Martin, N.; et al. Ikaros mediates the DNA methylation-independent silencing of MCJ/DNAJC15 gene expression in macrophages. Sci. Rep. 2015, 5, 14692. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Hatayama, T.; Yasuda, K.; Nishiyama, E. Characterization of high-molecular-mass heat shock proteins and 42 degrees C-specific heat shock proteins of murine cells. Biochem. Biophys. Res. Commun. 1994, 204, 357–365. [Google Scholar] [CrossRef]
- Fang, C.T.; Kuo, H.H.; Pan, T.S.; Yu, F.C.; Yih, L.H. HSP70 regulates the function of mitotic centrosomes. Cell Mol. Life Sci. 2016, 73, 3949–3960. [Google Scholar] [CrossRef]
- Pobre, K.F.R.; Poet, G.J.; Hendershot, L.M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 2019, 294, 2098–2108. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.H.; Chang, Y.J.; Lin, S.; Yang, W.Y. Hsc70/Stub1 promotes the removal of individual oxidatively stressed peroxisomes. Nat. Commun. 2020, 11, 5267. [Google Scholar] [CrossRef]
- Dulin, E.; Garcia-Barreno, P.; Guisasola, M.C. Extracellular heat shock protein 70 (HSPA1A) and classical vascular risk factors in a general population. Cell Stress Chaperones 2010, 15, 929–937. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, E.; Gehrmann, M.; Brunet, M.; Multhoff, G.; Garrido, C. Intracellular and extracellular functions of heat shock proteins: Repercussions in cancer therapy. J. Leukoc. Biol. 2007, 81, 15–27. [Google Scholar] [CrossRef]
- Alard, J.E.; Dueymes, M.; Mageed, R.A.; Saraux, A.; Youinou, P.; Jamin, C. Mitochondrial heat shock protein (HSP) 70 synergizes with HSP60 in transducing endothelial cell apoptosis induced by anti-HSP60 autoantibody. FASEB J. 2009, 23, 2772–2779. [Google Scholar] [CrossRef] [PubMed]
- Gvozdenov, Z.; Kolhe, J.; Freeman, B.C. The Nuclear and DNA-Associated Molecular Chaperone Network. Cold Spring Harb Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Fernandez, M.R.; Valpuesta, J.M. Hsp70 chaperone: A master player in protein homeostasis. F1000Reserch 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, C. GrpE, a nucleotide exchange factor for DnaK. Cell Stress Chaperones 2003, 8, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Bracher, A.; Verghese, J. The nucleotide exchange factors of Hsp70 molecular chaperones. Front. Mol. Biosci. 2015, 2, 10. [Google Scholar] [CrossRef]
- Kabbage, M.; Dickman, M.B. The BAG proteins: A ubiquitous family of chaperone regulators. Cell Mol. Life Sci. 2008, 65, 1390–1402. [Google Scholar] [CrossRef]
- Niu, L.; Lou, F.; Sun, Y.; Sun, L.; Cai, X.; Liu, Z.; Zhou, H.; Wang, H.; Wang, Z.; Bai, J.; et al. A micropeptide encoded by lncRNA MIR155HG suppresses autoimmune inflammation via modulating antigen presentation. Sci. Adv. 2020, 6, eaaz2059. [Google Scholar] [CrossRef]
- Kolb, H.; Burkart, V. Chaperones may cause the focus of diabetes autoimmunity on distinct (pro)insulin peptides. J. Autoimmun. 2019, 105, 102304. [Google Scholar] [CrossRef]
- Elson, C.J.; Thompson, S.J. Immunity, autoimmunity and immunotherapy: New frontiers in heat shock protein research. Clin. Exp. Immunol. 1994, 98, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Millar, D.G.; Garza, K.M.; Odermatt, B.; Elford, A.R.; Ono, N.; Li, Z.; Ohashi, P.S. Hsp70 promotes antigen-presenting cell function and converts T-cell tolerance to autoimmunity in vivo. Nat. Med. 2003, 9, 1469–1476. [Google Scholar] [CrossRef]
- Panayi, G.S.; Corrigall, V.M. BiP regulates autoimmune inflammation and tissue damage. Autoimmun. Rev. 2006, 5, 140–142. [Google Scholar] [CrossRef] [PubMed]
- van Herwijnen, M.J.; Wieten, L.; van der Zee, R.; van Kooten, P.J.; Wagenaar-Hilbers, J.P.; Hoek, A.; den Braber, I.; Anderton, S.M.; Singh, M.; Meiring, H.D.; et al. Regulatory T cells that recognize a ubiquitous stress-inducible self-antigen are long-lived suppressors of autoimmune arthritis. Proc. Natl. Acad. Sci. USA 2012, 109, 14134–14139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef] [Green Version]
- Gilfillan, S.; Ho, E.L.; Cella, M.; Yokoyama, W.M.; Colonna, M. NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat. Immunol. 2002, 3, 1150–1155. [Google Scholar] [CrossRef]
- Galazka, G.; Jurewicz, A.; Domowicz, M.; Cannella, B.; Raine, C.S.; Selmaj, K. HINT1 peptide/Hsp70 complex induces NK-cell-dependent immunoregulation in a model of autoimmune demyelination. Eur. J. Immunol. 2014, 44, 3026–3044. [Google Scholar] [CrossRef]
- Heneka, M.T.; Sharp, A.; Murphy, P.; Lyons, J.A.; Dumitrescu, L.; Feinstein, D.L. The heat shock response reduces myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis in mice. J. Neurochem. 2001, 77, 568–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deocharan, B.; Zhou, Z.; Antar, K.; Siconolfi-Baez, L.; Angeletti, R.H.; Hardin, J.; Putterman, C. Alpha-actinin immunization elicits anti-chromatin autoimmunity in nonautoimmune mice. J. Immunol. 2007, 179, 1313–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kottke, T.; Sanchez-Perez, L.; Diaz, R.M.; Thompson, J.; Chong, H.; Harrington, K.; Calderwood, S.K.; Pulido, J.; Georgopoulos, N.; Selby, P.; et al. Induction of hsp70-mediated Th17 autoimmunity can be exploited as immunotherapy for metastatic prostate cancer. Cancer Res. 2007, 67, 11970–11979. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, G.; Purcell, A.W.; Keech, C.L.; Farris, A.D.; McCluskey, J.; Gordon, T.P. Molecular chaperones are targets of autoimmunity in Ro(SS-A) immune mice. Clin. Exp. Immunol. 1999, 115, 268–274. [Google Scholar] [CrossRef]
- Purcell, A.W.; Todd, A.; Kinoshita, G.; Lynch, T.A.; Keech, C.L.; Gething, M.J.; Gordon, T.P. Association of stress proteins with autoantigens: A possible mechanism for triggering autoimmunity? Clin. Exp. Immunol. 2003, 132, 193–200. [Google Scholar] [CrossRef]
- Rodriguez-Iturbe, B.; Franco, M.; Tapia, E.; Quiroz, Y.; Johnson, R.J. Renal inflammation, autoimmunity and salt-sensitive hypertension. Clin. Exp. Pharmacol. Physiol. 2012, 39, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Petrakis, I.; Androvitsanea, A.; Stratakis, S.; Daphnis, E.; Stylianou, K. Intense immunostaining of heat shock protein 70 within renal interstitium associates with long-term renal survival in an ANCA-associated vasculitis cohort. Cell Stress Chaperones 2021, 26, 51–65. [Google Scholar] [CrossRef]
- Triantafilou, K.; Triantafilou, M.; Dedrick, R.L. A CD14-independent LPS receptor cluster. Nat. Immunol. 2001, 2, 338–345. [Google Scholar] [CrossRef]
- Sarkar, A.A.; Zohn, I.E. Hectd1 regulates intracellular localization and secretion of Hsp90 to control cellular behavior of the cranial mesenchyme. J. Cell Biol. 2012, 196, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Felts, S.J.; Owen, B.A.; Nguyen, P.; Trepel, J.; Donner, D.B.; Toft, D.O. The hsp90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. J. Biol. Chem. 2000, 275, 3305–3312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.; Goyal, S.; Jamal, S.; Singh, A.; Grover, A. Hsp90: Friends, clients and natural foes. Biochimie 2016, 127, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Beliu, G.; Helmerich, D.A.; Schubert, J.; Pearl, L.H.; Prodromou, C.; Neuweiler, H. Cooperation of local motions in the Hsp90 molecular chaperone ATPase mechanism. Nat. Chem. Biol. 2016, 12, 628–635. [Google Scholar] [CrossRef] [Green Version]
- Pearl, L.H. Review: The HSP90 molecular chaperone-an enigmatic ATPase. Biopolymers 2016, 105, 594–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Retzlaff, M.; Stahl, M.; Eberl, H.C.; Lagleder, S.; Beck, J.; Kessler, H.; Buchner, J. Hsp90 is regulated by a switch point in the C-terminal domain. EMBO Rep. 2009, 10, 1147–1153. [Google Scholar] [CrossRef] [Green Version]
- Chandawarkar, R.Y.; Wagh, M.S.; Kovalchin, J.T.; Srivastava, P. Immune modulation with high-dose heat-shock protein gp96: Therapy of murine autoimmune diabetes and encephalomyelitis. Int. Immunol. 2004, 16, 615–624. [Google Scholar] [CrossRef] [Green Version]
- Han, J.M.; Kwon, N.H.; Lee, J.Y.; Jeong, S.J.; Jung, H.J.; Kim, H.R.; Li, Z.; Kim, S. Identification of gp96 as a novel target for treatment of autoimmune disease in mice. PLoS ONE 2010, 5, e9792. [Google Scholar] [CrossRef]
- Ye, B.X.; Deng, X.; Shao, L.D.; Lu, Y.; Xiao, R.; Liu, Y.J.; Jin, Y.; Xie, Y.Y.; Zhao, Y.; Luo, L.F.; et al. Vibsanin B preferentially targets HSP90beta, inhibits interstitial leukocyte migration, and ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 2015, 194, 4489–4497. [Google Scholar] [CrossRef] [Green Version]
- Shalak, V.; Kaminska, M.; Mitnacht-Kraus, R.; Vandenabeele, P.; Clauss, M.; Mirande, M. The EMAPII cytokine is released from the mammalian multisynthetase complex after cleavage of its p43/proEMAPII component. J. Biol. Chem. 2001, 276, 23769–23776. [Google Scholar] [CrossRef] [Green Version]
- Han, J.M.; Park, S.G.; Liu, B.; Park, B.J.; Kim, J.Y.; Jin, C.H.; Song, Y.W.; Li, Z.; Kim, S. Aminoacyl-tRNA synthetase-interacting multifunctional protein 1/p43 controls endoplasmic reticulum retention of heat shock protein gp96: Its pathological implications in lupus-like autoimmune diseases. Am. J. Pathol. 2007, 170, 2042–2054. [Google Scholar] [CrossRef] [Green Version]
- Khalafalla, M.G.; Woods, L.T.; Camden, J.M.; Khan, A.A.; Limesand, K.H.; Petris, M.J.; Erb, L.; Weisman, G.A. P2X7 receptor antagonism prevents IL-1beta release from salivary epithelial cells and reduces inflammation in a mouse model of autoimmune exocrinopathy. J. Biol. Chem. 2017, 292, 16626–16637. [Google Scholar] [CrossRef] [Green Version]
- Kasperkiewicz, M.; Muller, R.; Manz, R.; Magens, M.; Hammers, C.M.; Somlai, C.; Westermann, J.; Schmidt, E.; Zillikens, D.; Ludwig, R.J.; et al. Heat-shock protein 90 inhibition in autoimmunity to type VII collagen: Evidence that nonmalignant plasma cells are not therapeutic targets. Blood 2011, 117, 6135–6142. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Liu, B.; Caudill, M.M.; Zheng, H.; Qiao, Y.; Podack, E.R.; Li, Z. Cell surface expression of heat shock protein gp96 enhances cross-presentation of cellular antigens and the generation of tumor-specific T cell memory. Cancer Immun. 2003, 3, 1. [Google Scholar] [PubMed]
- Soltys, B.J.; Gupta, R.S. Immunoelectron microscopic localization of the 60-kDa heat shock chaperonin protein (Hsp60) in mammalian cells. Exp. Cell Res. 1996, 222, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Tomasello, G.; Rodolico, V.; Zerilli, M.; Martorana, A.; Bucchieri, F.; Pitruzzella, A.; Marino Gammazza, A.; David, S.; Rappa, F.; Zummo, G.; et al. Changes in immunohistochemical levels and subcellular localization after therapy and correlation and colocalization with CD68 suggest a pathogenetic role of Hsp60 in ulcerative colitis. Appl. Immunohistochem. Mol. Morphol. 2011, 19, 552–561. [Google Scholar] [CrossRef] [Green Version]
- Soltys, B.J.; Gupta, R.S. Cell surface localization of the 60 kDa heat shock chaperonin protein (hsp60) in mammalian cells. Cell Biol. Int. 1997, 21, 315–320. [Google Scholar] [CrossRef]
- Ikawa, S.; Weinberg, R.A. An interaction between p21ras and heat shock protein hsp60, a chaperonin. Proc. Natl. Acad. Sci. USA 1992, 89, 2012–2016. [Google Scholar] [CrossRef] [Green Version]
- Shamaei-Tousi, A.; Stephens, J.W.; Bin, R.; Cooper, J.A.; Steptoe, A.; Coates, A.R.; Henderson, B.; Humphries, S.E. Association between plasma levels of heat shock protein 60 and cardiovascular disease in patients with diabetes mellitus. Eur. Heart J. 2006, 27, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.Y.; Kamuda, K.; Montoya, G.; Mesa, P. The TRiC/CCT Chaperonin and Its Role in Uncontrolled Proliferation. Adv. Exp. Med. Biol. 2020, 1243, 21–40. [Google Scholar] [CrossRef]
- Gomez-Llorente, Y.; Jebara, F.; Patra, M.; Malik, R.; Nisemblat, S.; Chomsky-Hecht, O.; Parnas, A.; Azem, A.; Hirsch, J.A.; Ubarretxena-Belandia, I. Structural basis for active single and double ring complexes in human mitochondrial Hsp60-Hsp10 chaperonin. Nat. Commun. 2020, 11, 1916. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.; Halilou, A.I.; Hu, L.; Cai, W.; Liu, J.; Huang, B. Heat shock protein 10 (Hsp10) in immune-related diseases: One coin, two sides. Int. J. Biochem. Mol. Biol. 2011, 2, 47–57. [Google Scholar] [PubMed]
- Horwich, A.L.; Fenton, W.A. Chaperonin-assisted protein folding: A chronologue. Q. Rev. Biophys 2020, 53, e4. [Google Scholar] [CrossRef] [PubMed]
- Qamra, R.; Mande, S.C. Crystal structure of the 65-kilodalton heat shock protein, chaperonin 60.2, of Mycobacterium tuberculosis. J. Bacteriol. 2004, 186, 8105–8113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butcher, M.J.; Zhu, J. Recent advances in understanding the Th1/Th2 effector choice. Fac. Rev. 2021, 10, 30. [Google Scholar] [CrossRef]
- Wick, G.; Perschinka, H.; Millonig, G. Atherosclerosis as an autoimmune disease: An update. Trends Immunol. 2001, 22, 665–669. [Google Scholar] [CrossRef]
- Parada, C.A.; Portaro, F.; Marengo, E.B.; Klitzke, C.F.; Vicente, E.J.; Faria, M.; Sant’Anna, O.A.; Fernandes, B.L. Autolytic Mycobacterium leprae Hsp65 fragments may act as biological markers for autoimmune diseases. Microb. Pathog. 2011, 51, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Puga Yung, G.L.; Fidler, M.; Albani, E.; Spermon, N.; Teklenburg, G.; Newbury, R.; Schechter, N.; van den Broek, T.; Prakken, B.; Billetta, R.; et al. Heat shock protein-derived T-cell epitopes contribute to autoimmune inflammation in pediatric Crohn’s disease. PLoS ONE 2009, 4, e7714. [Google Scholar] [CrossRef]
- van der Zee, R.; Anderton, S.M.; Prakken, A.B.; Liesbeth Paul, A.G.; van Eden, W. T cell responses to conserved bacterial heat-shock-protein epitopes induce resistance in experimental autoimmunity. Semin. Immunol. 1998, 10, 35–41. [Google Scholar] [CrossRef]
- Gavin, J.R., III; Alberti, K.G.M.M.; Davidson, M.B.; DeFronzo, R.A. Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 1997, 20, 1183–1197. [Google Scholar] [CrossRef]
- Elias, D.; Cohen, I.R. The hsp60 peptide p277 arrests the autoimmune diabetes induced by the toxin streptozotocin. Diabetes 1996, 45, 1168–1172. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.G.; Mathews, C.E.; Driver, J.P. The Role of NOD Mice in Type 1 Diabetes Research: Lessons from the Past and Recommendations for the Future. Front. Endocrinol. 2018, 9, 51. [Google Scholar] [CrossRef]
- Elias, D.; Meilin, A.; Ablamunits, V.; Birk, O.S.; Carmi, P.; Konen-Waisman, S.; Cohen, I.R. Hsp60 peptide therapy of NOD mouse diabetes induces a Th2 cytokine burst and downregulates autoimmunity to various beta-cell antigens. Diabetes 1997, 46, 758–764. [Google Scholar] [CrossRef]
- Bras, A.; Aguas, A.P. Diabetes-prone NOD mice are resistant to Mycobacterium avium and the infection prevents autoimmune disease. Immunology 1996, 89, 20–25. [Google Scholar] [CrossRef]
- Elias, D.; Prigozin, H.; Polak, N.; Rapoport, M.; Lohse, A.W.; Cohen, I.R. Autoimmune diabetes induced by the beta-cell toxin STZ. Immunity to the 60-kDa heat shock protein and to insulin. Diabetes 1994, 43, 992–998. [Google Scholar] [CrossRef]
- Grundtman, C.; Wick, G. The autoimmune concept of atherosclerosis. Curr. Opin. Lipidol. 2011, 22, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Erkkila, L.; Laitinen, K.; Haasio, K.; Tiirola, T.; Jauhiainen, M.; Lehr, H.A.; Aalto-Setala, K.; Saikku, P.; Leinonen, M. Heat shock protein 60 autoimmunity and early lipid lesions in cholesterol-fed C57BL/6JBom mice during Chlamydia pneumoniae infection. Atherosclerosis 2004, 177, 321–328. [Google Scholar] [CrossRef]
- Moudgil, K.D.; Durai, M. Regulation of autoimmune arthritis by self-heat-shock proteins. Trends Immunol. 2008, 29, 412–418. [Google Scholar] [CrossRef] [Green Version]
- Yokota, S.I.; Hirata, D.; Minota, S.; Higashiyama, T.; Kurimoto, M.; Yanagi, H.; Yura, T.; Kubota, H. Autoantibodies against chaperonin CCT in human sera with rheumatic autoimmune diseases: Comparison with antibodies against other Hsp60 family proteins. Cell Stress Chaperones 2000, 5, 337–346. [Google Scholar] [CrossRef] [Green Version]
- Horvath, L.; Czirjak, L.; Fekete, B.; Jakab, L.; Prohaszka, Z.; Cervenak, L.; Romics, L.; Singh, M.; Daha, M.R.; Fust, G. Levels of antibodies against C1q and 60 kDa family of heat shock proteins in the sera of patients with various autoimmune diseases. Immunol. Lett. 2001, 75, 103–109. [Google Scholar] [CrossRef]
- Menge, T.; Rzepka, R.; Melchers, I. Monoclonal autoantibodies from patients with autoimmune diseases: Specificity, affinity and crossreactivity of MAbs binding to cytoskeletal and nucleolar epitopes, cartilage antigens and mycobacterial heat-shock protein 60. Immunobiology 2002, 205, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zonneveld-Huijssoon, E.; van Wijk, F.; Roord, S.; Delemarre, E.; Meerding, J.; de Jager, W.; Klein, M.; Raz, E.; Albani, S.; Kuis, W.; et al. TLR9 agonist CpG enhances protective nasal HSP60 peptide vaccine efficacy in experimental autoimmune arthritis. Ann. Rheum. Dis. 2012, 71, 1706–1715. [Google Scholar] [CrossRef] [PubMed]
- Karopoulos, C.; Rowley, M.J.; Handley, C.J.; Strugnell, R.A. Antibody reactivity to mycobacterial 65 kDa heat shock protein: Relevance to autoimmunity. J. Autoimmun. 1995, 8, 235–248. [Google Scholar] [CrossRef]
- Moudgil, K.D. Diversification of response to hsp65 during the course of autoimmune arthritis is regulatory rather than pathogenic. Immunol. Rev. 1998, 164, 175–184. [Google Scholar] [CrossRef]
- Durai, M.; Kim, H.R.; Moudgil, K.D. The regulatory C-terminal determinants within mycobacterial heat shock protein 65 are cryptic and cross-reactive with the dominant self homologs: Implications for the pathogenesis of autoimmune arthritis. J. Immunol. 2004, 173, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Yonekura, K.; Yokota, S.; Tanaka, S.; Kubota, H.; Fujii, N.; Matsumoto, H.; Chiba, S. Prevalence of anti-heat shock protein antibodies in cerebrospinal fluids of patients with Guillain-Barre syndrome. J. Neuroimmunol. 2004, 156, 204–209. [Google Scholar] [CrossRef]
- Yoshida, Y.; Zhang, X.M.; Wang, H.; Machida, T.; Mine, S.; Kobayashi, E.; Adachi, A.; Matsutani, T.; Kamitsukasa, I.; Wada, T.; et al. Elevated levels of autoantibodies against DNAJC2 in sera of patients with atherosclerotic diseases. Heliyon 2020, 6, e04661. [Google Scholar] [CrossRef]
- Agius, M.A.; Kirvan, C.A.; Schafer, A.L.; Gudipati, E.; Zhu, S. High prevalence of anti-alpha-crystallin antibodies in multiple sclerosis: Correlation with severity and activity of disease. Acta Neurol. Scand. 1999, 100, 139–147. [Google Scholar] [CrossRef]
- Conroy, S.E.; Faulds, G.B.; Williams, W.; Latchman, D.S.; Isenberg, D.A. Detection of autoantibodies to the 90 kDa heat shock protein in systemic lupus erythematosus and other autoimmune diseases. Br. J. Rheumatol. 1994, 33, 923–926. [Google Scholar] [CrossRef]
- Chumpitazi, B.F.; Bouillet, L.; Drouet, M.T.; Kuhn, L.; Garin, J.; Zarski, J.P.; Drouet, C. Biological autoimmunity screening in hepatitis C patients by anti-HepG2 lysate and anti-heat shock protein 70.1 autoantibodies. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 137–146. [Google Scholar] [CrossRef]
- Zhu, J.; Quyyumi, A.A.; Rott, D.; Csako, G.; Wu, H.; Halcox, J.; Epstein, S.E. Antibodies to human heat-shock protein 60 are associated with the presence and severity of coronary artery disease: Evidence for an autoimmune component of atherogenesis. Circulation 2001, 103, 1071–1075. [Google Scholar] [CrossRef] [Green Version]
- Huittinen, T.; Leinonen, M.; Tenkanen, L.; Manttari, M.; Virkkunen, H.; Pitkanen, T.; Wahlstrom, E.; Palosuo, T.; Manninen, V.; Saikku, P. Autoimmunity to human heat shock protein 60, Chlamydia pneumoniae infection, and inflammation in predicting coronary risk. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 431–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyata, M.; Kogure, A.; Sato, H.; Kodama, E.; Watanabe, H.; Ohira, H.; Kuroda, M.; Takagi, T.; Sato, Y.; Kasukawa, R. Detection of antibodies to 65 KD heat shock protein and to human superoxide dismutase in autoimmune hepatitis-molecular mimicry between 65 KD heat shock protein and superoxide dismutase. Clin. Rheumatol. 1995, 14, 673–677. [Google Scholar] [CrossRef]
- Agashe, V.V.; Burlingham, W.J. Autoimmune Reactivity in Graft Injury: Player or Bystander? Curr. Transplant. Rep. 2015, 2, 211–221. [Google Scholar] [CrossRef]
- Barker, R.N.; Webb, G.R.; Thompson, S.J.; Ghoraishian, M.; Ponsford, F.M.; Elson, C.J. Differential effects of immunisation with mycobacterial 65 kD heat shock protein on two models of autoimmunity. Autoimmunity 1992, 14, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Lunin, S.M.; Khrenov, M.O.; Novoselova, T.V.; Parfenyuk, S.B.; Glushkova, O.V.; Fesenko, E.E.; Novoselova, E.G. Modulation of inflammatory response in mice with severe autoimmune disease by thymic peptide thymulin and an inhibitor of NF-kappaB signalling. Int. Immunopharmacol. 2015, 25, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Bonaguri, C.; Orsoni, J.G.; Zavota, L.; Monica, C.; Russo, A.; Pellistri, I.; Rubino, P.; Giovannelli, L.; Manzotti, F.; Piazza, F. Anti-68 kDa antibodies in autoimmune sensorineural hearing loss: Are these autoantibodies really a diagnostic tool? Autoimmunity 2007, 40, 73–78. [Google Scholar] [CrossRef]
- Youde, S.J.; Mower, J.; Moore, D.P.; Parkes, A.B. Stress protein expression in primary and immortalized cultures of human thyroid cells: A model system for the study of stress proteins in the pathogenesis of autoimmune thyroid disease. Cell Stress Chaperones 1998, 3, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Tukaj, S.; Zillikens, D.; Kasperkiewicz, M. Heat shock protein 90: A pathophysiological factor and novel treatment target in autoimmune bullous skin diseases. Exp. Dermatol. 2015, 24, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Marengo, E.B.; Commodaro, A.G.; Peron, J.P.; de Moraes, L.V.; Portaro, F.C.; Belfort, R., Jr.; Rizzo, L.V.; Sant’Anna, O.A. Administration of Mycobacterium leprae rHsp65 aggravates experimental autoimmune uveitis in mice. PLoS ONE 2009, 4, e7912. [Google Scholar] [CrossRef] [PubMed]
- Selli, M.E.; Wick, G.; Wraith, D.C.; Newby, A.C. Autoimmunity to HSP60 during diet induced obesity in mice. Int. J. Obes. 2017, 41, 348–351. [Google Scholar] [CrossRef] [Green Version]
- Haregewoin, A.; Singh, B.; Gupta, R.S.; Finberg, R.W. A mycobacterial heat-shock protein-responsive gamma delta T cell clone also responds to the homologous human heat-shock protein: A possible link between infection and autoimmunity. J. Infect. Dis. 1991, 163, 156–160. [Google Scholar] [CrossRef]
- Zonneveld-Huijssoon, E.; Albani, S.; Prakken, B.J.; van Wijk, F. Heat shock protein bystander antigens for peptide immunotherapy in autoimmune disease. Clin. Exp. Immunol. 2013, 171, 20–29. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Heat-Shock Protein (HSP) | Disease | Effect | References |
|---|---|---|---|
| HSP27 | Glaucoma—increased intraocular pressure | HSP27 serum auto-antibodies | [127] |
| Myasthenia gravis | Increased HSP27 phosphorylation | [18] | |
| T-cell neoplasia (thymoma, T-cell carcinoma) | Increased serum HSP27 protein, increased HSP27 tissue expression, patient subsets with reduced expression associated with worsened outcome | [45] | |
| Lung transplantation | Bronchioalveolar lavage HSP27 auto-antibodies associate with bronchiolitis obliterans | [49] | |
| Immunization of cancer patients (renal-, breast-, colon-carcinoma, melanoma, and astrocytoma) | Increased immunoreactivity following HSP27 vaccination | [128] | |
| Guillain Barret | HSP27 serum auto-antibodies | [129] | |
| HSP40 | Fibrillary glomerulonephritis | Colocalization of HSP40 with fibrils | [65,69] |
| Bullous pemphigoid, pemphigus vulgaris | HSP40 serum auto-antibodies | [66] | |
| Cigarette smoking and rheumatoid arthritis | HSP40 serum auto-antibodies, HSP40 increase in synovial fluid and worsened clinical course | [44] | |
| Stroke | HSP40 serum auto-antibodies | [50] | |
| Various arthritis phenotypes | Complex immunoregulatory or immunostimulatory action | [67] | |
| Atherosclerosis | Increased HSP40 in atheromatous lesions—implication in pathogenesis | [70] | |
| HSP70 | Thyroiditis | HSP70 serum auto-antibodies | [99] |
| Inner ear disease | HSP70 serum auto-antibodies, HSP70 associates with steroid responsiveness | [112,114] | |
| Diabetic microangiopathy | Association of HSP70 serum autoantibodies and disease severity | [110] | |
| HSP90 | SLE | HSP90 autoantibodies, HSP90 presence in peripheral blood monocytes | [130] |
| HCV infection | Interaction of HSP90 with HCV antigens | [131] | |
| HSP60/65 | Systemic lupus erythematosus(SLE), Sjögren syndrome, undifferentiated connective tissue disease, Bechcet’s disease, relapsing polychondritis | HSP60/65 auto-antibodies | [132,133,134] |
| Rheumatoid arthritis | HSP60/65 auto-antibodies, modification of immune response, T-cell expansion | [128,129,130] | |
| Coronary artery disease | Molecular mimicry, worsening of disease activity, presence of autoantibodies | [105,115,135,136] | |
| Heart transplantation | Worst prognosis co-related with serum autoantibodies | [125] | |
| Helicobacter pylori infection | Presence of autoantibodies | [87] | |
| Autoimmune hepatitis, hepatitis C virus (HCV) infection | Presence of autoantibodies, interaction with client proteins | [84] | |
| Renal transplantation | Increased renal HSP65 protein expression associated with Th2 cell shift. | [9] |
| Heat-Shock Protein (HSP) | Disease Model | Effect | Reference |
|---|---|---|---|
| HSP27 | NZBW mice—systemic lupus erythematosus | Lupus nephritis, mesangial cell activation | [30] |
| Rat model of glaucoma (increased intraocular pressure, IOP) | HSP27 auto-antibodies in cerebrospinal fluid | [127] | |
| HSP40 | Rheumatoid arthritis mouse model | HSP40 auto-antibodies, increased disease activity | [44] |
| HSP70 | Autoimmune arthritis mouse model | Suppression of T cells | [74] |
| Mouse model of experimental autoimmune encephalomyelitis(EAE) | Natural-killer-cell-induced immunoregulation, increased HSP70 mRNA associated with reduced inflammation, HSP70 induces a Th17 cell response | [79,137,138] | |
| Mouse model of salt-sensitive hypertension | Increased renal inflammatory infiltration | [139] | |
| HSP90 | Mouse model of type I diabetes mellitus | Immunization with HSP90 reduces autoimmunity | [88,90] |
| Mouse model of EAE | Reduction of autoimmune response | [90] | |
| Mouse models of bullous pemphigoid and pemphigus vulgaris | Reduction of autoimmune response | [140] | |
| Mouse model of autoimmune exocrinopathy | Increased autoimmunity | [93] | |
| Mouse model of anti-collagen VII autoimmunity | Increased infiltration of inflammatory cells | [94] | |
| Rat model of autoimmune arthritis | Immunization reduced arthritis activity, tolerogenicity induction | [124,141] | |
| Mouse model of hemolytic anemia | Immunization with HSP60/65 reduced autoantibodies against erythrocytes. | [124] | |
| Rat model of uveitis | Increased activity of uveitis | [122] | |
| HSP60/65 | Mouse model of type I diabetes (DM) | Immunization vs HSP60/65 reduced DM severity, immunization increased DM severity and autoimmune response | [116,118,142] |
| Mouse model of autoimmune arthritis | Immunization against HSP60/65 reduced arthritis activity, immunization against mycobacterial HSP65 increases arthritis severity | [107,108,143] | |
| Mouse model of atherosclerosis | Immunization against HSP60/65 increased inflammatory response in atheromatous vascular lesions | [144] | |
| Mouse model of intestinal autoimmune disease | Increase of intestinal autoimmune lesions | [121] | |
| Rat model of autoimmune arthritis | Immunization reduced arthritis activity, tolerogenicity induction | [124,141] | |
| Mouse model of hemolytic anemia | Immunization with HSP60/65 reduced autoantibodies against erythrocytes. | [124] | |
| Rat model of uveitis | Increased activity of uveitis | [122] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Androvitsanea, A.; Stylianou, K.; Drosataki, E.; Petrakis, I. The Pathophysiological Role of Heat Shock Response in Autoimmunity: A Literature Review. Cells 2021, 10, 2626. https://doi.org/10.3390/cells10102626
Androvitsanea A, Stylianou K, Drosataki E, Petrakis I. The Pathophysiological Role of Heat Shock Response in Autoimmunity: A Literature Review. Cells. 2021; 10(10):2626. https://doi.org/10.3390/cells10102626
Chicago/Turabian StyleAndrovitsanea, Ariadni, Kostas Stylianou, Eleni Drosataki, and Ioannis Petrakis. 2021. "The Pathophysiological Role of Heat Shock Response in Autoimmunity: A Literature Review" Cells 10, no. 10: 2626. https://doi.org/10.3390/cells10102626