
Targeting Chaperone/Co-Chaperone Interactions with Small Molecules: A Novel Approach to Tackle Neurodegenerative Diseases


Abstract
:1. Introduction
2. Hsp70 and Its Co-Chaperones
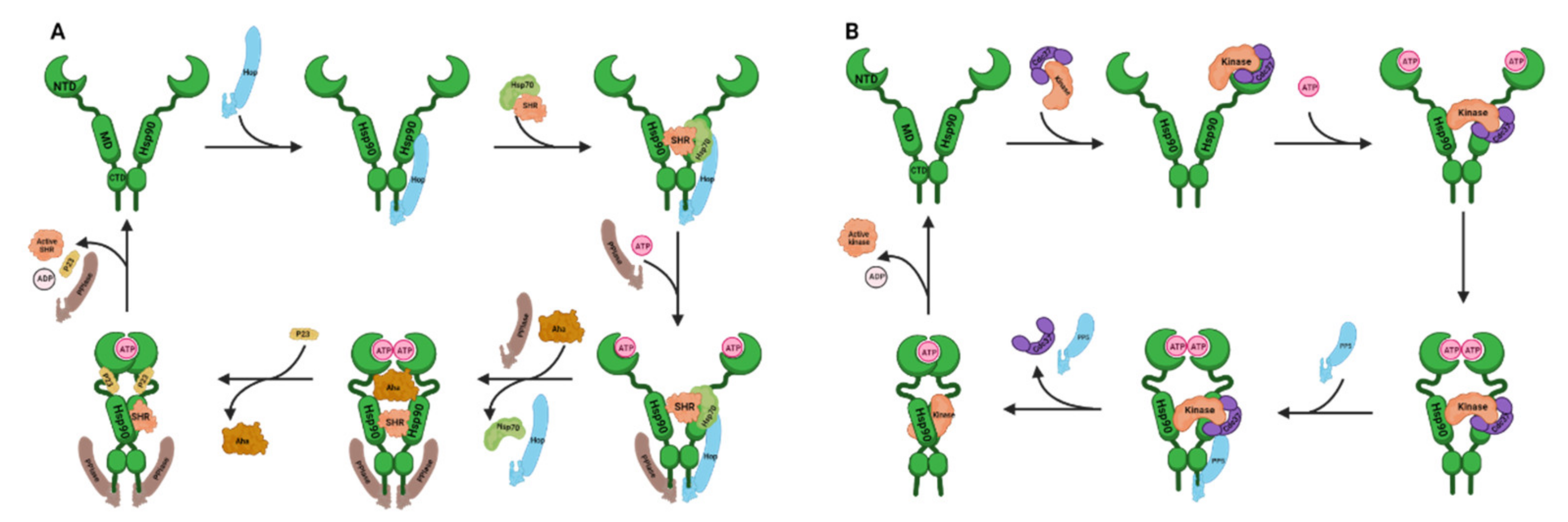
2.1. Hsp70 Structure and Chaperone Cycle
2.2. Hsp70 and Its Co-Chaperones in Neurodegenerative Diseases
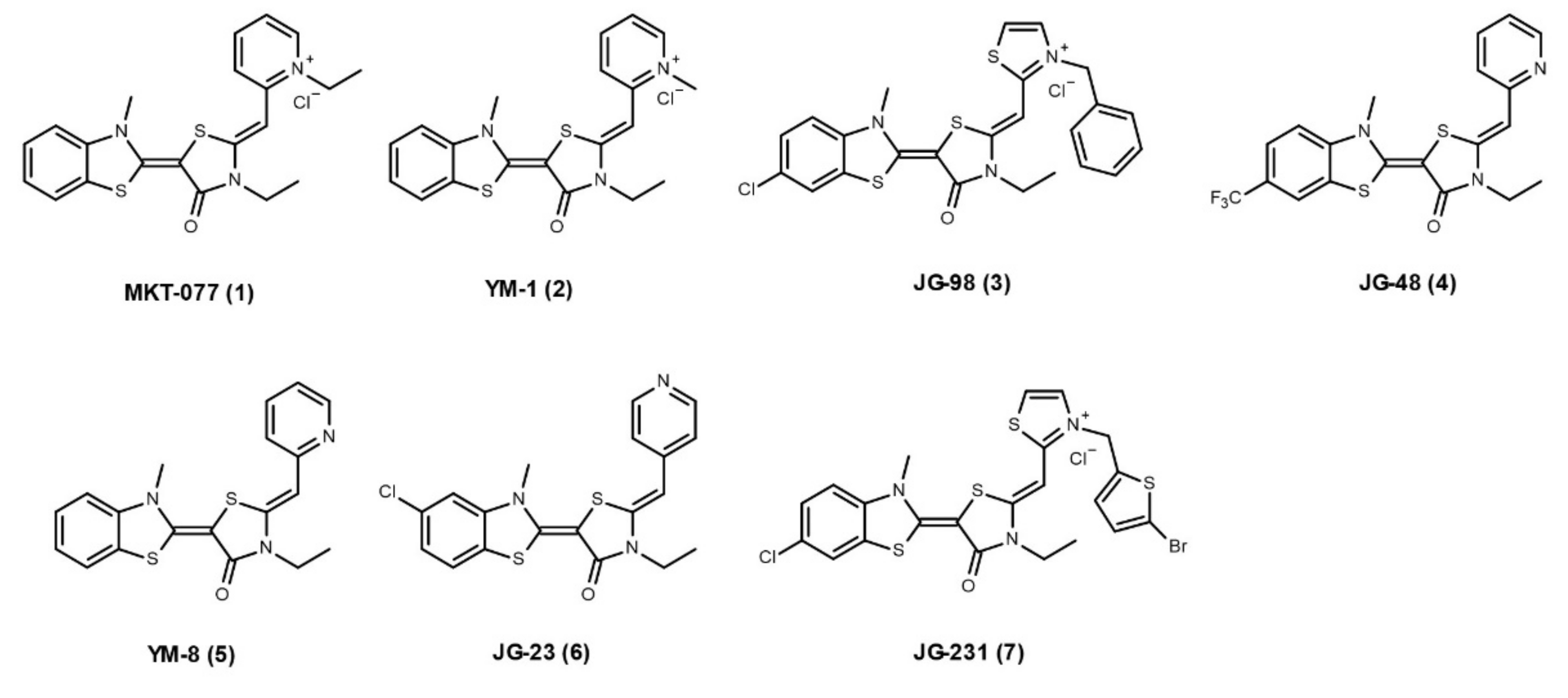
2.3. Hsp70 Co-Chaperone Interaction Inhibitors
3. Hsp90 and Its Co-Chaperones
3.1. Hsp90 Structure and Chaperone Cycles
3.2. Hsp90 and Its Co-Chaperones in Neurodegenerative Diseases and Their PPI Inhibitors
3.2.1. Hsp90–Cdc37 Interaction
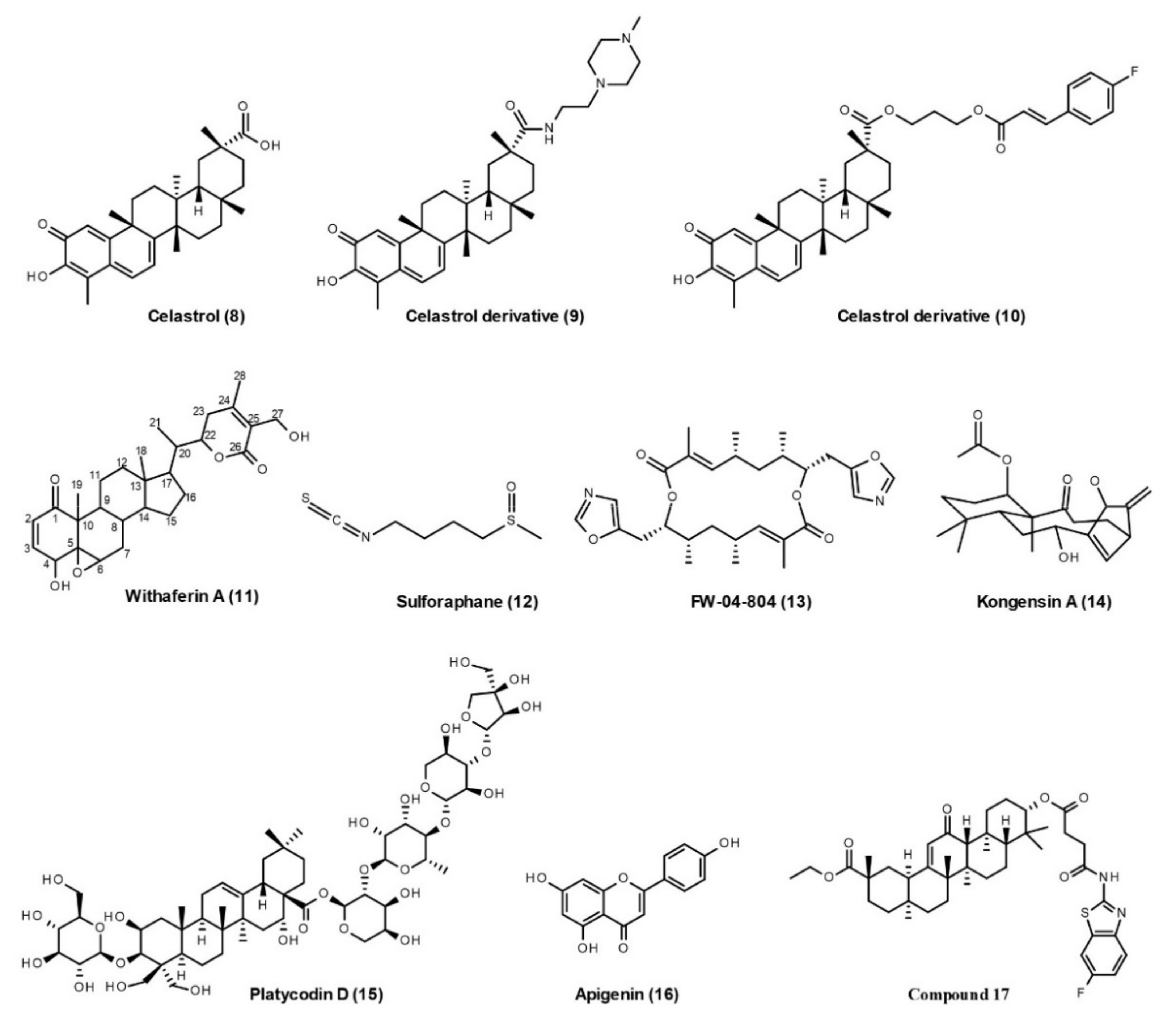
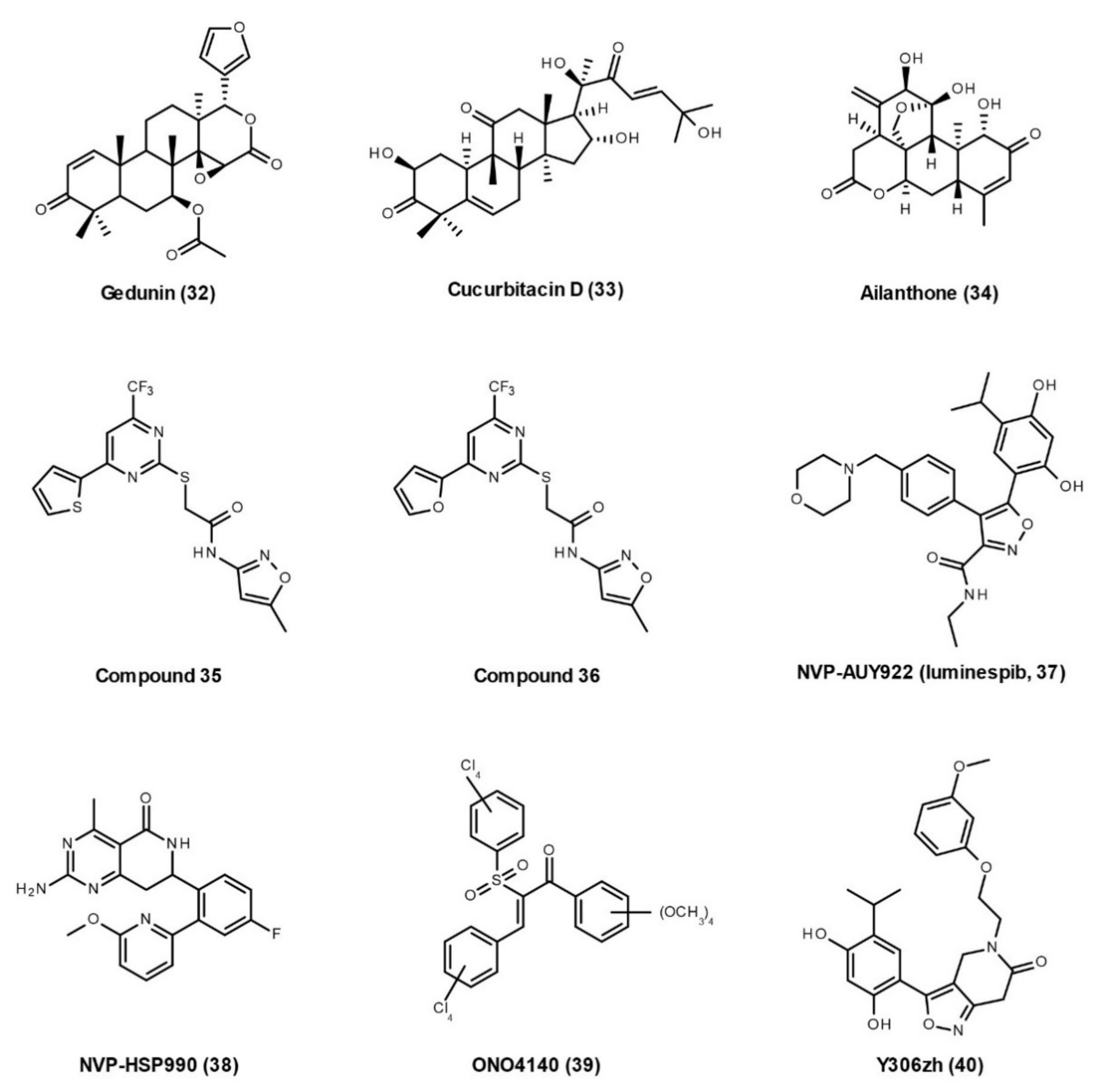
Natural Products
Cdc37 Peptides
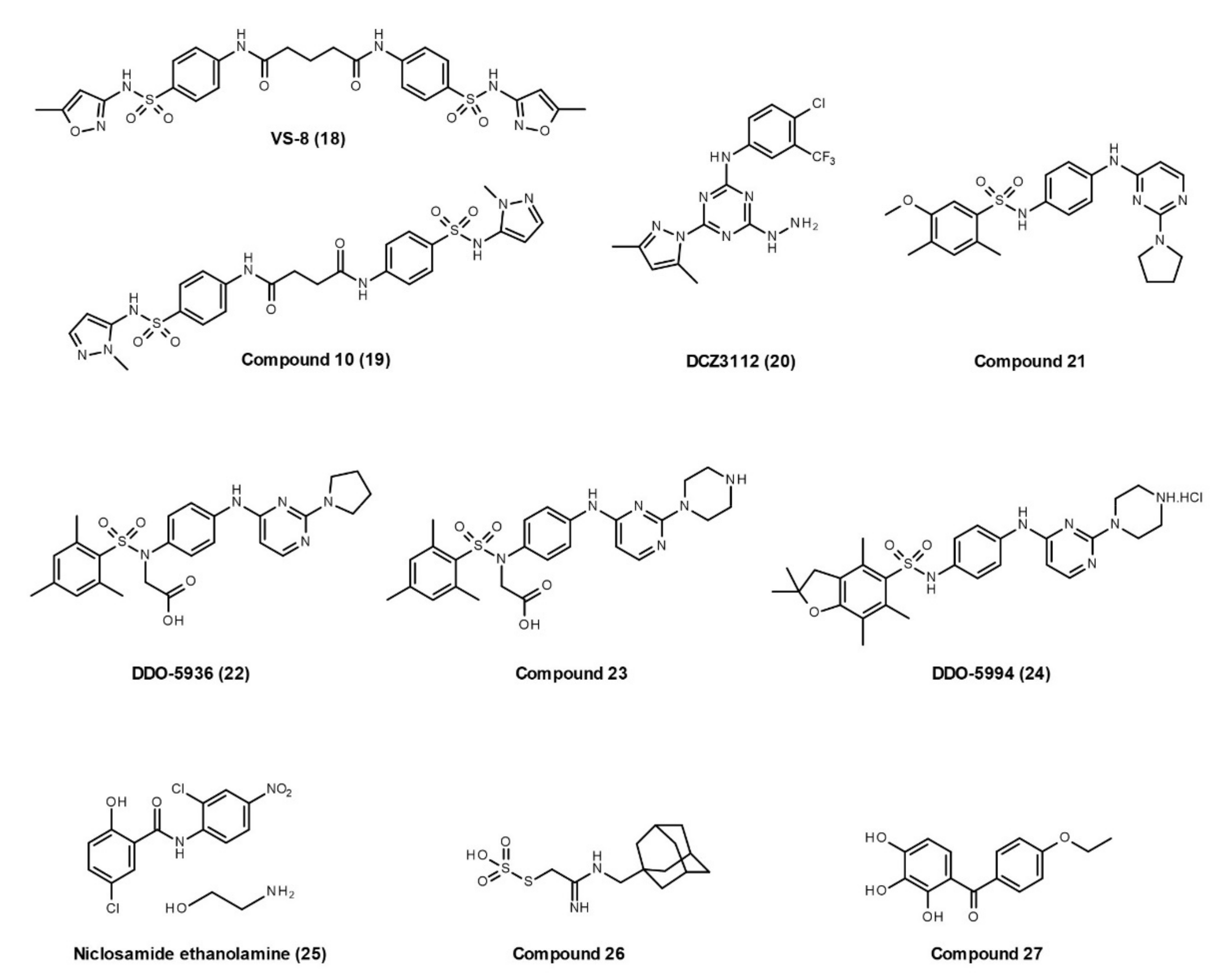
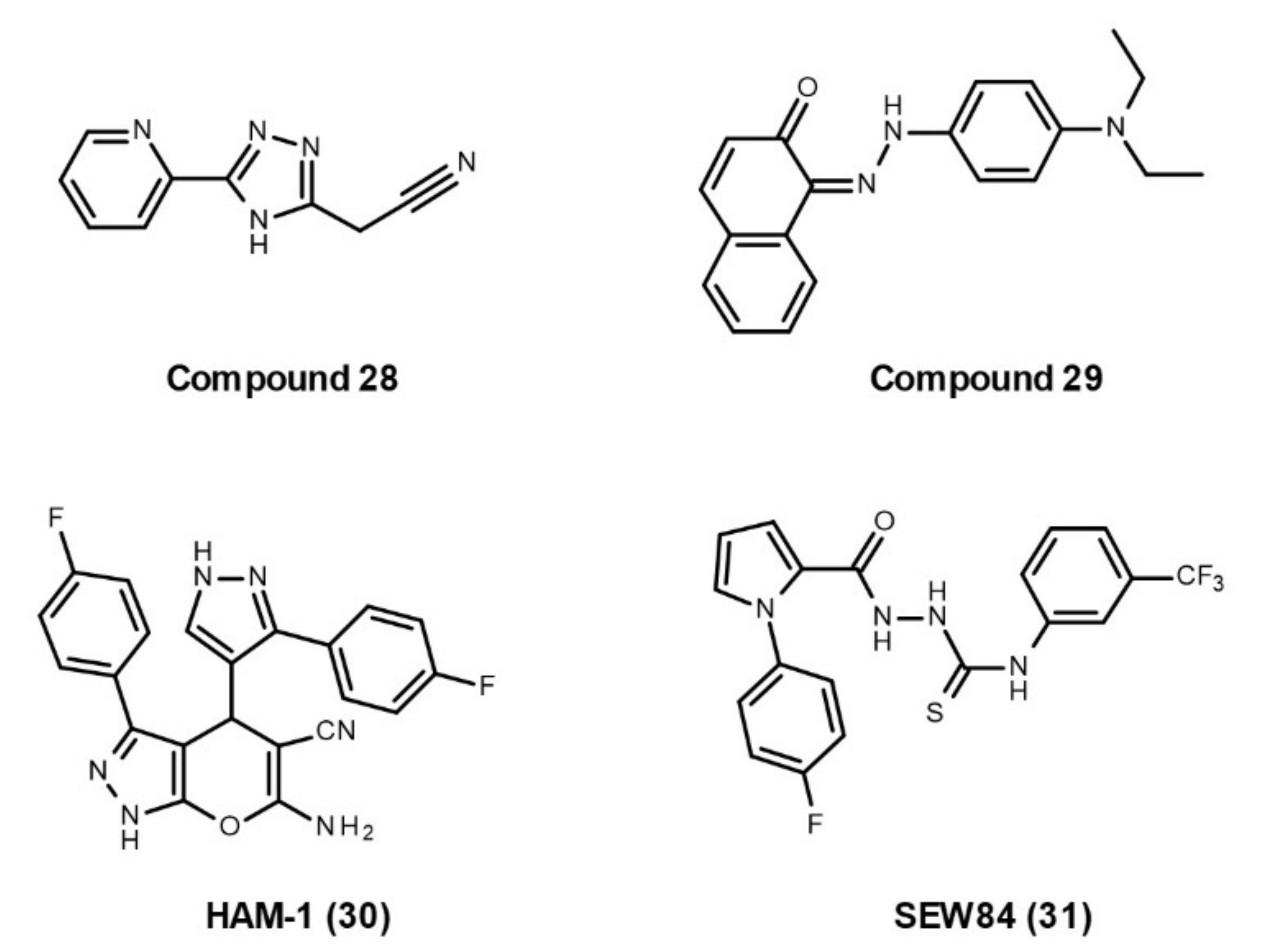
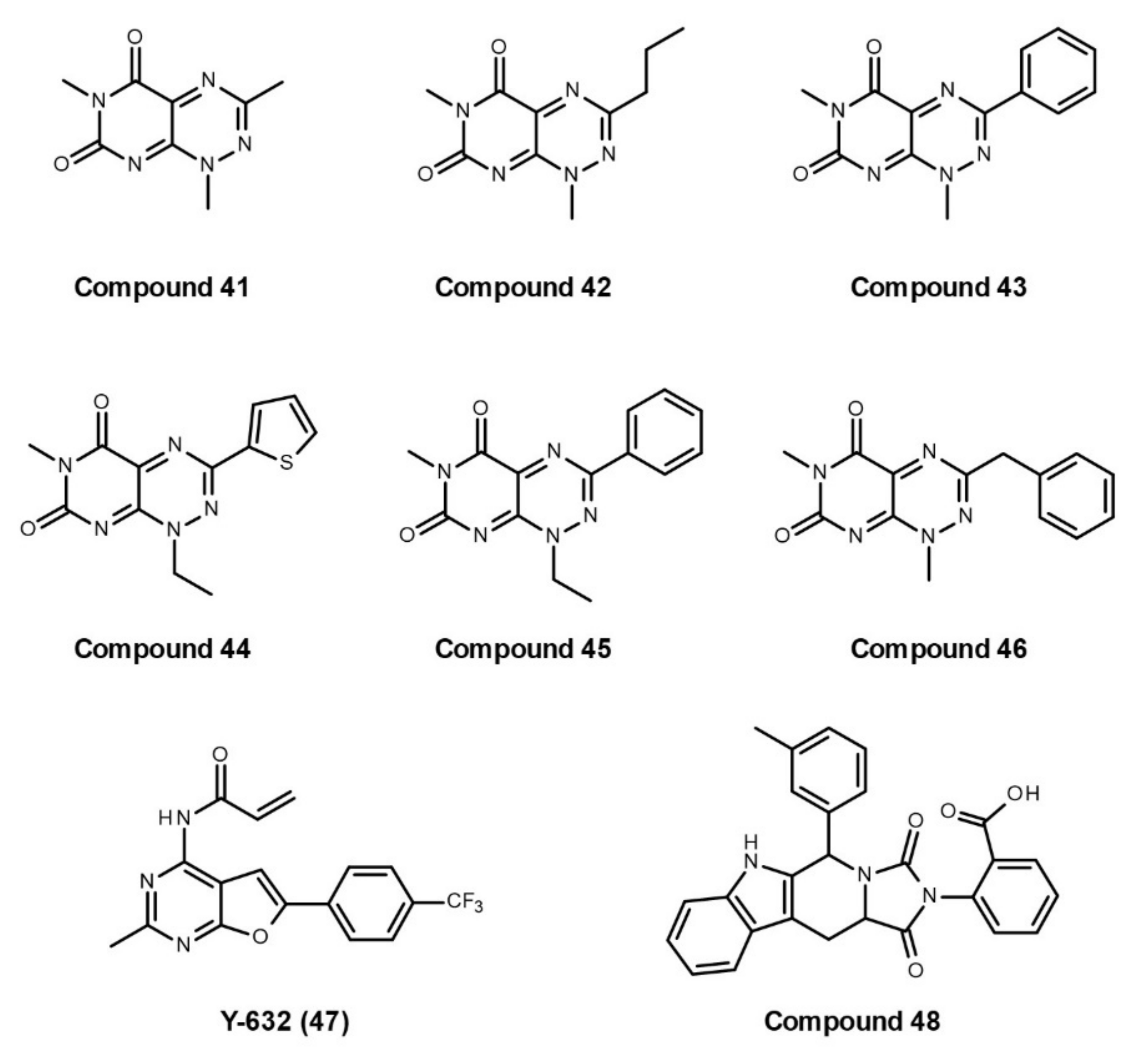
Small-Molecule Inhibitors
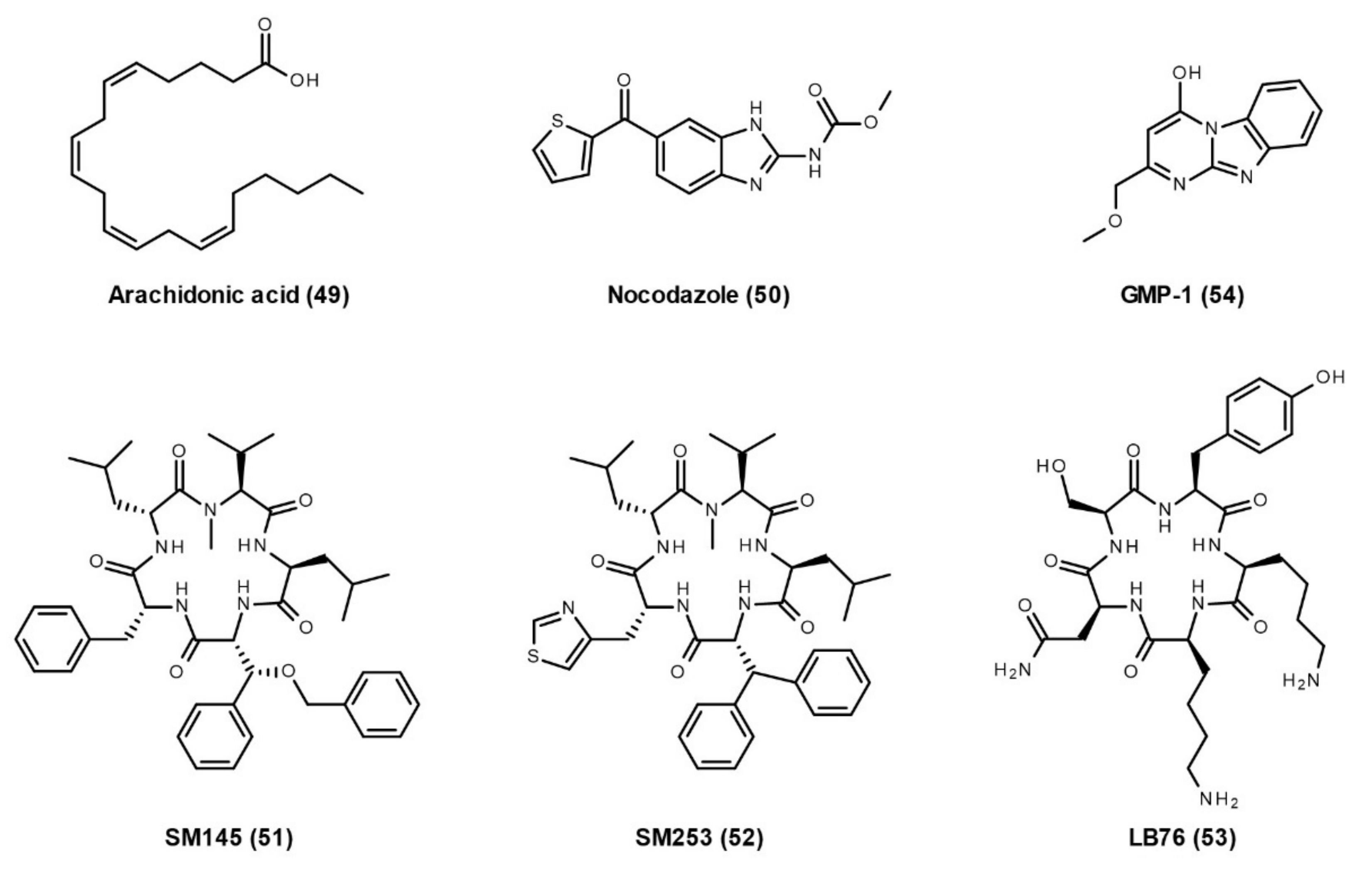
3.2.2. Hsp90–Aha1 Interaction
3.2.3. Hsp90–p23 Interaction
3.2.4. Hsp90–Hop Interaction
3.2.5. Hsp90 and Other TPR Co-Chaperones Interactions
Hsp90 TPR Co-Chaperones in Neurodegenerative Diseases
Hsp90–TPR Co-Chaperone Interaction Inhibitors
Hsp90/Hsp70 Interaction with Dicarboxylate Clamp TPR (dcTPR) Co-Chaperones
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [Green Version]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zatsepina, O.G.; Evgen’ev, M.B.; Garbuz, D.G. Role of a Heat Shock Transcription Factor and the Major Heat Shock Protein Hsp70 in Memory Formation and Neuroprotection. Cells 2021, 10, 1638. [Google Scholar] [CrossRef] [PubMed]
- Bohush, A.; Bieganowski, P.; Filipek, A. Hsp90 and Its Co-Chaperones in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 4976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiti, P.; Manna, J.; Veleri, S.; Frautschy, S. Molecular chaperone dysfunction in neurodegenerative diseases and effects of curcumin. BioMed Res. Int. 2014, 2014, 495091. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, A.J.; Chapman, E. Function, Therapeutic Potential, and Inhibition of Hsp70 Chaperones. J. Med. Chem. 2021, 64, 7060–7082. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Q.; You, Q. Targeting the HSP90-CDC37-kinase chaperone cycle: A promising therapeutic strategy for cancer. Med. Res. Rev. 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Bagatell, R.; Paine-Murrieta, G.D.; Taylor, C.W.; Pulcini, E.J.; Akinaga, S.; Benjamin, I.J.; Whitesell, L. Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 3312–3318. [Google Scholar]
- Bickel, D.; Gohlke, H. C-terminal modulators of heat shock protein of 90kDa (HSP90): State of development and modes of action. Bioorganic Med. Chem. 2019, 27, 115080. [Google Scholar] [CrossRef]
- Dutta Gupta, S.; Bommaka, M.K.; Banerjee, A. Inhibiting protein-protein interactions of Hsp90 as a novel approach for targeting cancer. Eur. J. Med. Chem. 2019, 178, 48–63. [Google Scholar] [CrossRef]
- Kasza, A.; Hunya, A.; Frank, Z.; Fulop, F.; Torok, Z.; Balogh, G.; Santha, M.; Balind, A.; Bernath, S.; Blundell, K.L.; et al. Dihydropyridine Derivatives Modulate Heat Shock Responses and have a Neuroprotective Effect in a Transgenic Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2016, 53, 557–571. [Google Scholar] [CrossRef] [Green Version]
- Roe, M.S.; Wahab, B.; Torok, Z.; Horvath, I.; Vigh, L.; Prodromou, C. Dihydropyridines Allosterically Modulate Hsp90 Providing a Novel Mechanism for Heat Shock Protein Co-induction and Neuroprotection. Front. Mol. Biosci. 2018, 5, 51. [Google Scholar] [CrossRef]
- Mak, O.W.; Sharma, N.; Reynisson, J.; Leung, I.K.H. Discovery of novel Hsp90 C-terminal domain inhibitors that disrupt co-chaperone binding. Bioorganic Med. Chem. Lett. 2021, 38, 127857. [Google Scholar] [CrossRef]
- Sharma, D.; Masison, D.C. Hsp70 structure, function, regulation and influence on yeast prions. Protein Pept. Lett. 2009, 16, 571–581. [Google Scholar] [CrossRef] [Green Version]
- Malinverni, D.; Jost Lopez, A.; De Los Rios, P.; Hummer, G.; Barducci, A. Modeling Hsp70/Hsp40 interaction by multi-scale molecular simulations and coevolutionary sequence analysis. eLife 2017, 6, e23471. [Google Scholar] [CrossRef]
- Kityk, R.; Kopp, J.; Mayer, M.P. Molecular Mechanism of J-Domain-Triggered ATP Hydrolysis by Hsp70 Chaperones. Mol. Cell 2018, 69, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Bracher, A.; Verghese, J. The nucleotide exchange factors of Hsp70 molecular chaperones. Front. Mol. Biosci. 2015, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Reviews. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinwal, U.K.; Akoury, E.; Abisambra, J.F.; O’Leary, J.C., 3rd; Thompson, A.D.; Blair, L.J.; Jin, Y.; Bacon, J.; Nordhues, B.A.; Cockman, M.; et al. Imbalance of Hsp70 family variants fosters tau accumulation. FASEB J.: Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 1450–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine, S.N.; Zheng, D.; Sabbagh, J.J.; Martin, M.D.; Chaput, D.; Darling, A.; Trotter, J.H.; Stothert, A.R.; Nordhues, B.A.; Lussier, A.; et al. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 2016, 35, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Nachman, E.; Wentink, A.S.; Madiona, K.; Bousset, L.; Katsinelos, T.; Allinson, K.; Kampinga, H.; McEwan, W.A.; Jahn, T.R.; Melki, R.; et al. Disassembly of Tau fibrils by the human Hsp70 disaggregation machinery generates small seeding-competent species. J. Biol. Chem. 2020, 295, 9676–9690. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shao, H.; Taylor, I.R.; Gestwicki, J.E. Targeting Allosteric Control Mechanisms in Heat Shock Protein 70 (Hsp70). Curr. Top. Med. Chem. 2016, 16, 2729–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colvin, T.A.; Gabai, V.L.; Gong, J.; Calderwood, S.K.; Li, H.; Gummuluru, S.; Matchuk, O.N.; Smirnova, S.G.; Orlova, N.V.; Zamulaeva, I.A.; et al. Hsp70-Bag3 interactions regulate cancer-related signaling networks. Cancer Res. 2014, 74, 4731–4740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Colvin, T.; Rauch, J.N.; Acosta-Alvear, D.; Kampmann, M.; Dunyak, B.; Hann, B.; Aftab, B.T.; Murnane, M.; Cho, M.; et al. Validation of the Hsp70-Bag3 protein-protein interaction as a potential therapeutic target in cancer. Mol. Cancer Ther. 2015, 14, 642–648. [Google Scholar] [CrossRef] [Green Version]
- Young, Z.T.; Rauch, J.N.; Assimon, V.A.; Jinwal, U.K.; Ahn, M.; Li, X.; Dunyak, B.M.; Ahmad, A.; Carlson, G.A.; Srinivasan, S.R.; et al. Stabilizing the Hsp70-Tau Complex Promotes Turnover in Models of Tauopathy. Cell Chem. Biol. 2016, 23, 992–1001. [Google Scholar] [CrossRef] [Green Version]
- Miyata, Y.; Li, X.; Lee, H.F.; Jinwal, U.K.; Srinivasan, S.R.; Seguin, S.P.; Young, Z.T.; Brodsky, J.L.; Dickey, C.A.; Sun, D.; et al. Synthesis and initial evaluation of YM-08, a blood-brain barrier permeable derivative of the heat shock protein 70 (Hsp70) inhibitor MKT-077, which reduces tau levels. ACS Chem. Neurosci. 2013, 4, 930–939. [Google Scholar] [CrossRef] [Green Version]
- Shao, H.; Li, X.; Hayashi, S.; Bertron, J.L.; Schwarz, D.M.C.; Tang, B.C.; Gestwicki, J.E. Inhibitors of heat shock protein 70 (Hsp70) with enhanced metabolic stability reduce tau levels. Bioorganic Med. Chem. Lett. 2021, 41, 128025. [Google Scholar] [CrossRef]
- Bengoechea, R.; Findlay, A.R.; Bhadra, A.K.; Shao, H.; Stein, K.C.; Pittman, S.K.; Daw, J.A.; Gestwicki, J.E.; True, H.L.; Weihl, C.C. Inhibition of DNAJ-HSP70 interaction improves strength in muscular dystrophy. J. Clin. Investig. 2020, 130, 4470–4485. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Reviews. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Eckl, J.M.; Richter, K. Functions of the Hsp90 chaperone system: Lifting client proteins to new heights. Int. J. Biochem. Mol. Biol. 2013, 4, 157–165. [Google Scholar]
- Jinwal, U.K.; Trotter, J.H.; Abisambra, J.F.; Koren, J., 3rd; Lawson, L.Y.; Vestal, G.D.; O’Leary, J.C., 3rd; Johnson, A.G.; Jin, Y.; Jones, J.R.; et al. The Hsp90 kinase co-chaperone Cdc37 regulates tau stability and phosphorylation dynamics. J. Biol. Chem. 2011, 286, 16976–16983. [Google Scholar] [CrossRef] [Green Version]
- Jinwal, U.K.; Abisambra, J.F.; Zhang, J.; Dharia, S.; O’Leary, J.C.; Patel, T.; Braswell, K.; Jani, T.; Gestwicki, J.E.; Dickey, C.A. Cdc37/Hsp90 protein complex disruption triggers an autophagic clearance cascade for TDP-43 protein. J. Biol. Chem. 2012, 287, 24814–24820. [Google Scholar] [CrossRef] [Green Version]
- Sonamoto, R.; Kii, I.; Koike, Y.; Sumida, Y.; Kato-Sumida, T.; Okuno, Y.; Hosoya, T.; Hagiwara, M. Identification of a DYRK1A Inhibitor that Induces Degradation of the Target Kinase using Co-chaperone CDC37 fused with Luciferase nanoKAZ. Sci. Rep. 2015, 5, 12728. [Google Scholar] [CrossRef] [PubMed]
- Narayan, M.; Zhang, J.; Braswell, K.; Gibson, C.; Zitnyar, A.; Lee, D.C.; Varghese-Gupta, S.; Jinwal, U.K. Withaferin A Regulates LRRK2 Levels by Interfering with the Hsp90- Cdc37 Chaperone Complex. Curr. Aging Sci. 2015, 8, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, Y.; Kim, Y.J.; Ido, Y.; Misawa, H.; Kawashima, K.; Endo, S.; Takahashi, R. L347P PINK1 mutant that fails to bind to Hsp90/Cdc37 chaperones is rapidly degraded in a proteasome-dependent manner. Neurosci. Res. 2008, 61, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Weihofen, A.; Ostaszewski, B.; Minami, Y.; Selkoe, D.J. Pink1 Parkinson mutations, the Cdc37/Hsp90 chaperones and Parkin all influence the maturation or subcellular distribution of Pink1. Hum. Mol. Genet. 2008, 17, 602–616. [Google Scholar] [CrossRef]
- Shelton, L.B.; Baker, J.D.; Zheng, D.; Sullivan, L.E.; Solanki, P.K.; Webster, J.M.; Sun, Z.; Sabbagh, J.J.; Nordhues, B.A.; Koren, J., 3rd; et al. Hsp90 activator Aha1 drives production of pathological tau aggregates. Proc. Natl. Acad. Sci. USA. 2017, 114, 9707–9712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criado-Marrero, M.; Gebru, N.T.; Blazier, D.M.; Gould, L.A.; Baker, J.D.; Beaulieu-Abdelahad, D.; Blair, L.J. Hsp90 co-chaperones, FKBP52 and Aha1, promote tau pathogenesis in aged wild-type mice. Acta Neuropathol. Commun. 2021, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Dickey, C.A.; Kamal, A.; Lundgren, K.; Klosak, N.; Bailey, R.M.; Dunmore, J.; Ash, P.; Shoraka, S.; Zlatkovic, J.; Eckman, C.B.; et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Investig. 2007, 117, 648–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rane, A.; Rajagopalan, S.; Ahuja, M.; Thomas, B.; Chinta, S.J.; Andersen, J.K. Hsp90 Co-chaperone p23 contributes to dopaminergic mitochondrial stress via stabilization of PHD2: Implications for Parkinson’s disease. Neurotoxicology 2018, 65, 166–173. [Google Scholar] [CrossRef]
- Wolfe, K.J.; Ren, H.Y.; Trepte, P.; Cyr, D.M. The Hsp70/90 cochaperone, Sti1, suppresses proteotoxicity by regulating spatial quality control of amyloid-like proteins. Mol. Biol. Cell 2013, 24, 3588–3602. [Google Scholar] [CrossRef] [PubMed]
- Ambegaokar, S.S.; Jackson, G.R. Functional genomic screen and network analysis reveal novel modifiers of tauopathy dissociated from tau phosphorylation. Hum. Mol. Genet. 2011, 20, 4947–4977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beraldo, F.H.; Arantes, C.P.; Santos, T.G.; Queiroz, N.G.; Young, K.; Rylett, R.J.; Markus, R.P.; Prado, M.A.; Martins, V.R. Role of alpha7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J. Biol. Chem. 2010, 285, 36542–36550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostapchenko, V.G.; Beraldo, F.H.; Mohammad, A.H.; Xie, Y.F.; Hirata, P.H.; Magalhaes, A.C.; Lamour, G.; Li, H.; Maciejewski, A.; Belrose, J.C.; et al. The prion protein ligand, stress-inducible phosphoprotein 1, regulates amyloid-beta oligomer toxicity. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 16552–16564. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Rossie, S.; Gong, C.X. Dephosphorylation of tau by protein phosphatase 5: Impairment in Alzheimer’s disease. J. Biol. Chem. 2005, 280, 1790–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Ortiz, E.; Hahm, B.K.; Armstrong, D.L.; Rossie, S. Protein phosphatase 5 protects neurons against amyloid-beta toxicity. J. Neurochem. 2009, 111, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Dickey, C.A.; Yue, M.; Lin, W.L.; Dickson, D.W.; Dunmore, J.H.; Lee, W.C.; Zehr, C.; West, G.; Cao, S.; Clark, A.M.; et al. Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 6985–6996. [Google Scholar] [CrossRef] [Green Version]
- Kalia, L.V.; Kalia, S.K.; Chau, H.; Lozano, A.M.; Hyman, B.T.; McLean, P.J. Ubiquitinylation of alpha-synuclein by carboxyl terminus Hsp70-interacting protein (CHIP) is regulated by Bcl-2-associated athanogene 5 (BAG5). PLoS ONE 2011, 6, e14695. [Google Scholar] [CrossRef]
- Shin, Y.; Klucken, J.; Patterson, C.; Hyman, B.T.; McLean, P.J. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J. Biol. Chem. 2005, 280, 23727–23734. [Google Scholar] [CrossRef] [Green Version]
- Bendifallah, M.; Redeker, V.; Monsellier, E.; Bousset, L.; Bellande, T.; Melki, R. Interaction of the chaperones alpha B-crystallin and CHIP with fibrillar alpha-synuclein: Effects on internalization by cells and identification of interacting interfaces. Biochem. Biophys. Res. Commun. 2020, 527, 760–769. [Google Scholar] [CrossRef]
- Ko, H.S.; Bailey, R.; Smith, W.W.; Liu, Z.; Shin, J.H.; Lee, Y.I.; Zhang, Y.J.; Jiang, H.; Ross, C.A.; Moore, D.J.; et al. CHIP regulates leucine-rich repeat kinase-2 ubiquitination, degradation, and toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 2897–2902. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Goldberg, M.S. Regulation of LRRK2 stability by the E3 ubiquitin ligase CHIP. PLoS ONE 2009, 4, e5949. [Google Scholar] [CrossRef]
- Jana, N.R.; Dikshit, P.; Goswami, A.; Kotliarova, S.; Murata, S.; Tanaka, K.; Nukina, N. Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J. Biol. Chem. 2005, 280, 11635–11640. [Google Scholar] [CrossRef] [Green Version]
- Miller, V.M.; Nelson, R.F.; Gouvion, C.M.; Williams, A.; Rodriguez-Lebron, E.; Harper, S.Q.; Davidson, B.L.; Rebagliati, M.R.; Paulson, H.L. CHIP suppresses polyglutamine aggregation and toxicity in vitro and in vivo. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 9152–9161. [Google Scholar] [CrossRef] [Green Version]
- Blair, L.J.; Nordhues, B.A.; Hill, S.E.; Scaglione, K.M.; O’Leary, J.C., 3rd; Fontaine, S.N.; Breydo, L.; Zhang, B.; Li, P.; Wang, L.; et al. Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Investig. 2013, 123, 4158–4169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinwal, U.K.; Koren, J., 3rd; Borysov, S.I.; Schmid, A.B.; Abisambra, J.F.; Blair, L.J.; Johnson, A.G.; Jones, J.R.; Shults, C.L.; O’Leary, J.C., 3rd; et al. The Hsp90 cochaperone, FKBP51, increases Tau stability and polymerizes microtubules. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boonying, W.; Joselin, A.; Huang, E.; Qu, D.; Safarpour, F.; Iyirhiaro, G.O.; Gonzalez, Y.R.; Callaghan, S.M.; Slack, R.S.; Figeys, D.; et al. Pink1 regulates FKBP5 interaction with AKT/PHLPP and protects neurons from neurotoxin stress induced by MPP(.). J. Neurochem. 2019, 150, 312–329. [Google Scholar] [CrossRef] [PubMed]
- Bailus, B.J.; Scheeler, S.M.; Simons, J.; Sanchez, M.A.; Tshilenge, K.T.; Creus-Muncunill, J.; Naphade, S.; Lopez-Ramirez, A.; Zhang, N.; Lakshika Madushani, K.; et al. Modulating FKBP5/FKBP51 and autophagy lowers HTT (huntingtin) levels. Autophagy 2021, 1–22, Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Giustiniani, J.; Chambraud, B.; Sardin, E.; Dounane, O.; Guillemeau, K.; Nakatani, H.; Paquet, D.; Kamah, A.; Landrieu, I.; Lippens, G.; et al. Immunophilin FKBP52 induces Tau-P301L filamentous assembly in vitro and modulates its activity in a model of tauopathy. Proc. Natl. Acad. Sci. USA 2014, 111, 4584–4589. [Google Scholar] [CrossRef] [Green Version]
- Giustiniani, J.; Guillemeau, K.; Dounane, O.; Sardin, E.; Huvent, I.; Schmitt, A.; Hamdane, M.; Buee, L.; Landrieu, I.; Lippens, G.; et al. The FK506-binding protein FKBP52 in vitro induces aggregation of truncated Tau forms with prion-like behavior. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 3171–3181. [Google Scholar] [CrossRef] [Green Version]
- Kamah, A.; Cantrelle, F.X.; Huvent, I.; Giustiniani, J.; Guillemeau, K.; Byrne, C.; Jacquot, Y.; Landrieu, I.; Baulieu, E.E.; Smet, C.; et al. Isomerization and Oligomerization of Truncated and Mutated Tau Forms by FKBP52 are Independent Processes. J. Mol. Biol. 2016, 428, 1080–1090. [Google Scholar] [CrossRef] [PubMed]
- Criado-Marrero, M.; Gebru, N.T.; Gould, L.A.; Blazier, D.M.; Vidal-Aguiar, Y.; Smith, T.M.; Abdelmaboud, S.S.; Shelton, L.B.; Wang, X.; Dahrendorff, J.; et al. FKBP52 overexpression accelerates hippocampal-dependent memory impairments in a tau transgenic mouse model. NPJ Aging Mech. Dis. 2021, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Giustiniani, J.; Sineus, M.; Sardin, E.; Dounane, O.; Panchal, M.; Sazdovitch, V.; Duyckaerts, C.; Chambraud, B.; Baulieu, E.E. Decrease of the immunophilin FKBP52 accumulation in human brains of Alzheimer’s disease and FTDP-17. J. Alzheimer’s Dis. JAD 2012, 29, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Sanokawa-Akakura, R.; Cao, W.; Allan, K.; Patel, K.; Ganesh, A.; Heiman, G.; Burke, R.; Kemp, F.W.; Bogden, J.D.; Camakaris, J.; et al. Control of Alzheimer’s amyloid beta toxicity by the high molecular weight immunophilin FKBP52 and copper homeostasis in Drosophila. PLoS ONE 2010, 5, e8626. [Google Scholar] [CrossRef] [Green Version]
- Gerard, M.; Deleersnijder, A.; Daniels, V.; Schreurs, S.; Munck, S.; Reumers, V.; Pottel, H.; Engelborghs, Y.; Van den Haute, C.; Taymans, J.M.; et al. Inhibition of FK506 binding proteins reduces alpha-synuclein aggregation and Parkinson’s disease-like pathology. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 2454–2463. [Google Scholar] [CrossRef]
- Labrador-Garrido, A.; Cejudo-Guillen, M.; Daturpalli, S.; Leal, M.M.; Klippstein, R.; De Genst, E.J.; Villadiego, J.; Toledo-Aral, J.J.; Dobson, C.M.; Jackson, S.E.; et al. Chaperome screening leads to identification of Grp94/Gp96 and FKBP4/52 as modulators of the alpha-synuclein-elicited immune response. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Roe, S.M.; Ali, M.M.; Meyer, P.; Vaughan, C.K.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. The Mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37). Cell 2004, 116, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Verba, K.A.; Wang, R.Y.; Arakawa, A.; Liu, Y.; Shirouzu, M.; Yokoyama, S.; Agard, D.A. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 2016, 352, 1542–1547. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Hamza, A.; Cao, X.; Wang, B.; Yu, S.; Zhan, C.G.; Sun, D. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer Ther. 2008, 7, 162–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Li, Y.; Yu, Y.; Zou, P.; Jiang, Y.; Sun, D. Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J. Biol. Chem. 2009, 284, 35381–35389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreeramulu, S.; Gande, S.L.; Gobel, M.; Schwalbe, H. Molecular mechanism of inhibition of the human protein complex Hsp90-Cdc37, a kinome chaperone-cochaperone, by triterpene celastrol. Angew. Chem. 2009, 48, 5853–5855. [Google Scholar] [CrossRef] [PubMed]
- Zanphorlin, L.M.; Alves, F.R.; Ramos, C.H. The effect of celastrol, a triterpene with antitumorigenic activity, on conformational and functional aspects of the human 90kDa heat shock protein Hsp90alpha, a chaperone implicated in the stabilization of the tumor phenotype. Biochim. Et Biophys. Acta 2014, 1840, 3145–3152. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Wang, H.J.; Bao, Q.C.; Wang, L.; Jin, Y.H.; Zhang, Q.; Jiang, D.; You, Q.D.; Xu, X.L. Optimization and biological evaluation of celastrol derivatives as Hsp90-Cdc37 interaction disruptors with improved druglike properties. Bioorganic Med. Chem. 2016, 24, 5431–5439. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Xu, M.; Wang, B.; Shi, Z.; Zhao, Z.; Tang, Y.; Wang, X.; Sun, J.; Chen, L. Discovery of Novel Celastrol Derivatives as Hsp90-Cdc37 Interaction Disruptors with Antitumor Activity. J. Med. Chem. 2019, 62, 10798–10815. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Li, J.; Xu, Z.; Chen, L.; Luo, R.; Zhang, C.; Gao, F.; Zhang, J.; Fu, C. Celastrol: A Review of Useful Strategies Overcoming its Limitation in Anticancer Application. Front. Pharmacol. 2020, 11, 558741. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Fu, R.J.; Zhang, S.; Yue, S.J.; Chen, Y.Y.; Xu, D.Q.; Tang, Y.P. Potential medicinal value of celastrol and its synthesized analogues for central nervous system diseases. Biomed. Pharmacother. Biomed. Pharmacother. 2021, 139, 111551. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Hamza, A.; Zhang, T.; Gu, M.; Zou, P.; Newman, B.; Li, Y.; Gunatilaka, A.A.; Zhan, C.G.; Sun, D. Withaferin A targets heat shock protein 90 in pancreatic cancer cells. Biochem. Pharmacol. 2010, 79, 542–551. [Google Scholar] [CrossRef] [Green Version]
- Grover, A.; Shandilya, A.; Agrawal, V.; Pratik, P.; Bhasme, D.; Bisaria, V.S.; Sundar, D. Hsp90/Cdc37 chaperone/co-chaperone complex, a novel junction anticancer target elucidated by the mode of action of herbal drug Withaferin A. BMC Bioinform. 2011, 12 (Suppl. S1), S30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, M.; Yu, Y.; Gunaherath, G.M.; Gunatilaka, A.A.; Li, D.; Sun, D. Structure-activity relationship (SAR) of withanolides to inhibit Hsp90 for its activity in pancreatic cancer cells. Investig. New Drugs 2014, 32, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Behl, T.; Sharma, A.; Sharma, L.; Sehgal, A.; Zengin, G.; Brata, R.; Fratila, O.; Bungau, S. Exploring the Multifaceted Therapeutic Potential of Withaferin A and Its Derivatives. Biomedicines 2020, 8, 571. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Karagoz, G.E.; Seo, Y.H.; Zhang, T.; Jiang, Y.; Yu, Y.; Duarte, A.M.; Schwartz, S.J.; Boelens, R.; Carroll, K.; et al. Sulforaphane inhibits pancreatic cancer through disrupting Hsp90-p50(Cdc37) complex and direct interactions with amino acids residues of Hsp90. J. Nutr. Biochem. 2012, 23, 1617–1626. [Google Scholar] [CrossRef] [Green Version]
- Kim, J. Pre-Clinical Neuroprotective Evidences and Plausible Mechanisms of Sulforaphane in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 2929. [Google Scholar] [CrossRef] [PubMed]
- Schepici, G.; Bramanti, P.; Mazzon, E. Efficacy of Sulforaphane in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8637. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Ye, M.; Zhang, L.R.; Wu, Q.D.; Zhang, M.; Xu, J.H.; Zheng, W. FW-04-806 inhibits proliferation and induces apoptosis in human breast cancer cells by binding to N-terminus of Hsp90 and disrupting Hsp90-Cdc37 complex formation. Mol. Cancer 2014, 13, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, F.A.; Parkkola, H.; Vukic, V.; Oetken-Lindholm, C.; Jaiswal, A.; Kiriazis, A.; Pavic, K.; Aittokallio, T.; Salminen, T.A.; Abankwa, D. Novel Small Molecule Hsp90/Cdc37 Interface Inhibitors Indirectly Target K-Ras-Signaling. Cancers 2021, 13, 927. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, C.; Li, L.; Chen, S.; Wang, L.; Li, Q.; Wang, X.; Lei, X.; Shen, Z. Natural Product Kongensin A is a Non-Canonical HSP90 Inhibitor that Blocks RIP3-dependent Necroptosis. Cell Chem. Biol. 2016, 23, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Lam, H.C.; Lei, X. Dissecting Programmed Cell Death with Small Molecules. Acc. Chem. Res. 2020, 53, 1034–1045. [Google Scholar] [CrossRef]
- Li, T.; Chen, X.; Dai, X.Y.; Wei, B.; Weng, Q.J.; Chen, X.; Ouyang, D.F.; Yan, R.; Huang, Z.J.; Jiang, H.L.; et al. Novel Hsp90 inhibitor platycodin D disrupts Hsp90/Cdc37 complex and enhances the anticancer effect of mTOR inhibitor. Toxicol. Appl. Pharmacol. 2017, 330, 65–73. [Google Scholar] [CrossRef]
- Patel, D.; Shukla, S.; Gupta, S. Apigenin and cancer chemoprevention: Progress, potential and promise (review). Int. J. Oncol. 2007, 30, 233–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayoobi, F.; Shamsizadeh, A.; Fatemi, I.; Vakilian, A.; Allahtavakoli, M.; Hassanshahi, G.; Moghadam-Ahmadi, A. Bio-effectiveness of the main flavonoids of Achillea millefolium in the pathophysiology of neurodegenerative disorders- a review. Iran. J. Basic Med. Sci. 2017, 20, 604–612. [Google Scholar] [CrossRef]
- Zhao, M.; Ma, J.; Zhu, H.Y.; Zhang, X.H.; Du, Z.Y.; Xu, Y.J.; Yu, X.D. Apigenin inhibits proliferation and induces apoptosis in human multiple myeloma cells through targeting the trinity of CK2, Cdc37 and Hsp90. Mol. Cancer 2011, 10, 104. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Huang, R.; Huang, X.; Zhang, B.; Ji, M.; Wang, H. Discovery of 18beta-glycyrrhetinic acid conjugated aminobenzothiazole derivatives as Hsp90-Cdc37 interaction disruptors that inhibit cell migration and reverse drug resistance. Bioorganic Med. Chem. 2018, 26, 1759–1775. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bao, Q.-C.; Xu, X.-L.; Jiang, F.; Gu, K.; Jiang, Z.-Y.; Zhang, X.-J.; Guo, X.-K.; You, Q.-D.; Sun, H.-P. Discovery and identification of Cdc37-derived peptides targeting the Hsp90–Cdc37 protein–protein interaction. RSC Adv. 2015, 5, 96138–96145. [Google Scholar] [CrossRef]
- Wang, L.; Li, L.; Fu, W.T.; Jiang, Z.Y.; You, Q.D.; Xu, X.L. Optimization and bioevaluation of Cdc37-derived peptides: An insight into Hsp90-Cdc37 protein-protein interaction modulators. Bioorganic Med. Chem. 2017, 25, 233–240. [Google Scholar] [CrossRef] [PubMed]
- D’Annessa, I.; Hurwitz, N.; Pirota, V.; Beretta, G.L.; Tinelli, S.; Woodford, M.; Freccero, M.; Mollapour, M.; Zaffaroni, N.; Wolfson, H.; et al. Design of Disruptors of the Hsp90-Cdc37 Interface. Molecules 2020, 25, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Bernard, D.; Yu, Y.; Xie, Y.; Zhang, T.; Li, Y.; Burnett, J.P.; Fu, X.; Wang, S.; Sun, D. Split Renilla luciferase protein fragment-assisted complementation (SRL-PFAC) to characterize Hsp90-Cdc37 complex and identify critical residues in protein/protein interactions. J. Biol. Chem. 2010, 285, 21023–21036. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, L.; Zhou, Z.H.; Jiang, Z.Y.; You, Q.D.; Xu, X.L. Structure-based virtual screening and optimization of modulators targeting Hsp90-Cdc37 interaction. Eur. J. Med. Chem. 2017, 136, 63–73. [Google Scholar] [CrossRef]
- Chen, X.; Liu, P.; Wang, Q.; Li, Y.; Fu, L.; Fu, H.; Zhu, J.; Chen, Z.; Zhu, W.; Xie, C.; et al. DCZ3112, a novel Hsp90 inhibitor, exerts potent antitumor activity against HER2-positive breast cancer through disruption of Hsp90-Cdc37 interaction. Cancer Lett. 2018, 434, 70–80. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, L.; Li, L.; Jiang, J.; Zheng, Z.; Shang, J.; Wang, C.; Chen, W.; Bao, Q.; Xu, X.; et al. Small-molecule inhibitor targeting the Hsp90-Cdc37 protein-protein interaction in colorectal cancer. Sci. Adv. 2019, 5, eaax2277. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Jiang, J.; Zhang, L.; Zhang, Q.; Zhou, J.; Li, L.; Xu, X.; You, Q. Discovery and Optimization of Small Molecules Targeting the Protein-Protein Interaction of Heat Shock Protein 90 (Hsp90) and Cell Division Cycle 37 as Orally Active Inhibitors for the Treatment of Colorectal Cancer. J. Med. Chem. 2020, 63, 1281–1297. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, X.; Zhou, J.; Zhang, L.; Xu, X.; Zhang, L.; You, Q.; Wang, L. Design, synthesis and bioevaluation of inhibitors targeting HSP90-CDC37 protein-protein interaction based on a hydrophobic core. Eur. J. Med. Chem. 2021, 210, 112959. [Google Scholar] [CrossRef]
- Chen, B.; Wei, W.; Ma, L.; Yang, B.; Gill, R.M.; Chua, M.S.; Butte, A.J.; So, S. Computational Discovery of Niclosamide Ethanolamine, a Repurposed Drug Candidate That Reduces Growth of Hepatocellular Carcinoma Cells In Vitro and in Mice by Inhibiting Cell Division Cycle 37 Signaling. Gastroenterology 2017, 152, 2022–2036. [Google Scholar] [CrossRef]
- Park, J.S.; Lee, Y.S.; Lee, D.H.; Bae, S.H. Repositioning of niclosamide ethanolamine (NEN), an anthelmintic drug, for the treatment of lipotoxicity. Free Radic. Biol. Med. 2019, 137, 143–157. [Google Scholar] [CrossRef]
- Han, P.; Weng, W.; Chen, Y.; Cai, Y.; Wang, Y.; Wang, M.; Zhan, H.; Yuan, C.; Yu, X.; Shao, M.; et al. Niclosamide ethanolamine attenuates systemic lupus erythematosus and lupus nephritis in MRL/lpr mice. Am. J. Transl. Res. 2020, 12, 5015–5031. [Google Scholar]
- Li, S.L.; Yan, J.; Zhang, Y.Q.; Zhen, C.L.; Liu, M.Y.; Jin, J.; Gao, J.L.; Xiao, X.L.; Shen, X.; Tai, Y.; et al. Niclosamide ethanolamine inhibits artery constriction. Pharmacol. Res. 2017, 115, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, F.A.; Parkkola, H.; Manoharan, G.B.; Abankwa, D. Medium-Throughput Detection of Hsp90/Cdc37 Protein-Protein Interaction Inhibitors Using a Split Renilla Luciferase-Based Assay. SLAS Discov. Adv. Life Sci. R D 2020, 25, 195–206. [Google Scholar] [CrossRef]
- Panaretou, B.; Siligardi, G.; Meyer, P.; Maloney, A.; Sullivan, J.K.; Singh, S.; Millson, S.H.; Clarke, P.A.; Naaby-Hansen, S.; Stein, R.; et al. Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone aha1. Mol. Cell 2002, 10, 1307–1318. [Google Scholar] [CrossRef] [Green Version]
- Wolmarans, A.; Lee, B.; Spyracopoulos, L.; LaPointe, P. The Mechanism of Hsp90 ATPase Stimulation by Aha1. Sci. Rep. 2016, 6, 33179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, P.; Prodromou, C.; Liao, C.; Hu, B.; Roe, S.M.; Vaughan, C.K.; Vlasic, I.; Panaretou, B.; Piper, P.W.; Pearl, L.H. Structural basis for recruitment of the ATPase activator Aha1 to the Hsp90 chaperone machinery. EMBO J. 2004, 23, 1402–1410. [Google Scholar] [CrossRef] [Green Version]
- Koulov, A.V.; LaPointe, P.; Lu, B.; Razvi, A.; Coppinger, J.; Dong, M.Q.; Matteson, J.; Laister, R.; Arrowsmith, C.; Yates, J.R., 3rd; et al. Biological and structural basis for Aha1 regulation of Hsp90 ATPase activity in maintaining proteostasis in the human disease cystic fibrosis. Mol. Biol. Cell 2010, 21, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Retzlaff, M.; Hagn, F.; Mitschke, L.; Hessling, M.; Gugel, F.; Kessler, H.; Richter, K.; Buchner, J. Asymmetric activation of the hsp90 dimer by its cochaperone aha1. Mol. Cell 2010, 37, 344–354. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, M.; Myasnikov, A.G.; Elnatan, D.; Delaeter, N.; Nguyenquang, M.; Agard, D. Cryo-EM structures reveal a multistep mechanism of Hsp90 activation by co-chaperone Aha1. Biorxiv 2020. [Google Scholar] [CrossRef]
- Oroz, J.; Blair, L.J.; Zweckstetter, M. Dynamic Aha1 co-chaperone binding to human Hsp90. Protein Sci. A Publ. Protein Soc. 2019, 28, 1545–1551. [Google Scholar] [CrossRef] [Green Version]
- Ihrig, V.; Obermann, W.M.J. Identifying Inhibitors of the Hsp90-Aha1 Protein Complex, a Potential Target to Drug Cystic Fibrosis, by Alpha Technology. SLAS Discov. Adv. Life Sci. R D 2017, 22, 923–928. [Google Scholar] [CrossRef] [Green Version]
- Stiegler, S.C.; Rubbelke, M.; Korotkov, V.S.; Weiwad, M.; John, C.; Fischer, G.; Sieber, S.A.; Sattler, M.; Buchner, J. A chemical compound inhibiting the Aha1-Hsp90 chaperone complex. J. Biol. Chem. 2017, 292, 17073–17083. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.K.; Hutt, D.M.; Tait, B.; Guy, N.C.; Sivils, J.C.; Ortiz, N.R.; Payan, A.N.; Komaragiri, S.K.; Owens, J.J.; Culbertson, D.; et al. Management of Hsp90-Dependent Protein Folding by Small Molecules Targeting the Aha1 Co-Chaperone. Cell Chem. Biol. 2020, 27, 292–305.e296. [Google Scholar] [CrossRef]
- Ali, M.M.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Yamout, M.A.; Venkitakrishnan, R.P.; Preece, N.E.; Kroon, G.; Wright, P.E.; Dyson, H.J. Localization of sites of interaction between p23 and Hsp90 in solution. J. Biol. Chem. 2006, 281, 14457–14464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagoz, G.E.; Duarte, A.M.; Ippel, H.; Uetrecht, C.; Sinnige, T.; van Rosmalen, M.; Hausmann, J.; Heck, A.J.; Boelens, R.; Rudiger, S.G. N-terminal domain of human Hsp90 triggers binding to the cochaperone p23. Proc. Natl. Acad. Sci. USA 2011, 108, 580–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, S.H.; Sobott, F.; Yao, Z.P.; Zhang, W.; Nielsen, P.R.; Grossmann, J.G.; Laue, E.D.; Robinson, C.V.; Jackson, S.E. The co-chaperone p23 arrests the Hsp90 ATPase cycle to trap client proteins. J. Mol. Biol. 2006, 356, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Walter, S.; Buchner, J. The Co-chaperone Sba1 connects the ATPase reaction of Hsp90 to the progression of the chaperone cycle. J. Mol. Biol. 2004, 342, 1403–1413. [Google Scholar] [CrossRef]
- Rehn, A.B.; Buchner, J. p23 and Aha1. Sub-Cell. Biochem. 2015, 78, 113–131. [Google Scholar] [CrossRef]
- Chadli, A.; Felts, S.J.; Wang, Q.; Sullivan, W.P.; Botuyan, M.V.; Fauq, A.; Ramirez-Alvarado, M.; Mer, G. Celastrol inhibits Hsp90 chaperoning of steroid receptors by inducing fibrillization of the Co-chaperone p23. J. Biol. Chem. 2010, 285, 4224–4231. [Google Scholar] [CrossRef] [Green Version]
- Patwardhan, C.A.; Fauq, A.; Peterson, L.B.; Miller, C.; Blagg, B.S.; Chadli, A. Gedunin inactivates the co-chaperone p23 protein causing cancer cell death by apoptosis. J. Biol. Chem. 2013, 288, 7313–7325. [Google Scholar] [CrossRef] [Green Version]
- Gorantla, N.V.; Das, R.; Chidambaram, H.; Dubey, T.; Mulani, F.A.; Thulasiram, H.V.; Chinnathambi, S. Basic Limonoid modulates Chaperone-mediated Proteostasis and dissolve Tau fibrils. Sci. Rep. 2020, 10, 4023. [Google Scholar] [CrossRef] [Green Version]
- Tom, S.; Rane, A.; Katewa, A.S.; Chamoli, M.; Matsumoto, R.R.; Andersen, J.K.; Chinta, S.J. Gedunin Inhibits Oligomeric Abeta1-42-Induced Microglia Activation Via Modulation of Nrf2-NF-kappaB Signaling. Mol. Neurobiol. 2019, 56, 7851–7862. [Google Scholar] [CrossRef]
- Yang, W.; Xie, J.; Qiang, Q.; Li, L.; Lin, X.; Ren, Y.; Ren, W.; Liu, Q.; Zhou, G.; Wei, W.; et al. Gedunin Degrades Aggregates of Mutant Huntingtin Protein and Intranuclear Inclusions via the Proteasomal Pathway in Neurons and Fibroblasts from Patients with Huntington’s Disease. Neurosci. Bull. 2019, 35, 1024–1034. [Google Scholar] [CrossRef]
- Nie, S.; Xu, Y.; Chen, G.; Ma, K.; Han, C.; Guo, Z.; Zhang, Z.; Ye, K.; Cao, X. Small molecule TrkB agonist deoxygedunin protects nigrostriatal dopaminergic neurons from 6-OHDA and MPTP induced neurotoxicity in rodents. Neuropharmacology 2015, 99, 448–458. [Google Scholar] [CrossRef]
- Hall, J.A.; Seedarala, S.; Rice, N.; Kopel, L.; Halaweish, F.; Blagg, B.S. Cucurbitacin D Is a Disruptor of the HSP90 Chaperone Machinery. J. Nat. Prod. 2015, 78, 873–879. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Peng, S.; Wang, J.; Chen, H.; Cong, X.; Chen, A.; Hu, M.; Qin, M.; Wu, H.; Gao, S.; et al. Ailanthone targets p23 to overcome MDV3100 resistance in castration-resistant prostate cancer. Nat. Commun. 2016, 7, 13122. [Google Scholar] [CrossRef]
- Chan, C.T.; Reeves, R.E.; Geller, R.; Yaghoubi, S.S.; Hoehne, A.; Solow-Cordero, D.E.; Chiosis, G.; Massoud, T.F.; Paulmurugan, R.; Gambhir, S.S. Discovery and validation of small-molecule heat-shock protein 90 inhibitors through multimodality molecular imaging in living subjects. Proc. Natl. Acad. Sci. USA 2012, 109, E2476–E2485. [Google Scholar] [CrossRef] [Green Version]
- Jensen, M.R.; Schoepfer, J.; Radimerski, T.; Massey, A.; Guy, C.T.; Brueggen, J.; Quadt, C.; Buckler, A.; Cozens, R.; Drysdale, M.J.; et al. NVP-AUY922: A small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Res. BCR 2008, 10, R33. [Google Scholar] [CrossRef] [Green Version]
- Menezes, D.L.; Taverna, P.; Jensen, M.R.; Abrams, T.; Stuart, D.; Yu, G.K.; Duhl, D.; Machajewski, T.; Sellers, W.R.; Pryer, N.K.; et al. The novel oral Hsp90 inhibitor NVP-HSP990 exhibits potent and broad-spectrum antitumor activities in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 730–739. [Google Scholar] [CrossRef] [Green Version]
- Eachkoti, R.; Reddy, M.V.; Lieu, Y.K.; Cosenza, S.C.; Reddy, E.P. Identification and characterisation of a novel heat shock protein 90 inhibitor ONO4140. Eur. J. Cancer 2014, 50, 1982–1992. [Google Scholar] [CrossRef] [Green Version]
- Xue, N.; Jin, J.; Liu, D.; Yan, R.; Zhang, S.; Yu, X.; Chen, X. Antiproliferative effect of HSP90 inhibitor Y306zh against pancreatic cancer is mediated by interruption of AKT and MAPK signaling pathways. Curr. Cancer Drug Targets 2014, 14, 671–683. [Google Scholar] [CrossRef]
- Blatch, G.L.; Lassle, M. The tetratricopeptide repeat: A structural motif mediating protein-protein interactions. BioEssays News Rev. Mol. Cell. Dev. Biol. 1999, 21, 932–939. [Google Scholar] [CrossRef]
- Scheufler, C.; Brinker, A.; Bourenkov, G.; Pegoraro, S.; Moroder, L.; Bartunik, H.; Hartl, F.U.; Moarefi, I. Structure of TPR domain-peptide complexes: Critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 2000, 101, 199–210. [Google Scholar] [CrossRef]
- Schmid, A.B.; Lagleder, S.; Grawert, M.A.; Rohl, A.; Hagn, F.; Wandinger, S.K.; Cox, M.B.; Demmer, O.; Richter, K.; Groll, M.; et al. The architecture of functional modules in the Hsp90 co-chaperone Sti1/Hop. EMBO J. 2012, 31, 1506–1517. [Google Scholar] [CrossRef] [Green Version]
- Lackie, R.E.; Maciejewski, A.; Ostapchenko, V.G.; Marques-Lopes, J.; Choy, W.Y.; Duennwald, M.L.; Prado, V.F.; Prado, M.A.M. The Hsp70/Hsp90 Chaperone Machinery in Neurodegenerative Diseases. Front. Neurosci. 2017, 11, 254. [Google Scholar] [CrossRef] [Green Version]
- Maciejewski, A.; Ostapchenko, V.G.; Beraldo, F.H.; Prado, V.F.; Prado, M.A.; Choy, W.Y. Domains of STIP1 responsible for regulating PrPC-dependent amyloid-beta oligomer toxicity. Biochem. J. 2016, 473, 2119–2130. [Google Scholar] [CrossRef]
- Yi, F.; Regan, L. A novel class of small molecule inhibitors of Hsp90. ACS Chem. Biol. 2008, 3, 645–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pimienta, G.; Herbert, K.M.; Regan, L. A compound that inhibits the HOP-Hsp90 complex formation and has unique killing effects in breast cancer cell lines. Mol. Pharm. 2011, 8, 2252–2261. [Google Scholar] [CrossRef] [PubMed]
- Horibe, T.; Kohno, M.; Haramoto, M.; Ohara, K.; Kawakami, K. Designed hybrid TPR peptide targeting Hsp90 as a novel anticancer agent. J. Transl. Med. 2011, 9, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, U.K.; Mahanta, S.; Paul, S. In silico design of small peptide-based Hsp90 inhibitor: A novel anticancer agent. Med Hypotheses 2013, 81, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, Y.; Zhao, Z.; Xie, C.; Xu, Y.; Hu, Y.; Quan, H.; Lou, L. Y-632 inhibits heat shock protein 90 (Hsp90) function by disrupting the interaction between Hsp90 and Hsp70/Hsp90 organizing protein, and exerts antitumor activity in vitro and in vivo. Cancer Sci. 2016, 107, 782–790. [Google Scholar] [CrossRef] [Green Version]
- Darby, J.F.; Vidler, L.R.; Simpson, P.J.; Al-Lazikani, B.; Matthews, S.J.; Sharp, S.Y.; Pearl, L.H.; Hoelder, S.; Workman, P. Solution structure of the Hop TPR2A domain and investigation of target druggability by NMR, biochemical and in silico approaches. Sci. Rep. 2020, 10, 16000. [Google Scholar] [CrossRef]
- Kang, H.; Sayner, S.L.; Gross, K.L.; Russell, L.C.; Chinkers, M. Identification of amino acids in the tetratricopeptide repeat and C-terminal domains of protein phosphatase 5 involved in autoinhibition and lipid activation. Biochemistry 2001, 40, 10485–10490. [Google Scholar] [CrossRef]
- Wandinger, S.K.; Suhre, M.H.; Wegele, H.; Buchner, J. The phosphatase Ppt1 is a dedicated regulator of the molecular chaperone Hsp90. EMBO J. 2006, 25, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Soroka, J.; Wandinger, S.K.; Mausbacher, N.; Schreiber, T.; Richter, K.; Daub, H.; Buchner, J. Conformational switching of the molecular chaperone Hsp90 via regulated phosphorylation. Mol. Cell 2012, 45, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Ballinger, C.A.; Connell, P.; Wu, Y.; Hu, Z.; Thompson, L.J.; Yin, L.Y.; Patterson, C. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol. 1999, 19, 4535–4545. [Google Scholar] [CrossRef] [Green Version]
- Edkins, A.L. CHIP: A co-chaperone for degradation by the proteasome. Sub-Cell. Biochem. 2015, 78, 219–242. [Google Scholar] [CrossRef]
- Storer, C.L.; Dickey, C.A.; Galigniana, M.D.; Rein, T.; Cox, M.B. FKBP51 and FKBP52 in signaling and disease. Trends Endocrinol. Metab. TEM 2011, 22, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambraud, B.; Daguinot, C.; Guillemeau, K.; Genet, M.; Dounane, O.; Meduri, G.; Pous, C.; Baulieu, E.E.; Giustiniani, J. Decrease of neuronal FKBP4/FKBP52 modulates perinuclear lysosomal positioning and MAPT/Tau behavior during MAPT/Tau-induced proteotoxic stress. Autophagy 2021, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Zeke, T.; Morrice, N.; Vazquez-Martin, C.; Cohen, P.T. Human protein phosphatase 5 dissociates from heat-shock proteins and is proteolytically activated in response to arachidonic acid and the microtubule-depolymerizing drug nocodazole. Biochem. J. 2005, 385, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Ardi, V.C.; Alexander, L.D.; Johnson, V.A.; McAlpine, S.R. Macrocycles that inhibit the binding between heat shock protein 90 and TPR-containing proteins. ACS Chem. Biol. 2011, 6, 1357–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConnell, J.R.; Alexander, L.A.; McAlpine, S.R. A heat shock protein 90 inhibitor that modulates the immunophilins and regulates hormone receptors without inducing the heat shock response. Bioorganic Med. Chem. Lett. 2014, 24, 661–666. [Google Scholar] [CrossRef] [Green Version]
- Koay, Y.C.; McConnell, J.R.; Wang, Y.; Kim, S.J.; Buckton, L.K.; Mansour, F.; McAlpine, S.R. Chemically accessible hsp90 inhibitor that does not induce a heat shock response. ACS Med. Chem. Lett. 2014, 5, 771–776. [Google Scholar] [CrossRef]
- Rahimi, M.N.; McAlpine, S.R. Protein-protein inhibitor designed de novo to target the MEEVD region on the C-terminus of Hsp90 and block co-chaperone activity. Chem. Commun. 2019, 55, 846–849. [Google Scholar] [CrossRef]
- Pavlov, P.F.; Hutter-Paier, B.; Havas, D.; Windisch, M.; Winblad, B. Development of GMP-1 a molecular chaperone network modulator protecting mitochondrial function and its assessment in fly and mice models of Alzheimer’s disease. J. Cell. Mol. Med. 2018, 22, 3464–3474. [Google Scholar] [CrossRef]
- Kumar, R.; Winblad, B.; Pavlov, P.F. Hsp90 as a Member of Dicarboxylate Clamp TPR Protein Interaction Network: Implication in Human Diseases and Prospect as a Drug Target. In Heat Shock Protein 90 in Human Diseases and Disorders; Kaur, P., Asea, A.A.A., Eds.; Springer: Cham, Switzerland, 2019; pp. 281–295. [Google Scholar]
- Kumar, R.; Moche, M.; Winblad, B.; Pavlov, P.F. Combined x-ray crystallography and computational modeling approach to investigate the Hsp90 C-terminal peptide binding to FKBP51. Sci. Rep. 2017, 7, 14288. [Google Scholar] [CrossRef] [Green Version]
- Bernadotte, A.; Kumar, R.; Winblad, B.; Pavlov, P.F. In silico identification and biochemical characterization of the human dicarboxylate clamp TPR protein interaction network. FEBS Open Bio. 2018, 8, 1830–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Kolos, J.M.; Voll, A.M.; Bauder, M.; Hausch, F. FKBP Ligands-Where We Are and Where to Go? Front. Pharmacol. 2018, 9, 1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Bergkvist, L.; Kumar, R.; Winblad, B.; Pavlov, P.F. Studies of Chaperone-Cochaperone Interactions using Homogenous Bead-Based Assay. J. Vis. Exp. JoVE 2021. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
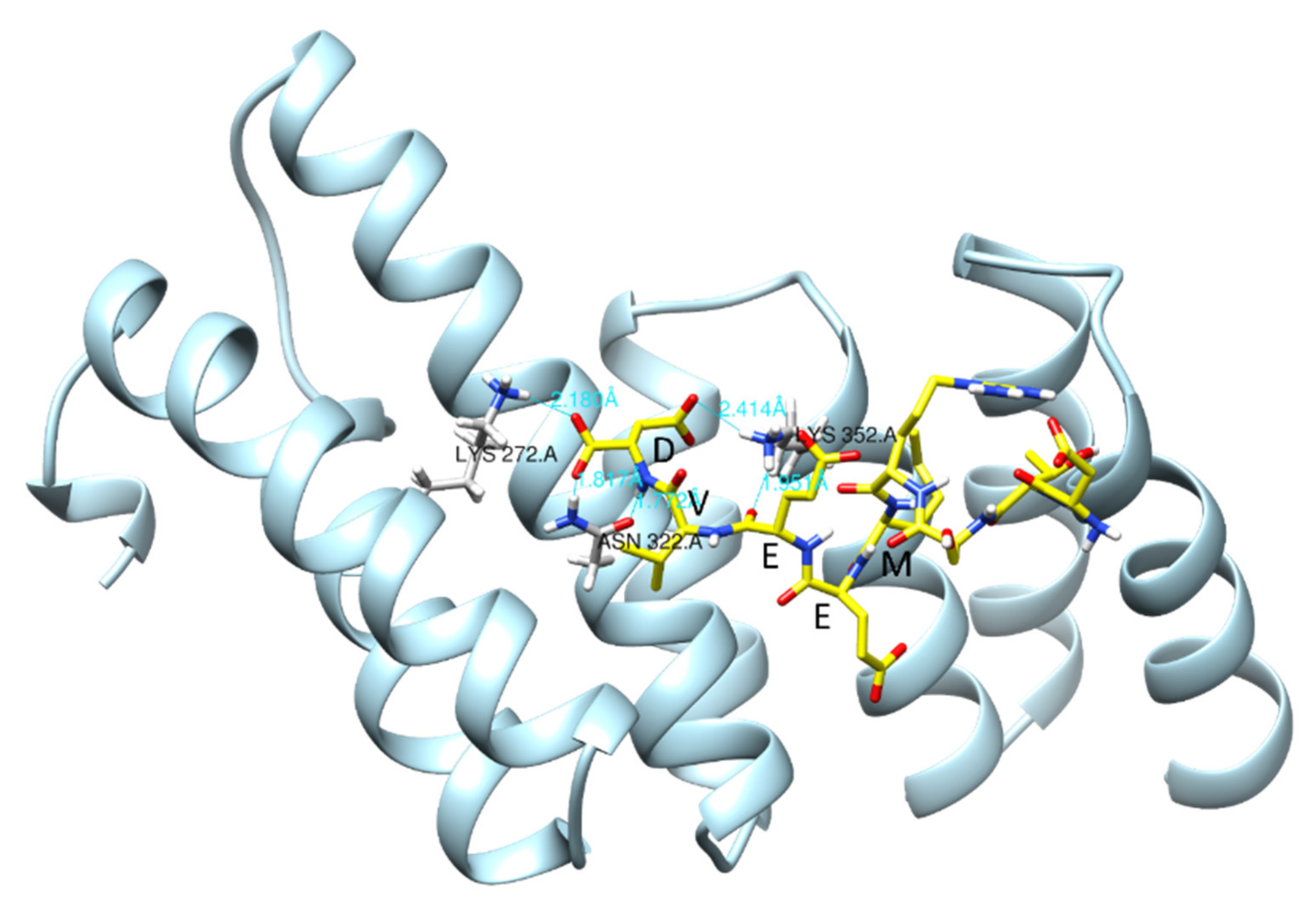{kind=link}
| Co-Chaperone | Full Name | Interacting Domain in Co-chaperone | Binding Site in HSP90 | Function | Disease | Cellular Processes |
|---|---|---|---|---|---|---|
| CDC37 | Cell division cycle 37 | MD, NTD | NTD, MD | Prevents closure of the “lid” in HSP90;Specific for maturation of kinases | AD, PD, ALS, FTLD | Stabilizes tau via Hsp90 and regulates the stability of distinct tau kinases, specifically Cdk5 and Akt [31]; Preserves TDP-43 [32]; Its client kinases include DYRK1A [33] Stabilizes LRRK2 [34]; Stabilizes PINK1 and influences its subcellular distribution [35,36] |
| Aha1 | Activator of Hsp90 ATPase homolog 1 | NTD, CTD | NTD, MD | Stimulates ATPase activity of HSP90 | AD | Increases tau fibril formation, Aha1 overexpression in rTg4510 mouse increases tau accumulation, leading to both neuron loss and cognitive deficits [37]. Aha1 overexpression in aged wild-type mice impairs associative learning and promotes tau phosphorylation [38]. |
| p23 (Sba1 in yeast) | Co-chaperone p23 | NTD | NTD, MD | Stabilizes the HSP90 closed 2 state; Inhibits Hsp90 ATPase activity | AD, PD | Knockdown of p23 reduces both total and phosphorylated tau levels [39]. Contributes to neurotoxicity in PD [40]. |
| Hop (Sti1) | Hsp70-Hsp90 organizing protein (stress-inducible phosphoprotein 1) | TPR | CTD, MD | Transfers clients from Hsp70 to Hsp90; Inhibits Hsp90 ATPase activity | HD, AD, Prion diseases | Hop overexpression in yeast inhibits the toxicity of HTT103Q and reorganizes small HTT103Q foci into larger assemblies [41]. Hop downregulation enhances tau toxicity in the fly model of tauopathy [42]. Binds to PrPC and promotes calcium influx through α7nAChRs [43]. Inhibits Aβ oligomers’ binding to PrPC and prevents synaptic loss, neuronal death, and depression of long-term potentiation induced by Aβ oligomers [44]. |
| PP5 (Ppt1 in yeast) | Protein phosphatase 5 | TPR | CTD | Dephosphorylates Hsp90; Dephosphorylates Cdc37 | AD | Dephosphorylates tau and its activity decreases in AD neocortex [45]. Protects primary neuron death induced by Aβ [46]. |
| CHIP | C terminus of Hsp70-interacting protein | TPR | CTD | E3 ubiquitin ligase | AD, PD, HD | Promotes the degradation of phosphorylated tau [39,47]. Reduces α-synuclein oligomerization and mediates α-synuclein degradation [48,49]. Reduces the uptake of α-synuclein fibrils by neuro-2a cells [50]. Promotes the degradation of LRRK2 [51,52]. CHIP overexpression promotes the degradation of polyglutamine-expanded HTT or ataxin-3 [53]. Suppresses polyglutamine aggregation and toxicity [54]. |
| FKBP51 | FK506 binding protein 51 kDa | TPR | CTD | Peptidyl-prolyl isomerase activity; Participates in Hsp90-steroid receptor complex; Generally regulates Hsp90 conformational cycle | AD, PD, HD | Enhances the production of tau oligomers and prevents tau degradation [55]. Increases with age in the mouse brain, and its expression is higher in AD patients [55,56]. Involved in Pink1′s regulation of AKT on neuronal survival [57]. FKBP51 downregulation reduces mutant HTT levels in HD models both in vitro and in vivo [58]. |
| FKBP52 | FK506 binding protein 52 kDa | TPR | CTD | Peptidyl-prolyl isomerase activity; Participates in Hsp90-steroid receptor complex; Generally regulates Hsp90 conformational cycle | AD, PD | Induces aggregation of multiple tau species in vitro [59,60,61]. FKBP52 overexpression in the hippocampus leads to cognitive impairments and neurotoxicity in aged wild-type mice and rTg4510 transgenic mice [38,62]. FKBP52 levels are abnormally low in the frontal cortex of AD brains [63]. Suppresses Aβ toxicity and increases the lifespan of Drosophila, which expresses Aβ peptides [64]. Accelerates α-synuclein aggregation and neuronal cell death [65]. Generates immune responses to α-synuclein-based immunizations in mice [66]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Bergkvist, L.; Kumar, R.; Winblad, B.; Pavlov, P.F. Targeting Chaperone/Co-Chaperone Interactions with Small Molecules: A Novel Approach to Tackle Neurodegenerative Diseases. Cells 2021, 10, 2596. https://doi.org/10.3390/cells10102596
Wang L, Bergkvist L, Kumar R, Winblad B, Pavlov PF. Targeting Chaperone/Co-Chaperone Interactions with Small Molecules: A Novel Approach to Tackle Neurodegenerative Diseases. Cells. 2021; 10(10):2596. https://doi.org/10.3390/cells10102596
Chicago/Turabian StyleWang, Lisha, Liza Bergkvist, Rajnish Kumar, Bengt Winblad, and Pavel F. Pavlov. 2021. "Targeting Chaperone/Co-Chaperone Interactions with Small Molecules: A Novel Approach to Tackle Neurodegenerative Diseases" Cells 10, no. 10: 2596. https://doi.org/10.3390/cells10102596